

SRC-1 proteolysis is increased by progestin agonist ligand during Progesterone Receptor-mediated transcriptional activation.
Abstract
The progesterone receptor (PR), a ligand-activated transcription factor, recruits the primary coactivator steroid receptor coactivator-1 (SRC-1) gene promoters. It is known that PR transcriptional activity is paradoxically coupled to its ligand-dependent down-regulation. However, despite its importance in PR function, the regulation of SRC-1 expression level during hormonal exposure is poorly understood. Here we report that SRC-1 expression level (but not other p160 family members) is down-regulated by the agonist ligand R5020 in a PR-dependent manner. In contrast, the antagonist RU486 fails to induce down-regulation of the coactivator and impairs PR agonist-dependent degradation of SRC-1. We show that SRC-1 proteolysis is a proteasome- and ubiquitin-mediated process that, predominantly but not exclusively, occurs in the cytoplasmic compartment in which SRC-1 colocalizes with proteasome antigens as demonstrated by confocal imaging. Moreover, SRC-1 was stabilized in the presence of leptomycin B or several proteasomal inhibitors. Two degradation motifs, amino-acids 2–16 corresponding to a PEST motif and amino acids 41–136 located in the basic helix loop helix domain of the coactivator, were identified and shown to control the stability as well as the hormone-dependent down-regulation of the coactivator. SRC-1 degradation is of physiological importance because the two nondegradable mutants that still interacted with PR as demonstrated by coimmunoprecipitation failed to stimulate transcription of exogenous and endogenous target genes, suggesting that concomitant PR/SRC-1 ligand-dependent degradation is a necessary step for PR transactivation activity. Collectively our findings are consistent with the emerging role of proteasome-mediated proteolysis in the gene-regulating process and indicate that the ligand-dependent down-regulation of SRC-1 is critical for PR transcriptional activity.
The progesterone receptor (PR), also known as NR3C3, plays a crucial role in the coordination of several aspects of female reproductive development and function (1). Invalidation of the PR gene in mice leads to pleiotropic reproductive abnormalities and demonstrates that PR orchestrates key events associated with the establishment and maintenance of pregnancy. From a pathophysiological perspective, accumulating evidence indicates that PR is involved in breast cancer cells proliferation and is implicated in the development and progression of breast cancer (2). Coregulators (coactivators or corepressors) are important nuclear receptor (NR)-recruited cofactors modulating NR-mediated transcription and leading to activation or repression of target specific genes (3). Steroid receptor coactivator-1 (SRC-1) is a PR coactivator belonging to the p160 gene family, which contains three homologous members (SRC-1, -2, and -3) serving as NR transcriptional coactivators (4). This family of coactivators is characterized by the presence of several conserved functional domains: a basic helix-loop-helix (bHLH)-Per-ARNT-Sim N-terminal domain, a cAMP response element-binding protein (CBP) interacting domain (AD1), a glutamine-rich region, a C-terminal activation domain (AD2), and several LXXLL boxes involved in NR binding. The p160 coactivators are defined as primary coactivators whose activity is regulated by posttranslational modifications (5–10). The current models indicate that p160 coactivators serve as a recruitment platform for other coactivator complexes carrying intrinsic enzymatic activities to specific enhancers/promoters resulting in the covalent modification of specific histones and/or other coregulators involved in the transcriptional machinery (11, 12).
Several experiments have revealed a tight association between the turnover rate of several NR and their transcriptional activity, showing that both aspects of NR function appear to be inversely related (13–18). Among the factors regulating PR levels are its ligands. It was initially shown that administration of progesterone to ovariectomized guinea pigs provoked a rapid fall in uterine receptor concentration (19). Hormone-dependent down-regulation of PR has been finally confirmed by several groups (20–22), but its biological significance is still unclear. Phosphorylation of PR on a key serine residue (Ser294) by MAPKs was shown to couple multiple receptor functions, including ligand-dependent PR down-regulation by the ubiquitin-proteasome pathway (13). The concept that transcriptional activation and ubiquitin-mediated proteolysis are interdependent processes is emerging as a potentially important control mechanism of transcription (16, 23). Although their significance remains to be defined, it appears that complex interactions between regulatory molecules governing both transcription and ubiquitination/degradation exist (24–26). However, little is known concerning the fate of coregulators during ligand-dependent NR down-regulation (27, 28).
In a previous study, we have shown that SRC-1 is exported from the nucleus to the cytoplasm and speculated that this export might be a regulatory mechanism controlling the termination of hormone action, possibly through its degradation (29). To establish a link between SRC-1 proteolysis and the PR-mediated transcription process, we studied the mechanism governing SRC-1 proteolysis at the steady-state level and questioned whether the ligand could modulate its turnover. In this study, we demonstrate that SRC-1 undergoes covalent modifications by ubiquitin, which targets the coactivator to the proteasome at the steady-state level. We identify two critical degron domains directly linked to the coactivator proteolysis. Aside from this ligand-independent stability regulation, we show that SRC-1 undergoes accelerated agonist-dependent and PR-mediated down-regulation via the ubiquitin-proteasome pathway. SRC-1 proteolysis occurs concomitantly of ligand-dependent PR degradation. Of note, the nature of the ligand is shown to be critical for this process because both PR and SRC-1 ligand-dependent proteolysis was inhibited in the presence of RU486, leading to dramatic loss of PR-transactivating capability.
Results
SRC-1 mainly colocalizes with cytoplasmic proteasome antigens
In our previous report about the regulatory mechanisms of SRC-1 subcellular trafficking, we have shown that SRC-1 localizes both in nuclear and cytoplasmic corpuscular structures (29). Several studies have reported coregulators localization in organelles (30, 31). We tried to identify the nature of these cytoplasmic and nuclear speckles by colocalization studies with various antigens and with fluorescent organelles markers. Because several NRs and coactivators such as SRC-3 have been shown to interact with the proteasome (32, 33), we used confocal microscopy to investigate whether proteasome components might also accumulate in SRC-1 speckles. By using antibodies directed against the human S7 subunit of the 19S (Rpt1) and the α/β-subunits of the 20S proteasome, we found that SRC-1 colocalized with both 26S proteasome antigens (Fig. 1A and Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). The fluorescence intensity profile indicates that colocalization was predominant in SRC-1 speckles: simultaneous fluorescence intensity increase was observed in cytoplasmic speckles but also in lesser extent in nuclear speckles (Fig. 1B), suggesting that SRC-1 is mainly but not exclusively proteolyzed in the cytoplasm. Similar intensity profiles were obtained for cells immunolabeled for SRC-1 and the 20S proteasome (data not shown). A partial colocalization of SRC-1 was also found with the promyelocytic leukemia protein in the typical nuclear domain (nuclear domain 10) (Supplemental Fig. 2A). Such an association has been already described (34). In contrast, nuclear speckles did not overlap with transcription sites as evidenced by the absence of colocalization with the SC-35/SRp30 spliceosome component (Supplemental Fig. 2B). Similarly, no colocalization of SRC-1 with organelles such as mitochondria, lysosomes, peroxisomes, or the Golgi apparatus could be observed (Supplemental Fig. 2, C–E, and data not shown).
Fig. 1.
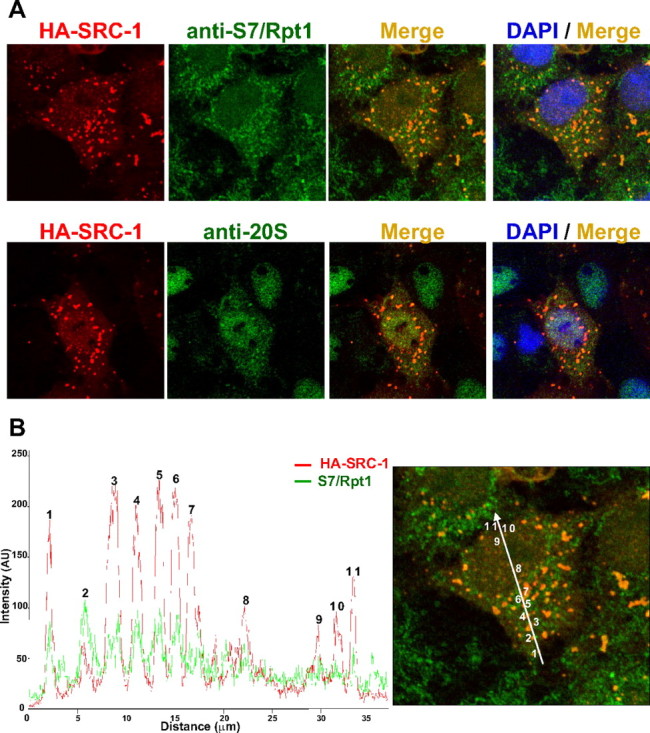
Colocalization of SRC-1 with the 26S proteasome by confocal microscopy. A, Colocalization analysis between HA-SRC-1 and endogenous proteasome antigens S7/Rpt1 and 20S subunits. COS-7 cells were transiently transfected with the expression vector encoding HA-SRC-1. Cells were fixed after 40 h, immunolabeled with anti-HA and either anti-S7/Rpt1 or anti-20S antibodies, and then observed by confocal microscopy. B, Validation of colocalization by scan of intensity profiles of a representative cell [expressed as arbitrary units (AU)]. Fluorescence intensity was calculated and plotted by drawing a line through the middle of the cell image in a distance covering several cytosolic and nuclear foci. Green lines represent the intensity profile for the proteasome antigen S7/Rpt1 signal, and the red lines represent the intensity profile for SRC-1 signal. Indicated numbers refer to identified speckles: cytoplasmic (1 to 7) or nuclear (8 to 11). Note that although the fluorescence intensity from the two channels is different, the peaks of both signals are overlapping.
SRC-1 is ubiquitinylated in vivo and is degraded by the proteasome
We next studied the mechanism of SRC-1 down-regulation. First, we investigated whether the coactivator was ubiquitinylated and targeted to the proteasome. COS-7 cells were transfected with the expression vector encoding the full-length SRC-1 and incubated in the presence of proteasome inhibitors, MG132, or epoxomicin. Consistent with previous reports (14, 35), both inhibitors increased SRC-1 protein level in comparison with cells treated with vehicle (Fig. 2A and Supplemental Fig. 3). To demonstrate that SRC-1 is polyubiquitinylated, COS-7 cells were transfected with SRC-1 expression vector in the presence or absence of a vector encoding His-tagged ubiquitin (His 6-Ub) and analyzed by Western blot. In the absence of His 6-Ub, the anti-SRC-1 antibody detected a major band of approximately 160 kDa (Fig. 2B, left panel, lane 1). In cells cotransfected with His 6-Ub expression vector, a moderate decrease in band intensity was observed with a slightly higher molecular weight smear, indicative of ubiquitinylated moieties (Fig. 2B, left panel, lane 2). His-tagged proteins were purified by chromatography on nickel-charged agarose beads (Ni-NTA) and analyzed by Western blot with an anti-SRC-1 antibody to show that these bands correspond to ubiquitinylated SRC-1 (Fig. 2B, right panel, lane 2).
Fig. 2.
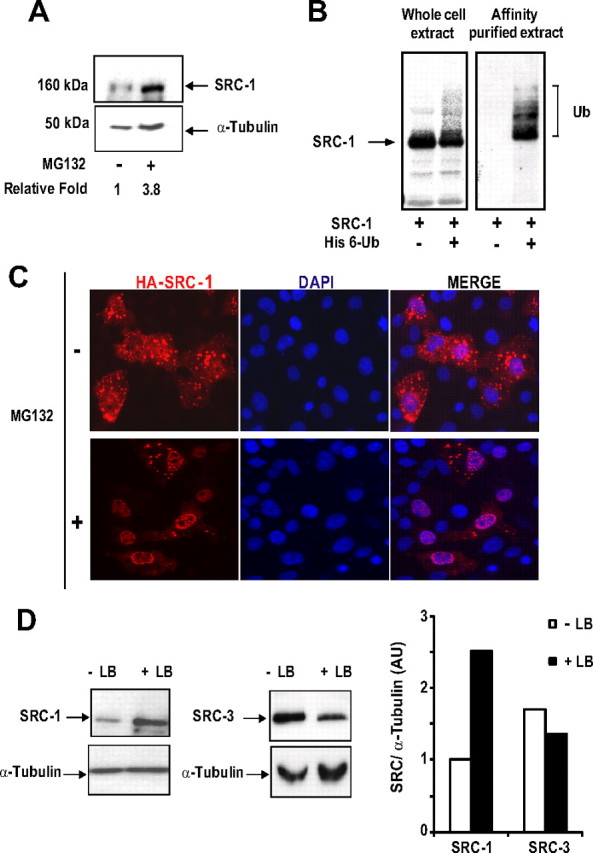
SRC-1 is proteolyzed by the 26S proteasome in a ubiquitin-dependent manner. A, COS-7 cells were transfected with the expression vector encoding SRC-1 and incubated in the absence or presence of MG132 (5 μm) during 15 h. Expression of SRC-1 was analyzed by Western blot using anti-SRC-1 and anti-α-tubulin antibodies. Band intensities corresponding to SRC-1 were quantified as described in Materials and Methods. B, CV-1 cells were transfected with the expression vector encoding HA-SRC-1 in the presence of the His6-tagged ubiquitin expression vector (His 6-Ub). Whole-cell extracts were analyzed by electrophoresis on 6.4% SDS-PAGE and immunoblotted with anti-HA monoclonal antibody. Alternatively, the same cotransfected CV-1 cells were lysed in buffer containing guanidium-HCl (Ni-NTA). The ubiquitin-modified proteins were purified using Ni-NTA agarose beads as described in Materials and Methods. Affinity-purified proteins were separated by electrophoresis, and His6-SRC-1 conjugates were detected by Western blot using the anti-HA monoclonal antibody. The ubiquitin conjugates of SRC-1 are indicated with brackets. C, COS-7 cells were transfected with the expression vector encoding HA-SRC-1. Twenty hours after transfection, cells were incubated during 24 h with MG132 (1 μm) or treated with vehicle (dimethylsulfoxide). Cells were then fixed and immunolabeled with anti-HA antibody. D, COS-7 cells were transfected with the expression vector encoding SRC-1 or SRC-3 and treated similarly than in C except that MG132 was replaced by LB treatment (20 ng/ml). Expression levels of SRC-1 and SRC-3 were analyzed by Western blot using anti-SRC-1 or anti-SRC-3 monoclonal antibodies as indicated. Band intensities representing the mean of at least two independent experiments were quantified as described in Materials and Methods.
Several cytoplasmic proteasome substrates have been shown to relocalize into the nucleus upon stabilization by proteasome inhibitors (36–38). We thus examined whether SRC-1 subcellular distribution was similarly modified in such conditions. Indeed, overnight treatment of cells with MG132 induces an obvious shift of the coactivator into the nucleus (Fig. 2C). This result suggests that escape from cytoplasmic proteolytic degradation stimulates the nuclear accumulation of SRC-1 (36). If our hypothesis is true, then inhibition of SRC-1 nuclear export should induce SRC-1 expression level stabilization. To verify this point, we followed the turnover rate of SRC-1Δ(NES), a mutant deleted of its nuclear export signal (NES) (29). The result shows a better stability of this mutant compared with the wild-type (wt) SRC-1 (Supplemental Fig. 4). In a similar approach, we used the nuclear export inhibitor leptomycin B (LB) to impede wt SRC-1 access to cytoplasm. In the presence of LB, SRC-1 not only relocalized into the nucleus (data not shown and Ref. 29), but its expression level also increased approximately 2.5 fold (Fig. 2D). However, SRC-1 stabilization with LB did not reach the level obtained with MG132 (data not shown; and compare quantification in Fig. 2A with Fig. 2D). Thus, the nuclear accumulation of the coactivator indicates a possibility of a partial degradation of SRC-1 in the nuclear compartment. Interestingly, similar experiments with the p160 coactivator SRC-3, which has been shown to be degraded mainly in the nucleus (39), showed no significant increase of SRC-3 expression level under LB treatment (Fig. 2D). Overall, our data show that SRC-1 turnover is a proteasome- and ubiquitin-mediated process that takes place, predominantly but not exclusively, in the cytoplasm.
Agonist ligand enhances concomitant proteolysis of PR and SRC-1
We next studied SRC-1 degradation in the context of PR activation. Progestins are known to induce PR proteolysis by the proteasome (22, 40). In addition, Li et al. (41) have shown that upon ligand treatment, PR preferentially interacts with SRC-1. We thus investigated whether SRC-1 down-regulation might be also modulated by PR ligands. As previously reported (22), immunocytochemical studies (Fig. 3A) and Western blot experiments (Supplemental Fig. 5) showed that the agonist ligand R5020 stimulates stably expressed endogenous PR proteolysis after 24 h treatment, whereas the antagonist ligand RU486 prevents PR proteolysis in Ishikawa cells stably expressing PR-B (Ishi-PR-B). To test the impact of ligands on SRC-1 expression level, Ishi-PR-B cells were transiently transfected with a SRC-1 expression vector and incubated overnight with R5020 or RU486. Western blot analyses revealed that SRC-1 and PR are concomitantly degraded in the presence of agonist R5020 and that RU486 prevents the degradation of both proteins (Fig. 3B). Similar results were obtained using different Ishi-PR-B subclones (data not shown). Real-time quantitative RT-PCR excluded the possibility of any ligand-dependent down-regulation of SRC-1 mRNA levels (Supplemental Fig. 6). MG132 exposure inhibited the agonist-dependent proteolysis of SRC-1 (Fig. 3B, lane 4), indicating that this stimulated down-regulation is mediated by the proteasome. Importantly, using antibodies specifically detecting endogenous SRC-1, we similarly observed agonist-dependent degradation of endogenous SRC-1 in Ishi-PR-B cells (Fig. 3, C and D). Of note, a 10-fold excess of antiprogestin RU486 abrogated the R5020-dependent degradation of endogenous SRC-1 and PR as shown in Fig. 3D (third lane), suggesting that SRC-1 degradation is tightly linked to the ligand-dependent PR activation. To further verify this hypothesis, we tested whether SRC-1 proteolysis could be stimulated in the absence of PR. We used the Ishikawa parental cell line (Ishi-PR-0) initially used to establish the Ishi-PR-B cell line and that lacks PR-B expression (42). Ishi-PR-0 cells were transfected with SRC-1 expression vector and incubated 24 h with R5020 or RU486. Under these conditions, both ligands did not affect the SRC-1 expression level, indicating that SRC-1 down-regulation requires the presence of PR-B (Fig. 3E). Finally, we determined whether other p160 coactivators such as SRC-2/transcription intermediary factor-2/glucocorticoid receptor-interacting protein-1 or SRC-3/ amplified in breast cancer 1 (AIB1), which are also known proteasome targets (14), could be degraded in response to R5020. None of these coactivators was significantly degraded under similar experimental conditions (Supplemental Fig. 7), suggesting a target-specific coactivator effect of PR.
Fig. 3.
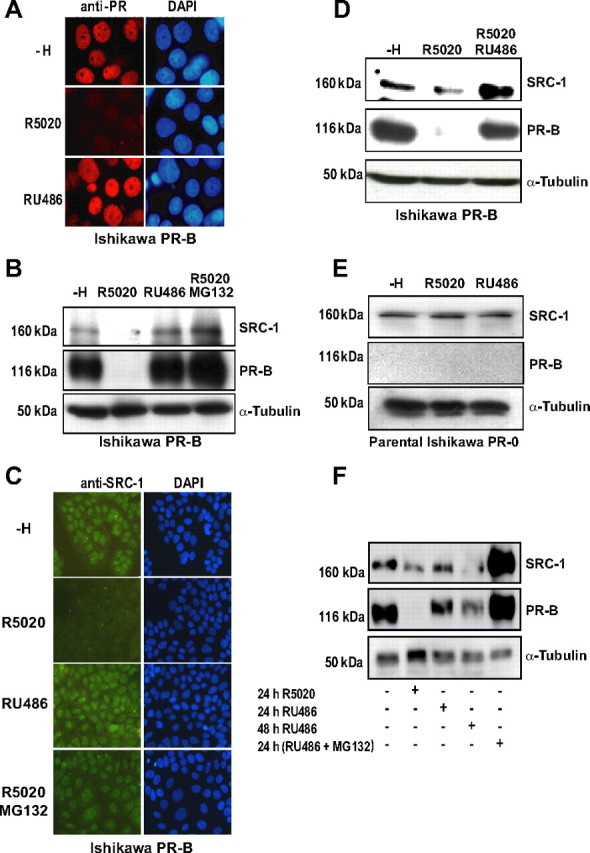
Ligand- and PR-dependent SRC-1 proteolysis. A, Ishi PR-B cells, a cell line stably expressing PR-B, were cultured 24 h in the absence or in the presence of either the agonist R5020 (10 nm) or the antagonist RU486 (10 nm). Cells were then treated for immunocytochemistry with anti-PR antibody (Let 126) and observed by fluorescence microscopy. DAPI, 4′,6′-Diamino-2-phenylindole. B, Ishi PR-B cells were transfected with the SRC-1 encoding vector. After 48 h, cells were cultured 15 h as indicated, either in the absence of ligand (control vehicle, −H), in the presence of R5020 (10 nm) or RU486 (10 nm), or in the presence of both R5020 (10 nm) and MG132 (5 μm). Whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies. C, Nontransfected Ishi PR-B cells were treated as in B. Cells were immunolabeled for endogenous SRC-1 using an anti-SRC-1 antibody. Note the agonist-ligand-dependent down-regulation of endogenous SRC-1. DAPI, 4′,6′-Diamino-2-phenylindole. D, Nontransfected Ishi PR-B cells were cultured 24 h in the absence of ligand (vehicle, −H) or in the presence of either the agonist R5020 (10 nm) alone or in combination with a 100× excess of the antagonist RU486 (1 μm). Whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted to detect endogenous SRC-1 and PR with the indicated antibodies. E, Ishi PR-0 cells (parental cell line, devoid of PR) were treated as in A. Cells were then analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies. F, Ishi PR-B cells were transfected with the SRC-1 encoding vector. After 24 h, cells were cultured in the absence of ligand (vehicle, −H), treated with R5020 (10 nm, 24 h) or RU486 (10 nm, 24 or 48 h) or RU486 (10 nm, 24 h) along with MG132 (1 μm). Whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies.
It has been initially proposed that antiprogestins are capable of inducing PR down-regulation but with much slower kinetics than agonists (22). We therefore tried a longer time point to check whether SRC-1 degradation was occurring in the presence of RU486. The result shows that, in contrast to 24 h incubation (Fig. 3F, lane 3), 48 h treatment with RU486 induced a significant reduction of both SRC-1 and PR (Fig. 3F, lane 4). More importantly, in the presence of MG132, RU486 treatment resulted in a dramatic accumulation of PR and SRC-1 (Fig. 3F, lane 5), showing that RU486-induced down-regulation is mediated by the proteasome. Thus, these results not only indicate that RU486 impairs the ligand-dependent down-regulation of PR and SRC-1 by slowing down their degradation but also confirm the concomitance of their ligand-dependent proteolysis. Collectively our results indicate that specific SRC-1 turnover is modulated in a ligand-dependent manner and requires PR expression.
Identification of SRC-1 domains involved in its degradation
To elucidate the mechanisms driving SRC-1 to the proteasome under basal conditions, we identified the domains involved in SRC-1 turnover. In silico analysis of SRC-1 primary sequence was carried out in search for putative PEST degradation motifs. The result indicated that the amino acids 2-16 of SRC-1 had a high score (+9.63) for this type of motif. We therefore focused our investigation on the N-terminal subdomain of the coactivator. A critical importance of the bHLH domain for AIB1/SRC-3 mediated proteolysis has been previously reported by Li et al. (39). Thus, we also explored the role of this domain in SRC-1 down-regulation. Two deletion mutants were generated, lacking either the PEST sequence or the bHLH domain, encompassing amino acids 2-16 [Δ(PEST)] and amino acids 41-136 [Δ(bHLH)], respectively (Fig. 4A).
Fig. 4.
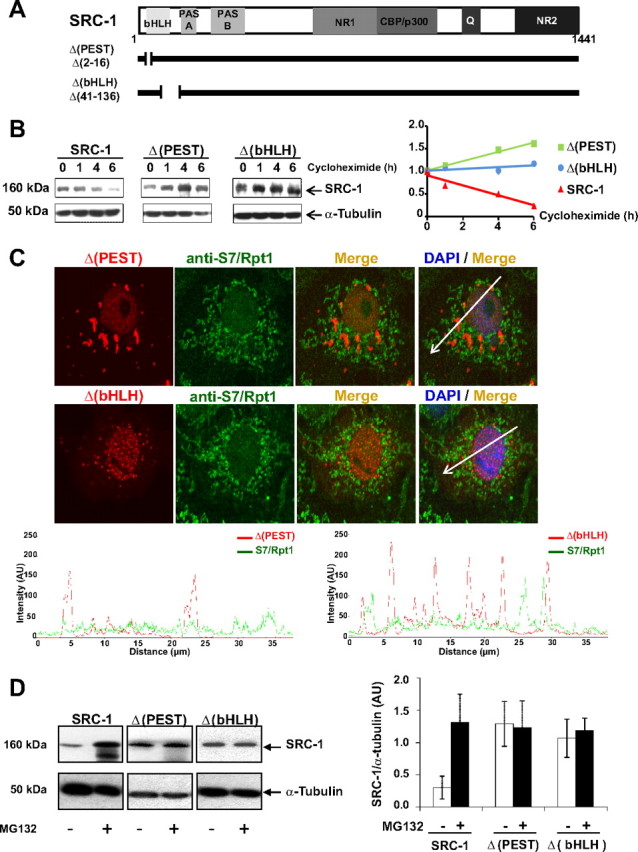
The N-terminal region of SRC-1 targets the coactivator to degradation. A, Schematic representation of the wild-type coactivator SRC-1 (1441 amino acids in length) with boxes corresponding to major functional domains: PAS, Per-ARNT-Sim motif; NR1 and NR2, NR-interacting domains 1 and 2, CBP/p300 interacting domain; Q, glutamine-rich domain. SRC-1 deletion mutants Δ(PEST) and Δ(bHLH) are represented below with a thick line interrupted by a gap corresponding to the deleted amino acids. B, COS-7 cells were transfected as indicated with SRC-1, Δ(PEST), or Δ(bHLH) encoding vectors. Seventy-two hours after transfection, cells were treated with cycloheximide (100 μg/ml) during 1, 4, or 6 h. Whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies. Band intensities (right panel) representing the mean of at least two independent experiments were quantified as described in Materials and Methods. C, Upper panel, Colocalization analysis of SRC-1 deletion mutants and S7/Rpt1. COS-7 cells were transiently transfected with Δ(PEST)- or Δ(bHLH)-encoding vectors. Cells were fixed after 40 h and immunolabeled with anti-HA and anti-Rpt1/S7 antibodies prior to analysis by confocal microscopy. Lower panel, Scan of intensity profiles expressed as arbitrary units (AU). Fluorescence intensity was calculated and plotted by drawing a line through the middle of the cell image in a distance covering several cytosolic and nuclear foci. Green lines represent the intensity profile for the proteasome antigen S7/Rpt1 signal, and the red lines represent the intensity profile for Δ(PEST) or Δ(bHLH) signals. Note the absence of significant peaks with overlapping signals. D, COS-7 cells were transfected with HA-SRC-1-, Δ(PEST)-, or Δ(bHLH)-encoding vectors. After 48 h, cells were incubated during 15 h with MG132 (5 μm) or vehicle. Whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies. The band intensities (right panel) were quantified as described in Materials and Methods.
To investigate whether these two motifs were involved in SRC-1 degradation, wt SRC-1, Δ(PEST), or Δ(bHLH) mutants were expressed in COS-7 cells, and cycloheximide was added to block protein neosynthesis. The decay of wt SRC-1 and mutant proteins was monitored and quantified by Western blot as a function of time. SRC-1 expression levels decreased after 1 h and almost disappeared after 6 h (Fig. 4B, left panel), indicating of a half-life of approximately 3 h. In contrast, both Δ(PEST) and Δ (bHLH) expression levels showed no decrease under the same experimental conditions (Fig. 4B, middle and right panels), showing that Δ(PEST) and Δ(bHLH) mutants are more stable than wt SRC-1.
To confirm that these motifs were involved in proteasome-mediated SRC-1 degradation, we compared both mutants and wt SRC-1 localization by immunocytochemistry and found that in contrast to the wild-type coactivator (Fig. 1) and the Δ(PEST) mutant, the Δ(bHLH) mutant localized predominantly in the nucleus (Fig. 4C). In contrast to the wild-type coactivator (Fig. 1), colocalization studies of both mutants with 19S proteasome antigens S7/Rpt1 and with the α/β-proteasome 20S subunits showed no significant overlap (Fig. 4C and data not shown).
Moreover, to investigate the involvement of these domains on SRC-1 protein stability, we compared the impact of MG132 on both mutants with wt SRC-1. Whereas SRC-1 protein levels were increased approximately 3-fold under 15 h MG132 treatment (Fig. 4D, left panel), the expression level of either Δ(PEST) or Δ(bHLH) remained unchanged under the same conditions (Fig. 4D, middle and right panels). Similarly, expression levels of both mutants were not increased in presence of epoxomicin (Supplemental Fig. 8). Of note, quantification comparison of band intensity (Fig. 4D, histograms) showed that both mutants were expressed to a greater extent than the wild-type coactivator, suggesting that the deletions may have indeed a stabilizing effect on these mutants. Taken together, our observations show that amino acids 2-16 and 41-136 are involved in SRC-1 down-regulation by targeting SRC-1 to proteasome degradation at the steady-state.
N-terminal degradation motifs of SRC-1 are necessary for its ligand-dependent down-regulation
To evaluate the contribution of the two degradation domains in the context of the hormonal activation, we transiently transfected Ishi-PR-B cells with wt SRC-1 or Δ(PEST) or Δ(bHLH) mutants. We hypothesized that if the two degradation motifs are also involved in hormone-stimulated down-regulation of SRC-1, then both mutants should not undergo proteolysis under hormone stimulation. As expected, after 24 h of R5020 treatment, wt SRC-1 was significantly down-regulated, whereas the expression level of both mutants showed no significant variation (Fig. 5A). Interestingly, the ligand-dependent down-regulation of PR still occurred in each condition, showing that the receptor down-regulation does not require SRC-1 degradation (Fig. 5A). To exclude the possibility that the two deletions may have impaired the interaction between the SRC-1 and PR, we conducted reciprocal coimmunoprecipitation experiments in cells transiently expressing PR and either wt SRC-1 or the deletion mutants. The result shown in Fig. 5B indicates that PR reciprocally coimmunoprecipitates with wt SRC-1 as well as with Δ(PEST) and Δ(bHLH) mutants. Taken together, these results indicate that under hormonal stimulation, SRC-1 ligand-dependent proteolysis requires both degradation signals.
Fig. 5.
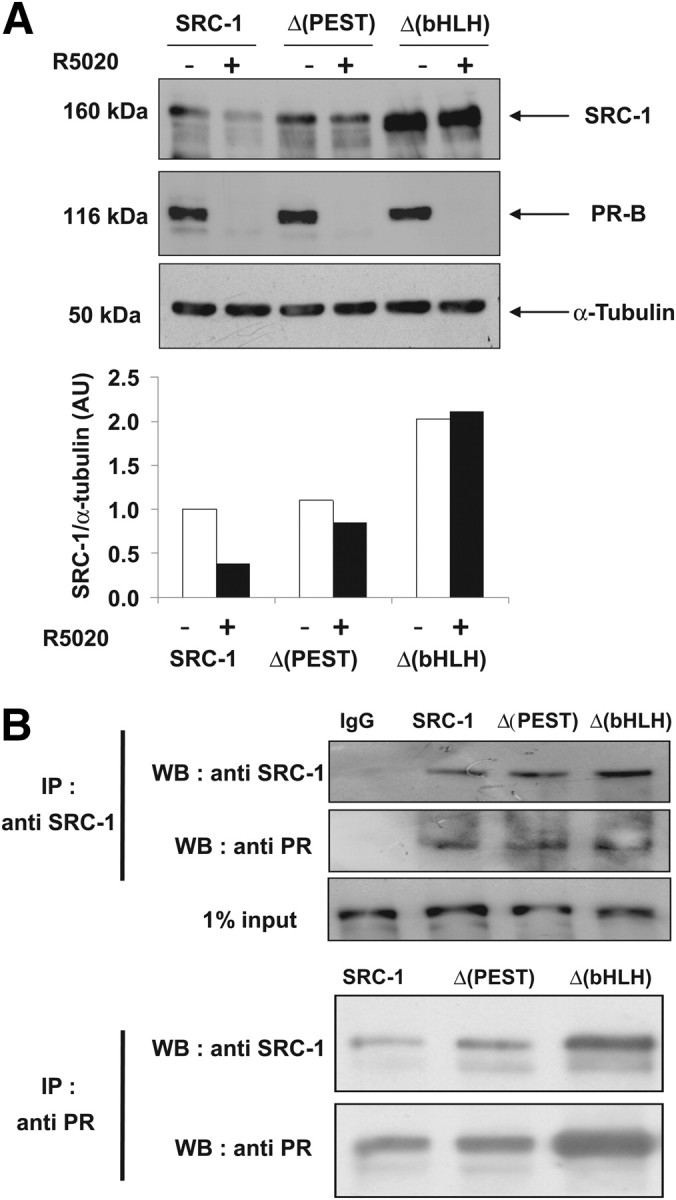
Ligand-dependent down-regulation of SRC-1 requires both degradation motifs of the coactivator. A, Ishi PR-B cells were transfected as indicated with HA-SRC-1-, Δ(PEST)-, or Δ(bHLH)-encoding vectors. After 48 h, cells were cultured in the absence of ligand (vehicle, −) or presence of the agonist R5020 (10 nm) during 24 h. The corresponding whole-cell extracts were analyzed by electrophoresis on 7.5% SDS-PAGE and immunoblotted with the indicated antibodies. The band intensities (lower panel) were quantified as described in Materials and Methods. B, HEK 293 cells were cotransfected with PR and the SRC-1-, Δ(PEST)-, or Δ(bHLH)-encoding vectors. Twenty four hours after transfection, cells were treated during 24 h with the agonist R5020 (10 nm). A coimmunoprecipitation assay was performed using the anti-SRC-1, the anti-PR, or the IgG1 control antibodies (IgG1). Purified proteins were separated on 7.5% SDS-PAGE. Coprecipitated complexes were identified with the indicated antibodies. IP, Immunoprecipitation; WB, Western blot.
Because we showed that SRC-1 could be partially proteolyzed in the cytoplasm in which it colocalized in speckles with the proteasome (Fig. 1), we next wondered whether PR will colocalize in the same cytoplasmic speckles. This may specially be the case if we consider the work of Qiu et al. (43), who have shown that PR down-regulation under hormone treatment occurs in the cytoplasm. Our result shows that in the absence of hormone, SRC-1 is expectedly cytonuclear and does not colocalize with PR (Fig. 6A). Eight hours of hormonal treatment (in the presence of cycloheximide) induces the nuclear accumulation of both PR and SRC-1, indicative of their interaction during the nuclear import (29). Interestingly, the ligand also induces the colocalization of PR and its coactivator in cytoplasmic speckles (Fig. 6A), suggesting that PR/SRC-1 complexes might be exported back to the cytoplasm. In contrast, in the presence of R5020, Δ(PEST), and Δ(bHLH) mutants were efficiently accumulated in the nucleus, consistent with our coimmunoprecipitation data showing that they do interact with PR in the presence of ligand, but did not colocalize with PR in cytoplasmic speckles (Fig. 6, B–D). Overall, this experiment suggests that PR and SRC-1 could be proteolyzed as a PR/SRC-1 complex through the same proteasome.
Fig. 6.
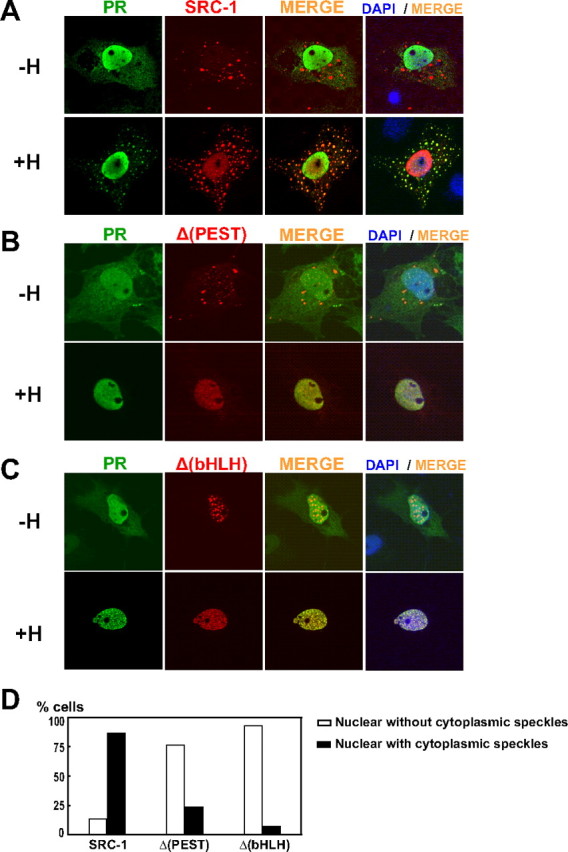
Colocalization of PR and SRC-1 in cytoplasmic speckles. A, COS-7 cells were transiently transfected with the expression vector encoding HA-SRC-1 and PR. Twenty-four hours after transfection, cells were incubated or not for 8 h with R5020 in presence of cycloheximide (100 μg/ml) prior to fixation. Cells were immunolabeled with anti-PR (Let 126) and anti-HA antibodies. DAPI, 4′,6′-Diamino-2-phenylindole. B and C, cells were treated as in A, except that PR was transfected as indicated with Δ(PEST) and Δ(bHLH), respectively. D, Quantification of cells treated as described in A–C. Percent of cells treated with R5020 shows nuclear localization with or without cytoplasmic speckles. At least 100 cells were counted.
Ligand-dependent proteolysis of SRC-1 is necessary for PR-mediated gene transactivation
To examine the functional link between SRC-1 degradation and its coactivating function, we investigated the impact of coactivator proteolysis on PR-mediated transcription. To this aim, we first analyzed whether the proteasome function was required for efficient PR transcriptional activation. Cotransfection of PRE2-TATA-luc reporter gene with the PR encoding vector was performed in parental Ishi-PR-0 cells (devoid of PR), either alone or in combination with the vector encoding SRC-1. Twenty-four hours after transfection, cells were treated for 24 h with R5020 alone or in combination with MG132. To exclude the possibility that the cellular toxicity of MG132 might affect general transcription in Ishikawa cells, we used a 500-nm concentration of the inhibitor, a dose compatible with cell survival of endometrial carcinoma cell lines (44). We show that MG132 drastically attenuates ligand-dependent PR transactivation (Fig. 7A), confirming previous observations made by Dennis et al. (45). Interestingly, SRC-1-potentiated PR-mediated transcription was also abolished by the proteasome inhibitor (Fig. 7A). This result suggests that the proteasome-mediated degradation is required not only for PR transcriptional activity but also for SRC-1 potentiation of PR. To further explore the relationship between coactivator degradation and the functional consequences on PR-mediated transcription, we used the two nondegradable mutants Δ(PEST) and Δ(bHLH) in cotransfection experiments with PR (Fig. 7B). Because these two mutants are not efficiently degraded by the proteasome (see Fig. 4, B and D), we predicted that they might not exert efficient potentiation of PR transactivation. Indeed, in the presence of R5020, SRC-1 strongly coactivated PR, whereas both Δ(PEST) and Δ(bHLH) mutants were unable to enhance PR-mediated transactivation as compared with wt SRC-1 (Fig. 7B). These results suggest that the concomitant degradation of SRC-1 and PR is necessary for efficient transcriptional activity of the receptor. Finally, to determine whether the functional link between SRC-1 proteolysis and its coactivating properties were also relevant for human endogenous gene activation, we quantified the level of the progesterone-induced amphiregulin gene that we have previously studied (46). Parental Ishi-PR-0 cells were transfected with PR alone or in combination with wt or SRC-1 mutants. Amphiregulin mRNA levels were significantly increased upon R5020-dependent PR activation and were further enhanced in the presence of SRC-1 (Fig. 7C). Conversely, coexpression of PR with either Δ(PEST) or Δ(bHLH) mutant significantly reduced amphiregulin expression (P < 0.001). Taken together, our results demonstrate that hormone-induced degradation of SRC-1 is physiologically relevant for potentiation of PR-mediated transcriptional events.
Fig. 7.
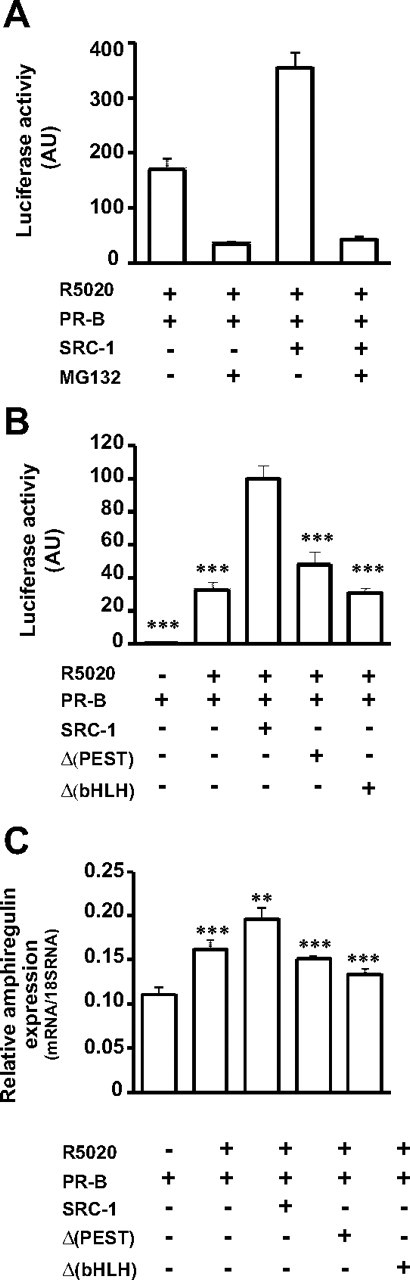
SRC-1 degradation is necessary for PR transcriptional activity. A, Ishi PR-0 cells were cotransfected as indicated with expression vectors encoding PR and SRC-1 together with the reporter gene PRE2-TATA-luc and the internal control pRS-β-gal. Cells were incubated with R5020 (10 nm) and treated or not with MG132 (500 nm) during 24 h. Luciferase activity was quantified and normalized by β-galactosidase activity. Data represent means ± sem of at least three independent determinations. B, COS-7 cells were cotransfected as indicated with HA-SRC1-, Δ(PEST)-, or Δ(bHLH)-encoding vectors together with expression vector encoding PR, the reporter gene PRE2-TATA-luc, and the internal control pRS-β-gal. Cells were treated during 24 h with R5020 (10 nm) or vehicle (control, −). Luciferase activity was quantified and normalized by β-galactosidase activity. Data represent means ± sem of four independent determinations performed in triplicate. C, Ishi PR-0 cells were cotransfected as indicated with HA-SRC-1-, Δ(PEST)-, or Δ(bHLH)-encoding vectors together with PR encoding vector and were treated with the agonist R5020 10 nm for 3 h. Total RNAs were extracted and relative expression of amphiregulin gene was quantified by quantitative RT-PCR. Results, normalized by the amplification of 18S RNA, are mean ± sem of three independent determinations. ***, Statistical significance, P < 0.001 vs. wild-type SRC-1 used as reference.
Discussion
In this study, we investigated the impact of SRC-1 proteolysis on PR-mediated transcription. We provided evidence that the agonist-dependent degradation of SRC-1 is pivotal for PR-mediated transcription. We have established that agonist ligand R5020, but not antagonist RU486, induces the concomitant degradation of endogenous or ectopic PR and SRC-1. Interestingly, SRC-1 turnover requires the presence of PR. Both basal and induced SRC-1 down-regulation are mediated through the proteasome pathway and seem to occur at least in part, in the cytoplasmic compartment. Two regions located in the N-terminal part of SRC-1 (i.e. a PEST motif and amino acids 41-136 of the bHLH domain) were identified as two degron motifs. Both signals were shown to be responsible for basal- and hormone-induced degradation of SRC-1. Deletion of each of these domains [Δ(PEST) and Δ(bHLH) mutants] leads to nondegradable SRC-1 mutants insensitive to proteasome inhibitors. By comparing the biological functions of these two mutants with wt SRC-1, we found that they were incapable of potentiating PR-mediated transactivation on a synthetic PR response-element but also on amphiregulin, an endogenous PR target gene. The HAT motif and the CBP-interacting domain of SRC-1 are known to regulate the transcriptional activity of SRC-1 (47, 48). Both regions are present in Δ(PEST) and Δ(bHLH) mutants (Fig. 4A), and therefore, the reduced PR-dependent transactivation of the mutants is not due to an alteration of these regulatory domains but rather to a defect in down-regulation. Thus, our results are indicative of a functional link between proteasome-mediated down-regulation of SRC-1 and its coactivating property.
We have previously shown that SRC-1 is a transcriptional coactivator whose localization is hormonally regulated in the presence of PR (29). Mainly functioning in the nuclear compartment, this coactivator may also be present in the cytoplasm, predominantly concentrated in cytoplasmic speckles (29). Several studies have also demonstrated that p160 coregulators might be localized in the cytoplasm (7, 30, 31). Although the concentration of SRC-1 in cytoplasmic speckles was initially reported to be linked to overexpression (49), it has been also observed for endogenous p160 coactivators (50), and more importantly, a recent study correlated this archetypical distribution with the cytoplasmic sequestration of SRC-1 by SRC-interacting protein (51). During our primary search to identify the nature of these speckles, we initially observed a colocalization between SRC-1 and proteasome antigens, indicating that SRC-1 cytoplasmic speckles are enriched of proteasome components (Fig. 1A). Similar subcellular distribution studies already reported SRC-2 colocalization with proteasome antigens but specifically at the nuclear level (34, 52). Coactivator/proteasome interaction also have been described at the biochemical level for the p160 coactivators (33, 53), as well as NR such as the thyroid receptor, the retinoic acid receptor-α and retinoid X receptor, the estrogen receptor (ER)-α, or the vitamin D receptor (32). We detected a strong colocalization in the cytoplasmic compartment, although a weaker colocalization in speckles was also observed in the nuclear compartment (Fig. 1B) indicative of a predominant but not exclusive proteolysis of the coactivator in the cytoplasmic compartment. Interestingly, nuclear export of SRC-3 has been shown to be required for its proteasomal degradation (54). However, our finding is not consistent with the work of Li et al. (39), who recently showed that proteasome-dependent turnover of SRC-3 occurs specifically in the nucleus. Although we could not completely exclude that nuclear degradation also occurs for SRC-1 (see colocalization profiles in Fig. 1B), this discrepancy between SRC-1 and SRC-3 argues for the fact that each SRC family member has different and specific physiological functions (55).
We have shown that the ubiquitin-proteasome pathway mediates selective degradation of SRC-1 and regulates the steady-state expression level of the coactivator. Similarly, Yan et al. (35) have shown that several SR coactivators were degraded through the ubiquitin-proteasome dependent pathway and that SRC-1 proteolysis occurs specifically through the ubiquitin-conjugating enzyme 2. The half-life regulation of p160 coactivators has been extensively investigated since the discovery of their prototype SRC-1, and several studies have demonstrated the physiological and pathophysiological importance of regulating SRC-1 expression levels (56–58). SRC-1 is an important modulator of PR-mediated gene transcription, and to accurately exert its physiological function, its level must be therefore tightly regulated in vivo. In this context, Han et al. (59) used an original transgenic mouse model in which SRC-1 levels were shown to influence the compartment specific corepressor to coactivator ratio to modulate PR activity in the uterus. Cell regulation of SRC-1 levels seems to be also critical for tumorigenesis, and studies have demonstrated that SRC-1 expression is significantly increased in breast tumors and positively correlates with disease recurrence and poor disease-free survival (55). Consistent with this finding, SRC-1 level is up-regulated during mammary tumor progression (60), and the role of this coactivator in promoting mammary tumor cell invasion was recently demonstrated in vivo (57, 58).
Besides the regulation of SRC-1 proteolysis at basal level, the present study also analyzes ligand-stimulated down-regulation of the coactivator. Similarly to other rapidly turned over transcription factors, engagement of PR in transactivation has been shown to be coupled to PR degradation by the ubiquitin-proteasome pathway (13). However, the functional impact of the SRC-1 coactivator on PR-mediated transactivation has never been clearly established. We demonstrate for the first time that concomitantly to PR degradation, SRC-1 proteolysis is dramatically increased in the presence of the agonist ligand R5020 and that this process is mediated through the proteasome. Similarly to PR (22), this down-regulation is necessary for PR-mediated transcription. Recent advances in molecular biology have redefined the role of proteasome as a regulatory system that influences the fate of many cellular processes, such as cell proliferation, apoptosis, and more recently gene transcription. Despite the disparate nature of the later process, a growing body of evidences indicates that ubiquitin and the proteasome are intimately involved in NR-mediated gene control (45, 61, 62). Steroid hormone receptors and their coactivators cycle onto and off steroid-responsive promoters in a ligand-dependent manner, and it is now believed that the ubiquitin-proteasome functions in promoting the turnover of transcription complexes, thereby facilitating proper gene transcription (16, 63). Dennis et al. (45) have proposed the existence of a transcriptional mechanism that links the proteasome function with the continued recruitment of RNA polymerase II to sustain the transcriptional response. Consistent with these observations is the fact that a number of ubiquitin pathway enzymes and components of the proteasome have been found to act as modulators of NR function (24, 26, 64) and that enzymes and components of the proteasome are recruited to the promoters of NR-responsive genes (16, 63).
Despite this, it is difficult to conceive how a coactivator will be paradoxically part of a coactivating complex positively modulating gene activation and at the same time a specific target of the ubiquitin-proteasome pathway. Thus, the coupling of PR/SRC-1 proteolysis and efficient transcriptional activation is counterintuitive and rather puzzling but could be a general phenomenon occurring during transcription (65). Consistent with this, is the fact that neither PR nor its coactivator was down-regulated in presence of the antagonist RU486. This result may suggest that RU486 indirectly prevents recruitment of the proteasome machinery, thereby inhibiting transcription. The same observation was made with ERα and the partial antagonist tamoxifen, although it may not be considered as a general phenomenon for steroid receptor because the pure antagonist faslodex dramatically stabilize ER in similar conditions (49). It is not the first example of a hormonal regulatory mechanism implicated in specific coregulators proteolysis: indeed, SRC-2 is down-regulated through the activation of the cAMP-dependent protein kinase pathway (52). More importantly, Gianni et al. (27) showed that SRC-3, but not SRC-1 or SRC-2, is phosphorylated by p38MAPK in a retinoic acid-dependent manner and then degraded by the proteasome pathway. In this case, phosphorylation of SRC-3 has a biphasic effect on retinoic acid receptor-α transactivation with facilitation followed by restriction of transcription.
Because the presence of PR is required for SRC-1 degradation, two important remaining questions concern the identification of the key player responsible for SRC-1 degradation and whether this factor is involved in both basal and ligand-induced SRC-1 down-regulation. Shao et al. used RNA interference to knock-down SRC-3 that consequently abolishes ERα ligand-dependent degradation, suggesting that the coactivator itself regulates ERα degradation (66). Conversely, because the two nondegradable mutants did not impede the ligand-induce PR down-regulation (Fig. 5A), our results do not converge toward a link between the recruitment of a common E3-ligase by SRC-1 which will in turn induce the ligand-dependent degradation of the PR/SRC-1 complex. The signal that targets PR and SRC-1 to progress from transcription to degradation may also involve post-translational modifications operating like a molecular signature such as phosphorylation, ubiquitinylation or sumoylation (9, 67, 68). Alternatively, direct recruitment of ligase in the vicinity of the coactivator complex or directly at the enhancer level may be also implicated in SRC-1 turnover along with PR. A good candidate would be the PR-B coactivator/ubiquitin ligase E6-AP because this coactivator plays a major role in controlling the regulated degradation of SRC-3 and PR-B isoform (54, 69). Alternatively, the colocalization with proteasome antigens observed in our study (Fig. 1) might also be linked to the direct interaction observed between SRC-1 and the proteasome through the low molecular mass polypeptide 2 proteasome subunit (53). Such a direct ligand-dependent interaction may drive the coactivator to proteolysis. Another potential candidate for PR and SRC-1 degradation might be Jun-activation domain-binding protein 1 (Jab1), a coactivator involved in ER degradation (70). We are currently investigating this hypothesis because we have shown in a previous study that Jab1 is a coactivator of PR, inducing the formation of a PR/SRC-1/Jab1 ternary complex during the transcription process (71).
In summary, we demonstrate in the present study that SRC-1 expression level is hormonally regulated by the ligand. Whereas in the presence of an agonist, the PR/SRC-1 complex is proteolyzed to achieve transcription, an antagonist such as RU486 impairs the ligand-dependent degradation of PR/SRC-1 and consequently the transactivation process. Our data indicate that the expression level of SRC-1 coactivator is critical for PR transcriptional activity. These findings are consistent with the emerging role of the 26S proteasome in the gene regulation process (72). P160 family members are certainly not the only coactivators implicated in such processes, and it will be interesting to elucidate the sequential progression of each coregulator degradation during gene regulation.
Materials and Methods
Hormone and inhibitors
Cycloheximide, epoxomicin, MG132, and LB were purchased from Sigma (St. Louis, MO). Agonist R5020 (17,21-dimethyl-19-norpregna-4,9-dien-3,20-dione) and antagonist RU486 (Sigma) were used at a concentration of 10 nm, except where indicated.
Plasmids
Nomenclature
Derivatives denoted with a Δ lack the protein segment delineated by the numbered amino acids. Plasmids encoding the wild-type human progesterone receptor (pSG5-PR) and coactivator SRC-1 (pSG5-SRC-1, pSG5-HA-SRC-1, pSG5-HA-GFP-SRC-1) have previously been described (29). PCR-based site-directed mutagenesis of pSG5-HA-SRC-1 was used to create deletion mutants: pSG5-HA-SRC-1-Δ (2–16) [named Δ(PEST)], pSG5-SRC-1-Δ (41-136) [named Δ(bHLH)], and pSG5-HA-SRC-1Δ(990-1060) [named Δ(NES), (29)]. The plasmid pPRE2-TATA-Luc has been previously described (71). Plasmid pSG5-His6-Ub is a gift of D. Bohmann (Laboratory EMBL, Heidelberg, Germany). Plasmids pSG5-SRC-2 and pCR3.1-SRC-3 have been described previously (7, 73), and GFP-peroxisome targeting signal expression vector was purchased (CLONTECH, Mountain View, CA).
Cell culture and DNA transfection
Human endometrial Ishikawa cells (parental cell line, Ishi-PR-0, and stable, Ishi-PR-B) were provided by Dr. L. J. Blok (Erasmus University, Rotterdam, The Netherlands) (74). COS-7, human embryonic kidney (HEK) 293, Ishi-PR-0, and Ishi-PR-B were grown in DMEM containing 10% fetal bovine serum (Biowest, Miami, FL) and supplemented with l-glutamine and antibiotics (penicillin/streptomycin; PAA Laboratories Les Mureaux, France). For hormonal regulation experiments, cells were grown in the presence of 10% steroid-depleted fetal bovine serum prior (24 h) and during transfection experiments. Transfections were performed with the indicated expression vectors using LipofectAMINE 2000 according to the manufacturer's recommendations (Invitrogen, Carlsbad, CA).
Antibodies
Monoclonal anti-PR antibodies used in the study were the Let126 (0.5 μg/ml) (75), the monoclonal anti-PR from Novocastra (NCL-L-PGR-312/2; Newcastle-upon-Tyne, UK) or the rabbit polyclonal anti-PR (sc-538) from Santa Cruz Biotechnology (Santa Cruz, CA), used for immunoprecipitation. Anti-SRC-1 mouse monoclonal antibody (Millipore, Billerica, MA) was used for Western blot and immunocytochemistry (1 μg/ml). Endogenous SRC-1 was detected with anti-SRC-1 (sc-6096) purchased from Santa Cruz Biotechnology. Anti-HA 3F10 (200 ng/ml) was from Roche Applied Science (Indianapolis, IN). Rabbit polyclonal antibody directed against human S7/Rpt1 and 20S proteasome subunits and KAT13C/nuclear receptor coactivator 2/SRC-2 were purchased from Abcam (Cambridge, MA) and used at 1:1000 dilution. Anti-α-tubulin (1:10,000) and anti-SC-35 (1 μg/ml) were purchased from Sigma. Antipromyelocytic leukemia protein was provided by H. de Thé (Institut Universitaire d'Hématologie, Paris, France). Anti-SRC3/AIB1 antibody was purchased from BD Biosciences (San Diego, CA) and was used at 0.5 μg/ml. Secondary antibodies (1:4000) included the following: antimouse, antirat, and antirabbit antibodies conjugated to Alexa 488 (green) or 595 (red) or Dylight 549 (red) were from Invitrogen and Jackson ImmunoResearch Laboratories (West Grove, PA). Secondary peroxidase-conjugated antimouse (Calbiochem, San Diego, CA) and antirabbit (Vector Laboratories Inc., Burlingame, CA) antibodies were used at 1:15,000 dilution.
Luciferase reporter gene assays
COS-7 cells were cultured in free steroid medium and reverse transfected in 96-well plates with 4 ng PR, 100 ng PRE2-TATA-Luciferase, 100 ng SRC-1 (wild type or mutants), and 5 ng β-galactosidase (internal control). The pBlue-Script plasmid was used to equally adjust DNA quantity. After 24 h transfection, cells were incubated with or without 10 nm R5020 for 24 h. Cells were collected with the passive lysis buffer (Promega, Madison, WI), and luciferase activity was measured with a luminometer (Victor; PerkinElmer, Waltham, MA). Luciferase activity was normalized with β-galactosidase activity. The results are means ± se of four independent experiments.
Immunocytochemistry
Cells were seeded in 24-well plates and processed as previously described (7). Briefly, cells were fixed with 4% paraformaldehyde and permeabilized for 30 min with a 0.5% solution of Triton X-100 diluted in PBS. Cells were then incubated with primary antibody overnight at 4 C, followed by the appropriate fluorochrome-coupled secondary antibody (Alexa 488 or 595; Invitrogen; or Dylight 549, Jackson ImmunoResearch Laboratories) for 30 min. Nuclear counterstaining was performed with 0.5 μg/ml 4′,6′-diamidino-2-phenylindole, and coverslips were mounted on slides with ProLong Gold mounting medium (Invitrogen). For standard microscopy (see Figs. 2 and 3), fluorescent cells were observed with an Olympus Provis AX70 (Rungis, France), and images were acquired with Qcapture Pro version 5.1 (Q Imaging Inc., Surrey, British Columbia, Canada) using an Evolution VF Monochrome camera (Media Cybernetics Inc., Bethesda, MD).
Confocal microscopy
For some of the figures (see Figs. 1, 4, and 6), a Zeiss LSM-510 confocal laser scanning microscope (Carl Zeiss, Thornwood, NY) was used for fluorescence acquisition. Images of fixed cells were collected from equatorial planes of cells with a pinhole setting of approximately 1.0 airy unit (optical thickness of 0.8 μm) using a ×63:1.4NA oil immersion plan-apochromat objective with ×8 frame averaging accumulation. To exclude cross talk artifacts, both red and green fluorescence emission were acquired sequentially in separated channels. The confocal microscope settings were kept the same for all scans. To validate colocalization of proteins (see Figs. 1 and 4), line scans of intensity profiles across the cells were generated with the LSM browser software (76). This function associates the merge images with an intensity profile of each channel, measured along a freely positioned line. To obtain an average representative intensity profile expressed as arbitrary units (AU), lines were drawn through the middle of each cell images in a distance covering the cytosol and the nucleus. Green lines represent the intensity profile for the proteasome antigen S7/Rpt1 signal and the red lines represent the intensity profile for SRC-1 signal.
Western blot and immunoprecipitation
Cells were lysed in lysis buffer [50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Triton X-100, 2 mm EDTA, 0.2 mm NaF, 0.2 mm Na3VO4, protease inhibitor cocktail] for 15 min, and the debris was cleared by centrifugation at 14,000 × g for 15 min at 4 C. Samples were resolved by 7.5% sodium dodecyl sulfate gel electrophoresis and transferred onto nitrocellulose membranes. The indicated antibodies were diluted in Tris-buffered saline and 0.1% Tween 20 buffer supplemented with 5% nonfat milk and added to the membranes for 1 h 30 min at room temperature or overnight at 4 C followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies for 45 min at room temperature. All proteins were detected with ECL Plus detection reagents (Amersham Biosciences Corp., Piscataway, NJ) and visualized by chemiluminescence. For the normalization, the membrane was stripped, probed with anti-α-tubulin antibody diluted to 1:1000 (Sigma). The bands were quantified after digitalization on a gel scanner using Image J software (National Institutes of Health, Bethesda, MD). Results, mean of three independent experiments (except Fig. 5A), are presented as the ratio SRC-1 (or PR) to α-tubulin and are expressed as fold induction above the value measured for wild-type SRC1 in the absence of MG132 arbitrarily set at 1. For coimmunoprecipitation, HEK 293 cells were transfected in a 100-mm plate with wild-type SRC-1, Δ(PEST), or Δ(bHLH) plasmids and cultured in presence of 10−8 m R5020 for 24 h. Cells were lysed at 4 C in 500 μl lysis buffer, and cell debris was pelleted by centrifugation (14,000 rpm, 15 min, 4 C). Immunoprecipitation of the supernatant with anti-SRC-1 or with the rabbit polyclonal anti-PR or with IgG control were performed with Protein G magnetics beads (Millipore) according to the manufacturer's instructions. Bound immunocomplexes were boiled in Laemmli buffer, separated by 7.5% SDS-PAGE, blotted nitrocellulose membranes with anti-1 μg/ml SRC-1 (Millipore) and anti-PR-B (Let 126, 0.5 μg/ml) antibodies, detected with ECL Plus detection reagents (Amersham Biosciences), and visualized by chemiluminescence.
Real-time RT-PCR
The Ishikawa cell line expressing PR-B or not was transfected by the indicated plasmids by Polyfect reagent (QIAGEN, Valencia, CA) in six-well plates (six wells per condition). After a 2 h-treatment by R5020 10 nm, cells were washed and lysed by Trizol reagent (Life Technologies, Gaithersburg, MD). Total RNA was extracted as described by the manufacturer. One microgram of each sample was treated by deoxyribonuclease I and was reverse transcribed using random primers as previously described (77). Real-time quantitative PCR of the amphiregulin gene was performed as described (46) using the Power SYBR Green master mix (Applied Biosystems, Carlsbad, CA) in duplicate with 1:20 fraction of each cDNA sample and the corresponding primers, using an ABI Prism 7300 apparatus(Applied Biosystems). For each sample, the mRNA concentration was extrapolated from standard curve and averaged cycle threshold value was divided by that of the corresponding reverse-transcribed 18S RNA (relative mRNA).
Statistical analysis
Data are expressed as the mean ± sem. The Mann-Whitney U test was used to determine significant differences between the two groups. For multiple comparisons, the Kruskal-Wallis test followed by Dunn's posttest was performed using the computer software Prism 4 (GraphPad Software, San Diego, CA). Statistical significance is indicated at P < 0.05, 0.01, and 0.001.
Acknowledgments
The authors are indebted to Youssef Alj for the construction of the Δ(PEST) and Δ(bHLH) SRC-1 mutants. We thank C. Massaad and B. W. O'Malley for kindly providing SRC-2 and SRC-3 plasmids, respectively. We gratefully acknowledge Luc Outin for excellent technical support and Geri Meduri for critical and thorough reading of the manuscript. We are thankful to Meriem Messina for her help in plasmid preparation.
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale, the Université Paris-Sud 11. L.A. and R.K.T. were recipients of a fellowship from the Fondation pour la Recherche Médicale. A.R. is a recipient of a fellowship from the Université Paris-Sud 11. J.A.K. is on study leave from the University of Agriculture, Faisalabad, Pakistan, and is a recipient of a doctoral scholarship from the Higher Education Commission, Pakistan.
This work was supported by grants from Inserm, the Université Paris-Sud 11 and the Académie Nationale de Médecine.
Present address for A.C.: Institut National de la Santé et de la Recherche Médicale Unité 981, Institut de Cancérologie Gustave Roussy, 114 Rue Edouard Vaillant, Villejuif F-94805, France.
Present address for R.K.T.: Special Centre for Molecular Medicine, Jawaharlal Nehru University, New Delhi 110067, India.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AIB1
- Amplified in breast cancer 1
- AU
- arbitary unit
- bHLH
- basic helix-loop-helix
- CBP
- cAMP response element-binding protein
- ER
- estrogen receptor
- HEK
- human embryonic kidney
- Jab1
- Jun-activation domain-binding protein 1
- LB
- leptomycin B
- NES
- nuclear export signal
- NR
- nuclear receptor
- PR
- progesterone receptor
- SRC-1
- steroid receptor coactivator-1
- wt
- wild type.
References
- 1. Li X , O'Malley BW. 2003. Unfolding the action of progesterone receptors. J Biol Chem 278:39261–39264 [DOI] [PubMed] [Google Scholar]
- 2. Lange CA. 2008. Challenges to defining a role for progesterone in breast cancer. Steroids 73:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lonard DM , O'Malley BW. 2006. The expanding cosmos of nuclear receptor coactivators. Cell 125:411–414 [DOI] [PubMed] [Google Scholar]
- 4. Xu J , Li Q. 2003. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol 17:1681–1692 [DOI] [PubMed] [Google Scholar]
- 5. Chen D , Ma H , Hong H , Koh SS , Huang SM , Schurter BT , Aswad DW , Stallcup MR. 1999. Regulation of transcription by a protein methyltransferase. Science 284:2174–2177 [DOI] [PubMed] [Google Scholar]
- 6. Torchia J , Rose DW , Inostroza J , Kamei Y , Westin S , Glass CK , Rosenfeld MG. 1997. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387:677–684 [DOI] [PubMed] [Google Scholar]
- 7. Amazit L , Pasini L , Szafran AT , Berno V , Wu RC , Mielke M , Jones ED , Mancini MG , Hinojos CA , O'Malley BW , Mancini MA. 2007. Regulation of SRC-3 intercompartmental dynamics by estrogen receptor and phosphorylation. Mol Cell Biol 27:6913–6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naeem H , Cheng D , Zhao Q , Underhill C , Tini M , Bedford MT , Torchia J. 2007. The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol Cell Biol 27:120–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rowan BG , Weigel NL , O'Malley BW. 2000. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem 275:4475–4483 [DOI] [PubMed] [Google Scholar]
- 10. Wu RC , Qin J , Hashimoto Y , Wong J , Xu J , Tsai SY , Tsai MJ , O'Malley BW. 2002. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by IκB kinase. Mol Cell Biol 22:3549–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKenna NJ , O'Malley BW. 2002. Minireview: nuclear receptor coactivators—an update. Endocrinology 143:2461–2465 [DOI] [PubMed] [Google Scholar]
- 12. Rosenfeld MG , Lunyak VV , Glass CK. 2006. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428 [DOI] [PubMed] [Google Scholar]
- 13. Shen T , Horwitz KB , Lange CA. 2001. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol 21:6122–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lonard DM , Nawaz Z , Smith CL , O'Malley BW. 2000. The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol Cell 5:939–948 [DOI] [PubMed] [Google Scholar]
- 15. Wijayaratne AL , McDonnell DP. 2001. The human estrogen receptor-α is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 276:35684–35692 [DOI] [PubMed] [Google Scholar]
- 16. Reid G , Hübner MR , Métivier R , Brand H , Denger S , Manu D , Beaudouin J , Ellenberg J , Gannon F. 2003. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11:695–707 [DOI] [PubMed] [Google Scholar]
- 17. Yokota K , Shibata H , Kobayashi S , Suda N , Murai A , Kurihara I , Saito I , Saruta T. 2004. Proteasome-mediated mineralocorticoid receptor degradation attenuates transcriptional response to aldosterone. Endocr Res 30:611–616 [DOI] [PubMed] [Google Scholar]
- 18. Tirard M , Almeida OF , Hutzler P , Melchior F , Michaelidis TM. 2007. Sumoylation and proteasomal activity determine the transactivation properties of the mineralocorticoid receptor. Mol Cell Endocrinol 268:20–29 [DOI] [PubMed] [Google Scholar]
- 19. Milgrom E , Thi L , Atger M , Baulieu EE. 1973. Mechanisms regulating the concentration and the conformation of progesterone receptor(s) in the uterus. J Biol Chem 248:6366–6374 [PubMed] [Google Scholar]
- 20. Mullick A , Katzenellenbogen BS. 1986. Progesterone receptor synthesis and degradation in MCF-7 human breast cancer cells as studied by dense amino acid incorporation. Evidence for a non-hormone binding receptor precursor. J Biol Chem 261:13236–13246 [PubMed] [Google Scholar]
- 21. Wei LL , Krett NL , Francis MD , Gordon DF , Wood WM , O'Malley BW , Horwitz KB. 1988. Multiple human progesterone receptor messenger ribonucleic acids and their autoregulation by progestin agonists and antagonists in breast cancer cells. Mol Endocrinol 2:62–72 [DOI] [PubMed] [Google Scholar]
- 22. Lange CA , Shen T , Horwitz KB. 2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA 97:1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lipford JR , Deshaies RJ. 2003. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat Cell Biol 5:845–850 [DOI] [PubMed] [Google Scholar]
- 24. Masuyama H , MacDonald PN. 1998. Proteasome-mediated degradation of the vitamin D receptor (VDR) and a putative role for SUG1 interaction with the AF-2 domain of VDR. J Cell Biochem 71:429–440 [PubMed] [Google Scholar]
- 25. Nawaz Z , Lonard DM , Dennis AP , Smith CL , O'Malley BW. 1999. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA 96:1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verma S , Ismail A , Gao X , Fu G , Li X , O'Malley BW , Nawaz Z. 2004. The ubiquitin-conjugating enzyme UBCH7 acts as a coactivator for steroid hormone receptors. Mol Cell Biol 24:8716–8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gianni M , Parrella E , Raska I , Gaillard E , Nigro EA , Gaudon C , Garattini E , Rochette-Egly C. 2006. P38MAPK-dependent phosphorylation and degradation of SRC-3/AIB1 and RARα-mediated transcription. EMBO J 25:739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han SJ , Tsai SY , Tsai MJ , O'Malley BW. 2007. Distinct temporal and spatial activities of RU486 on progesterone receptor function in reproductive organs of ovariectomized mice. Endocrinology 148:2471–2486 [DOI] [PubMed] [Google Scholar]
- 29. Amazit L , Alj Y , Tyagi RK , Chauchereau A , Loosfelt H , Pichon C , Pantel J , Foulon-Guinchard E , Leclerc P , Milgrom E , Guiochon-Mantel A. 2003. Subcellular localization and mechanisms of nucleocytoplasmic trafficking of steroid receptor coactivator-1. J Biol Chem 278:32195–32203 [DOI] [PubMed] [Google Scholar]
- 30. Chen Y , Chen PL , Chen CF , Sharp ZD , Lee WH. 1999. Thyroid hormone, T3-dependent phosphorylation and translocation of Trip230 from the Golgi complex to the nucleus. Proc Natl Acad Sci USA 96:4443–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grenier J , Trousson A , Chauchereau A , Cartaud J , Schumacher M , Massaad C. 2006. Differential recruitment of p160 coactivators by glucocorticoid receptor between Schwann cells and astrocytes. Mol Endocrinol 20:254–267 [DOI] [PubMed] [Google Scholar]
- 32. Ferrell K , Wilkinson CR , Dubiel W , Gordon C. 2000. Regulatory subunit interactions of the 26S proteasome, a complex problem. Trends Biochem Sci 25:83–88 [DOI] [PubMed] [Google Scholar]
- 33. Yi P , Feng Q , Amazit L , Lonard DM , Tsai SY , Tsai MJ , O'Malley BW. 2008. Atypical protein kinase C regulates dual pathways for degradation of the oncogenic coactivator SRC-3/AIB1. Mol Cell 29:465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baumann CT , Ma H , Wolford R , Reyes JC , Maruvada P , Lim C , Yen PM , Stallcup MR , Hager GL. 2001. The glucocorticoid receptor interacting protein 1 (GRIP1) localizes in discrete nuclear foci that associate with ND10 bodies and are enriched in components of the 26S proteasome. Mol Endocrinol 15:485–500 [DOI] [PubMed] [Google Scholar]
- 35. Yan F , Gao X , Lonard DM , Nawaz Z. 2003. Specific ubiquitin-conjugating enzymes promote degradation of specific nuclear receptor coactivators. Mol Endocrinol 17:1315–1331 [DOI] [PubMed] [Google Scholar]
- 36. Chen F , Chang D , Goh M , Klibanov SA , Ljungman M. 2000. Role of p53 in cell cycle regulation and apoptosis following exposure to proteasome inhibitors. Cell Growth Differ 11:239–246 [PubMed] [Google Scholar]
- 37. Davarinos NA , Pollenz RS. 1999. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytoplasmic proteasome following nuclear export. J Biol Chem 274:28708–28715 [DOI] [PubMed] [Google Scholar]
- 38. Tomoda K , Kubota Y , Kato J. 1999. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398:160–165 [DOI] [PubMed] [Google Scholar]
- 39. Li C , Wu RC , Amazit L , Tsai SY , Tsai MJ , O'Malley BW. 2007. Specific amino acid residues in the basic helix-loop-helix domain of SRC-3 are essential for its nuclear localization and proteasome-dependent turnover. Mol Cell Biol 27:1296–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Syvälä H , Vienonen A , Zhuang YH , Kivineva M , Ylikomi T , Tuohimaa P. 1998. Evidence for enhanced ubiquitin-mediated proteolysis of the chicken progesterone receptor by progesterone. Life Sci 63:1505–1512 [DOI] [PubMed] [Google Scholar]
- 41. Li X , Wong J , Tsai SY , Tsai MJ , O'Malley BW. 2003. Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol Cell Biol 23:3763–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smid-Koopman E , Blok LJ , Kühne LC , Burger CW , Helmerhorst TJ , Brinkmann AO , Huikeshoven FJ. 2003. Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J Soc Gynecol Investig 10:49–57 [PubMed] [Google Scholar]
- 43. Qiu M , Olsen A , Faivre E , Horwitz KB , Lange CA. 2003. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol 17:628–642 [DOI] [PubMed] [Google Scholar]
- 44. Dolcet X , Llobet D , Encinas M , Pallares J , Cabero A , Schoenenberger JA , Comella JX , Matias-Guiu X. 2006. Proteasome inhibitors induce death but activate NF-κB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem 281:22118–22130 [DOI] [PubMed] [Google Scholar]
- 45. Dennis AP , Lonard DM , Nawaz Z , O'Malley BW. 2005. Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J Steroid Biochem Mol Biol 94:337–346 [DOI] [PubMed] [Google Scholar]
- 46. Georgiakaki M , Chabbert-Buffet N , Dasen B , Meduri G , Wenk S , Rajhi L , Amazit L , Chauchereau A , Burger CW , Blok LJ , Milgrom E , Lombès M , Guiochon-Mantel A , Loosfelt H. 2006. Ligand-controlled interaction of histone acetyltransferase binding to ORC-1 (HBO1) with the N-terminal transactivating domain of progesterone receptor induces steroid receptor coactivator 1-dependent coactivation of transcription. Mol Endocrinol 20:2122–2140 [DOI] [PubMed] [Google Scholar]
- 47. Liu Z , Wong J , Tsai SY , Tsai MJ , O'Malley BW. 1999. Steroid receptor coactivator-1 (SRC-1) enhances ligand-dependent and receptor-dependent cell-free transcription of chromatin. Proc Natl Acad Sci USA 96:9485–9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith CL , Oñate SA , Tsai MJ , O'Malley BW. 1996. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc Natl Acad Sci USA 93:8884–8888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stenoien DL , Patel K , Mancini MG , Dutertre M , Smith CL , O'Malley BW , Mancini MA. 2001. FRAP reveals that mobility of oestrogen receptor-α is ligand- and proteasome-dependent. Nat Cell Biol 3:15–23 [DOI] [PubMed] [Google Scholar]
- 50. Chen SL , Wang SC , Hosking B , Muscat GE. 2001. Subcellular localization of the steroid receptor coactivators (SRCs) and MEF2 in muscle and rhabdomyosarcoma cells. Mol Endocrinol 15:783–796 [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y , Zhang H , Liang J , Yu W , Shang Y. 2007. SIP, a novel ankyrin repeat containing protein, sequesters steroid receptor coactivators in the cytoplasm. EMBO J 26:2645–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoang T , Fenne IS , Cook C , Borud B , Bakke M , Lien EA , Mellgren G. 2004. cAMP-dependent protein kinase regulates ubiquitin-proteasome-mediated degradation and subcellular localization of the nuclear receptor coactivator GRIP1. J Biol Chem 279:49120–49130 [DOI] [PubMed] [Google Scholar]
- 53. Zhang H , Sun L , Liang J , Yu W , Zhang Y , Wang Y , Chen Y , Li R , Sun X , Shang Y. 2006. The catalytic subunit of the proteasome is engaged in the entire process of estrogen receptor-regulated transcription. EMBO J 25:4223–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mani A , Oh AS , Bowden ET , Lahusen T , Lorick KL , Weissman AM , Schlegel R , Wellstein A , Riegel AT. 2006. E6AP mediates regulated proteasomal degradation of the nuclear receptor coactivator amplified in breast cancer 1 in immortalized cells. Cancer Res 66:8680–8686 [DOI] [PubMed] [Google Scholar]
- 55. Xu J , Wu RC , O'Malley BW. 2009. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer 9:615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fleming FJ , Myers E , Kelly G , Crotty TB , McDermott EW , O'Higgins NJ , Hill AD , Young LS. 2004. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer: a predictive role for SRC-1. J Clin Pathol 57:1069–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang S , Yuan Y , Liao L , Kuang SQ , Tien JC , O'Malley BW , Xu J. 2009. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Natl Acad Sci USA 106:151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qin L , Liu Z , Chen H , Xu J. 2009. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res 69:3819–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han SJ , Jeong J , Demayo FJ , Xu J , Tsai SY , Tsai MJ , O'Malley BW. 2005. Dynamic cell type specificity of SRC-1 coactivator in modulating uterine progesterone receptor function in mice. Mol Cell Biol 25:8150–8165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hudelist G , Czerwenka K , Kubista E , Marton E , Pischinger K , Singer CF. 2003. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat 78:193–204 [DOI] [PubMed] [Google Scholar]
- 61. Muratani M , Tansey WP. 2003. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol 4:192–201 [DOI] [PubMed] [Google Scholar]
- 62. Collins GA , Tansey WP. 2006. The proteasome: a utility tool for transcription? Curr Opin Genet Dev 16:197–202 [DOI] [PubMed] [Google Scholar]
- 63. Perissi V , Aggarwal A , Glass CK , Rose DW , Rosenfeld MG. 2004. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell 116:511–526 [DOI] [PubMed] [Google Scholar]
- 64. Nawaz Z , Lonard DM , Smith CL , Lev-Lehman E , Tsai SY , Tsai MJ , O'Malley BW. 1999. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol 19:1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Molinari E , Gilman M , Natesan S. 1999. Proteasome-mediated degradation of transcriptional activators correlates with activation domain potency in vivo. EMBO J 18:6439–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shao W , Keeton EK , McDonnell DP , Brown M. 2004. Coactivator AIB1 links estrogen receptor transcriptional activity and stability. Proc Natl Acad Sci USA 101:11599–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Salghetti SE , Caudy AA , Chenoweth JG , Tansey WP. 2001. Regulation of transcriptional activation domain function by ubiquitin. Science 293:1651–1653 [DOI] [PubMed] [Google Scholar]
- 68. Chauchereau A , Amazit L , Quesne M , Guiochon-Mantel A , Milgrom E. 2003. Sumoylation of the progesterone receptor and of the steroid receptor coactivator SRC-1. J Biol Chem 278:12335–12343 [DOI] [PubMed] [Google Scholar]
- 69. Ramamoorthy S , Dhananjayan SC , Demayo FJ , Nawaz Z. 2010. Isoform-specific degradation of PR-B by E6-AP is critical for normal mammary gland development. Mol Endocrinol 24:2099–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Calligé M , Kieffer I , Richard-Foy H. 2005. CSN5/Jab1 is involved in ligand-dependent degradation of estrogen receptor α by the proteasome. Mol Cell Biol 25:4349–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chauchereau A , Georgiakaki M , Perrin-Wolff M , Milgrom E , Loosfelt H. 2000. JAB1 interacts with both the progesterone receptor and SRC-1. J Biol Chem 275:8540–8548 [DOI] [PubMed] [Google Scholar]
- 72. Kwak J , Workman JL , Lee D. 17 August 2010. The proteasome and its regulatory roles in gene expression. Biochim Biophys Acta 10.1016/j.bbagrm.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 73. Grenier J , Trousson A , Chauchereau A , Amazit L , Lamirand A , Leclerc P , Guiochon-Mantel A , Schumacher M , Massaad C. 2004. Selective recruitment of p160 coactivators on glucocorticoid-regulated promoters in Schwann cells. Mol Endocrinol 18:2866–2879 [DOI] [PubMed] [Google Scholar]
- 74. Smid-Koopman E , Kuhne LC , Hanekamp EE , Gielen SC , De Ruiter PE , Grootegoed JA , Helmerhorst TJ , Burger CW , Brinkmann AO , Huikeshoven FJ , Blok LJ. 2005. Progesterone-induced inhibition of growth and differential regulation of gene expression in PRA- and/or PRB-expressing endometrial cancer cell lines. J Soc Gynecol Investig 12:285–292 [DOI] [PubMed] [Google Scholar]
- 75. Lorenzo F , Jolivet A , Loosfelt H , Thu vu Hai M , Brailly S , Perrot-Applanat M , Milgrom E. 1988. A rapid method of epitope mapping. Application to the study of immunogenic domains and to the characterization of various forms of rabbit progesterone receptor. Eur J Biochem 176:53–60 [DOI] [PubMed] [Google Scholar]
- 76. Potokar M , Kreft M , Chowdhury HH , Vardjan N , Zorec R. 2006. Subcellular localization of Apaf-1 in apoptotic rat pituitary cells. Am J Physiol Cell Physiol 290:C672–C677 [DOI] [PubMed] [Google Scholar]
- 77. Viengchareun S , Kamenicky P , Teixeira M , Butlen D , Meduri G , Blanchard-Gutton N , Kurschat C , Lanel A , Martinerie L , Sztal-Mazer S , Blot-Chabaud M , Ferrary E , Cherradi N , Lombès M. 2009. Osmotic stress regulates mineralocorticoid receptor expression in a novel aldosterone-sensitive cortical collecting duct cell line. Mol Endocrinol 23:1948–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]