

Background: MitoNEET functions as a [2Fe-2S] cluster transfer protein, and the anti-type II diabetes drug pioglitazone can inhibit cluster transfer.
Results: Binding of NADPH inhibits transfer of the [2Fe-2S] cluster to apo-acceptor proteins.
Conclusion: NADPH, through interactions with Asp-84, inhibits the cluster transfer ability of mitoNEET.
Significance: In the cellular environment, NADPH may act as a key regulator of mitoNEET [2Fe-2S] cluster transfer.
Keywords: Diabetes, Iron-Sulfur Protein, Mitochondrial Diseases, Oxidation-Reduction, Oxidative Stress, Reactive Oxygen Species (ROS), CDGSH, MitoNEET, Pioglitazone, Thiazolidinedione
Abstract
MitoNEET (mNT) is the founding member of the recently discovered CDGSH family of [2Fe-2S] proteins capable of [2Fe-2S] cluster transfer to apo-acceptor proteins. It is a target of the thiazolidinedione (TZD) class of anti-diabetes drugs whose binding modulate both electron transfer and cluster transfer properties. The [2Fe-2S] cluster in mNT is destabilized upon binding of NADPH, which leads to loss of the [2Fe-2S] cluster to the solution environment. Because mNT is capable of transferring [2Fe-2S] clusters to apo-acceptor proteins, we sought to determine whether NADPH binding also affects cluster transfer. We show that NADPH inhibits transfer of the [2Fe-2S] cluster to an apo-acceptor protein with an inhibition constant (Ki) of 200 μm, which reflects that of NADPH concentrations expected under physiological conditions. In addition, we determined that the strictly conserved cluster interacting residue Asp-84 in the CDGSH domain is necessary for the NADPH-dependent inhibition of [2Fe-2S] cluster transfer. The most critical cellular function of NADPH is in the maintenance of a pool of reducing equivalents, which is essential to counteract oxidative damage. Taken together, our findings suggest that NADPH can regulate both mNT [2Fe-2S] cluster levels in the cell as well as the ability of the protein to transfer [2Fe-2S] clusters to cytosolic or mitochondrial acceptors.
Introduction
Iron-sulfur (FeS)2 cluster-containing proteins are key players in many essential processes, such as photosynthesis, respiration, and nitrogen fixation (1, 2). They appear in various compositions and confer upon FeS proteins the ability to accept or donate single electrons and/or iron, catalyze enzymatic reactions, or even function as regulatory proteins (3, 4). Disruption of FeS cluster biogenesis is deleterious to vital cell processes in humans, leading to diseases such as Friedreich ataxia (4, 5), X-linked sideroblastic anemia with ataxia (XLSA/A) (6), and a form of sideroblastic anemia associated with a deletion in the GLRX5 gene (7). The accumulation of iron in mitochondria, which leads to misdistribution of the metal (8) and mismanagement of cellular iron regulatory properties (9, 10), is a hallmark of various diseases. These observations are consistent with the localization of the FeS cluster biogenesis machinery and key FeS protein metabolic functions in mitochondria (2, 9). Moreover, because mitochondria are the primary energy providers of mammalian cells and key players in a large variety of metabolic processes (11), they have been implicated in metabolic diseases such as type II diabetes (12–15).
Human mitoNEET (mNT) is a newly discovered FeS-containing protein that is a target (16) of the insulin-sensitizing thiazolidinedione (TZD) class of type II diabetes drugs (17, 18). The interaction of TZD drugs with mNT has been proposed to be of therapeutic importance (16–20). This is the first FeS protein to be directly targeted by drug binding (16, 20). Crystallization of human mNT revealed that the protein contains two [2Fe-2S] clusters (20–22) (Fig. 1), which are more labile than typical [2Fe-2S] cluster-containing proteins (23). TZD binding to mNT both stabilizes the [2Fe-2S] cluster against release (20) and shifts the redox potential by as much as −100 mV (24). Recently, we discovered that mNT is a cluster transfer protein both in vitro and in vivo (25). TZD binding blocks this process (25). Because NADPH binding was recently found to destabilize the [2Fe-2S] cluster in mNT (26), it poses an interesting question: does NADPH binding alter the cluster transfer or redox properties of mNT?
FIGURE 1.

Surface representation of mNT structure (Protein Data Bank code 2QH7) is shown in gray with [2Fe-2S] cluster as red and yellow spheres. The unusual 3-Cys-1-His ligand coordination found in mNT is shown as blue sticks. Asp-84, part of the CDGSH domain, is shown as well (blue sticks). Residues that show chemical shift changes upon binding of NADPH (26) are shown in magenta. Cluster binding region is shown as a green surface. The sequence (amino acids 33–108) of the mNT construct used in this study is shown to the right.
In this study we used the unique visible spectrum of the [2Fe-2S] cluster in mNT to investigate interactions between the protein and NADPH. We found that NADPH inhibits transfer of the [2Fe-2S] cluster in mNT to an apo-acceptor protein, in this case apo-ferredoxin (a-Fd), at concentrations typically found in cells. We also found that NADPH binding causes a negative shift in the redox potential of mNT in a manner similar to that observed for binding of TZDs, suggesting involvement of His-87 in NADPH binding. Finally, we found that the conserved Asp-84 in the CDGSH domain is essential for inhibition of cluster transfer by NADPH, which suggests a role for the strict conservation of this residue across CDGSH family members. Taken together, these findings suggest a potential role for NADPH as a cellular regulator of the [2Fe-2S] cluster transfer from mNT to apo-acceptor proteins.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Overexpression and purification of the soluble fragment of mNT (amino acids 33–108) as well as a-Fd were performed as outlined previously (25).
Optical Spectroscopy, Cluster Transfer Kinetics, Potentiometric Redox Titrations, and Cluster Stability Measurements
All UV-visible absorption spectra were measured from the near UV to the near IR (300–800 nm) on a Cary50 spectrophotometer (Varian, Palo Alto, CA) equipped with a temperature-controlled cell. The methods employed in this study for monitoring cluster transfer kinetics are identical to those reported previously on mNT (25). Further details on experimental conditions used in FeS cluster transfer studies can be found Wu and Cowan (27). In this study, cluster transfer experiments were performed aerobically at 35 °C at pH 8.0 with and without varying concentrations of NADPH (Fisher Scientific) using 100 μm mNT or D84S mutant and 100 μm a-Fd in 50 mm BisTris, 100 mm NaCl. The samples were covered with mineral oil (Hampton Research) to prevent losses due to evaporation. Transfer rates were obtained by following the (423 nm)/(458 nm) ratio corresponding to loss of the mNT 458 nm peak and emergence of the holo-ferredoxin 423-nm peak with time as described previously (25). Data were fit to a single exponential decay, and transfer rates were determined by taking the slope of the exponential fit after the first 15 min. Transfer rates were then fit to a standard Michaelis-Menten curve.
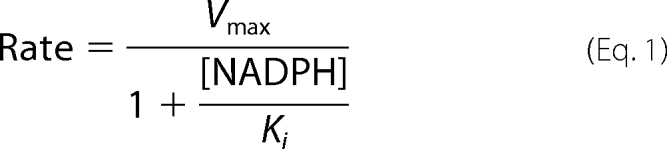 |
In the equation, Vmax is the maximum transfer rate in the presence of 100 μm mNT and a-Fd in the absence of any NADPH (measured to be 225 m−1 min−1), Ki is the inhibition constant for the inhibitor NADPH.
A detailed explanation of optical potentiometric redox titration methods can be found elsewhere (28). For this study, optical potentiometric redox titrations were performed as outlined previously (29) but with the addition of 10 mm NADPH where noted. Briefly, experiments were performed anaerobically at 25 °C under an argon atmosphere with and without 10 mm NADPH using 50 μm mNT in 50 mm BisTris, 50 mm Tris, 100 mm NaCl (to span the pH range 6.0–9.0) at varying pH values in the presence of mediators. Sodium dithionite (Sigma-Aldrich) was titrated in via syringe to reduce the [2Fe-2S] clusters, and once fully reduced, the clusters were reoxidized by titrating in fixed amounts of ambient oxygen through a syringe. Optical scans (300–700 nm) were performed with each addition of dithionite or oxygen. Optical potentiometric redox titration data were fit to the Nernst equation, and redox potentials were plotted as a function of pH as described previously (29). From this fit the variables pKox and Eacid could be determined in the presence and absence of NADPH.
Cluster stability measurements were performed aerobically at 35 °C at pH 6.0 using 50 μm D84S or H87C mutant with and without 10 mm NADPH in 100 mm BisTris, 100 mm NaCl. Cluster loss was measured over time as a decrease in absorbance at 458 nm.
RESULTS
NADPH Inhibits Transfer of [2Fe-2S] Clusters from mNT to an Apo-acceptor Protein
NADPH is the first identified physiological coenzyme that binds to mNT (26), as recently shown by measuring NMR chemical shift perturbations in specific residues in mNT in the presence of NADPH (26). However, NMR methods are not easily amenable to monitor the FeS cluster directly because of the paramagnetic broadening of residues surrounding the cluster (Fig. 1). To determine whether NADPH binding affects [2Fe-2S] cluster transfer of to an acceptor protein (25) we tested for cluster transfer in the presence of NADPH. NADPH inhibits transfer of the [2Fe-2S] cluster to a-Fd in a concentration-dependent manner (Fig. 2A). Fitting a standard Michaelis-Menten curve to the data (Fig. 2B) yields an inhibition constant (Ki) equal to 200 ± 60 μm. This value falls within the expected physiological concentration of NADPH, which varies between different organisms as well as different human tissues but is estimated to be between 100 and 200 μm (30, 31).
FIGURE 2.

Binding of NADPH inhibits transfer of [2Fe-2S] cluster from mNT to a-Fd. A, oxidized mNT was incubated with a-Fd, and cluster transfer was observed with time as reported previously (25) (black circles). To test the effect of NADPH on the cluster transfer rate, oxidized mNT was incubated with a-Fd and varying concentrations of NADPH: 0.1 mm (red triangles), 0.5 mm (blue triangles), 1 mm (green squares), and 10 mm (purple diamonds). Cluster transfer experiments were performed at 35 °C at pH 8.0 with and without varying concentrations of NADPH using 100 μm mNT and 100 μm a-Fd in 50 mm Tris, 100 mm NaCl. B, cluster transfer rates were plotted as a function of NADPH concentration. Rates were fit to a Michaelis-Menten curve, giving inhibition constant (Ki) = 200 ± 60 μm.
Binding of NADPH Leads to Shift in the pH-dependent Redox Potential
In this work we used visible spectroscopy (Fig. 3A) to determine the effect of the coenzyme on mNT cluster properties (29, 32–34) such as redox potential. In addition, we tested the coenzyme sensitivity of mutant mNT in which mutations were made in the CDGSH domain (Fig. 3B). Upon addition of NADPH, mNT is not reduced even after a 25-h incubation (Fig. 3A, blue line). No reduction of the [2Fe-2S] cluster is observed even though the redox potential for NADPH is −320 mV, which is significantly lower than that of the [2Fe-2S clusters] of mNT (∼0 mV) (24). Thus, the reduction of mNT is clearly kinetically inhibited. To determine whether binding of NADPH affects the redox potential of mNT we performed optical potentiometric titrations (28, 29) on the protein in the presence of 10 mm NADPH and mediators (Fig. 3C). We observed a decrease in redox potential of −40 mV at pH 6.0. As a control we found that redox changes were not observed in the presence of NADH (Fig. 3D), which does not bind mNT (26). Thus, the additional phosphate group of NADPH is essential for the binding of NADPH to mNT.
FIGURE 3.

Optical potentiometric titrations in the presence of NADPH lead to changes in redox properties of mNT. A, visible spectrum of mNT (black line). Addition of NADPH in the absence of mediators does not reduce mNT even after 25 h (blue line). Visible spectrum of NADPH (red line) shows that there is no overlap at 458 nm where mNT has a λmax that is used for monitoring the [2Fe-2S] cluster. B, directly coordinating ligands: Cys-72, Cys-74, Cys-83, and His-87 as well as cluster-interacting residue Asp-84. C and D, redox potential of mNT measured by titrating dithionite into a solution of oxidized mNT (50 μm) with or without 10 mm NADPH or NADH in the presence of 100 mm BisTris, 100 mm NaCl, pH 6.0, and mediators.
Because the redox properties of mNT are pH-dependent (24) we wished to explore whether NADPH binding affects the pH profile. The pH-dependent redox potential of the [2Fe-2S] cluster in mNT can be adequately described as being coupled to a single titrating group. Two terms describe the pH-dependent redox potential for mNT; they are Eacid and pKox, which have been described in more detail previously (29). Briefly, Eacid is the redox potential of the protein when a single redox-coupled group is in the fully protonated state, whereas pKox is the pH at which the redox-coupled group is half-protonated. Eacid for mNT was previously shown to be +40 mV and pKox to be 6.8. The redox-coupled group which undergoes protonation upon reduction of the [2Fe-2S] cluster was attributed to the noncluster coordinating Nϵ of His-87 (29). The redox shift for mNT in the presence of NADPH is more dramatic at pH <7.0 and less so at pH >7.0 (Fig. 4, black and white circles), indicating that binding of NADPH shifts pKox from pH 6.8 to pH 7.8. It is also clear that Eacid has been shifted −40 mV (Fig. 4). We attribute this negative change in Eacid to the addition of multiple negative charges from the phosphate groups of NADPH. Placement of a negative charge near the [2Fe-2S] cluster should discourage the further addition of negative charge resulting from a single electron reduction, which consequently results in a lower measured value for Eacid. Having previously proposed that pKox is due to protonation of His-87 (29), these results suggest that binding of NADPH either directly or indirectly involves His-87.
FIGURE 4.

Binding of NADPH shifts redox properties (Eacid and pKox) of [2Fe-2S] cluster. In the presence of 10 mm NADPH, Eacid drops from +40 mV (24) to ∼0 mV. In addition, pKox changes from 6.8 (29) to 7.8. Experiments were performed at 25 °C with and without 10 mm NADPH using 50 μm mNT in 50 mm BisTris, 50 mm Tris, 100 mm NaCl at varying pH in the presence of mediators.
Accelerated [2Fe-2S] Cluster Loss as Result of NADPH Binding in Ferredoxin-like 4-Cys mNT Mutant (H87C)
The single-coordinating histidine, His-87, is critical for [2Fe-2S] cluster stability (35), cluster transfer (25), and redox properties (24). These cluster properties in the mutant protein are intriguing in light of the fact that the H87C mutant maintains a structure nearly identical to that of the WT protein (35) (Fig. 5A). Accelerated loss of the [2Fe-2S] cluster by NADPH (t½ = 700 min) was still observed in the H87C mutant (t½ = 2200 min H87C alone) (Fig. 5B) despite the fact that optical potentiometric titrations on the H87C mutant show no redox changes in the presence of 10 mm NADPH (Fig. 5C). Taken together, the results suggest that His-87 is partly responsible for the observed pKox shift induced by NADPH binding in WT mNT, yet this strictly conserved residue is not essential for binding to NADPH and accelerating cluster loss.
FIGURE 5.

NADPH binding leads to accelerated cluster loss in 4-Cys mNT (H87C) in the absence of a redox shift. A, structure of H87C mutant (Protein Data Bank code 3LPQ) showing 4-Cys coordination (35). Cys-87 is found in two conformations in the crystal structure, and both conformations are shown (35). B, optical decay experiments showing time-dependent decrease in absorbance at 458 nm, indicating loss of [2Fe-2S] cluster for H87C mutant in the presence (black triangles) and absence of NADPH (white triangles). C, redox measurements showing no shift in redox potential at pH 6.0 in the presence (black triangles) and absence of NADPH (white triangles). Decay experiments were performed at 35 °C at pH 6.0 using 50 μm H87C mutant with and without 10 mm NADPH in 100 mm BisTris, 100 mm NaCl. Redox experiments were performed under similar conditions but at 25 °C.
[2Fe-2S] Cluster-interacting Residue Asp-84 in CDGSH Domain Is Necessary for Inhibition of [2Fe-2S] Cluster Transfer by NADPH
The next residue to examine in the CDGSH cluster binding region is the strictly conserved Asp-84, which is within hydrogen bond distance of one of the sulfurs in the [2Fe-2S] cluster but invisible in previous NMR studies. Replacement of conserved Asp-84 with Ser resulted in no accelerated cluster loss at pH 6.0 in the presence of NADPH (t½ ∼10 min in both cases) (Fig. 6, black and white circles). To determine whether the natural decay of the cluster in the D84S mutant at pH 6.0 is significantly faster than any accelerated cluster loss induced by NADPH binding, the experiment was performed at pH 7.5, where the [2Fe-2S] cluster is an order of magnitude more stable in the D84S mutant. No increased cluster loss of the D84S mutant by NADPH (t½ ∼1000 min in both cases) was observed (Fig. 6A, black and white triangles). Having observed that the D84S mutant was no longer destabilized in the presence of NADPH, we decided to test whether cluster transfer was inhibited for the D84S mutant in the presence of NADPH. Replacement of Asp-84 with Ser likely decreases the binding affinity of mNT for NADPH because cluster transfer was not noticeably inhibited (Fig. 6B) even at 10 mm NADPH, which is significantly higher than physiological concentrations (∼200 μm).
FIGURE 6.

Asp-84 necessary for inhibition of [2Fe-2S] cluster transfer and accelerated cluster loss by NADPH. A, replacement of Asp-84 with Ser (D84S) prevents accelerated loss of the cluster in the presence of NADPH. Experiments were performed at 35 °C at pH 6.0 (black and white squares) or 7.5 (black and white squares) using 50 μm protein with or without 10 mm NADPH in the presence of 100 mm BisTris, 100 mm NaCl. B, transfer of [2Fe-2S] cluster from D84S mutant to a-Fd in the presence (white squares) and absence (black squares) of 10 mm NADPH. Transfer experiments were performed using 100 μm D84S mutant and 100 μm a-Fd in 50 mm BisTris, 100 mm NaCl with and without 10 mm NADPH.
Our examination of the CDGSH cluster binding domain of this [2Fe-2S] protein implicated in type II diabetes using optical methods shows that the strictly conserved cluster-interacting residue Asp-84 plays a direct role in NADPH binding and inhibition of [2Fe-2S] cluster transfer. Taken together, our results show that NADPH binding alters cluster stability and cluster transfer properties in this protein, and this likely has physiological consequences.
DISCUSSION
The recent discovery that NADPH binds to mNT and affects the stability of the [2Fe-2S] cluster led us to investigate whether the redox and cluster transfer properties of mNT are perturbed by interaction with this ubiquitous cofactor. The NMR methods previously showed specific residues that interact with NADPH, but the technical limitation of the paramagnetic broadening NMR technology imposed the difficulty of observing chemical shifts for residues in close proximity to the paramagnetic [2Fe-2S] cluster, specifically regarding the conserved CDGSH residues Asp-84 and His-87. The latter left open the possibility that residues in this region could be critical for facilitating both NADPH binding and the corresponding effects on cluster properties, e.g. cluster stability, cluster redox potential, and cluster transfer. Our investigation led us to the discovery that binding of NADPH to mNT inhibits the [2Fe-2S] cluster from being transferred to a-Fd (Fig. 2B). This inhibition occurs even though the [2Fe-2S] cluster in mNT is oxidized in these experiments, when transfer is expected to occur readily. In addition, the conserved Asp-84 in the CDGSH domain of the protein is critical for inhibition of [2Fe-2S] cluster transfer by NADPH.
A functional role for Asp-84 in the binding of NADPH may explain why this residue is highly conserved through evolution and in all CDGSH proteins. Because NADPH plays a very important functional role in providing strong reducing equivalents in the cytosolic environment of all organisms, the inhibition of [2Fe-2S] cluster transfer in mNT at physiological concentrations of ∼200 μm suggests a functional importance to this binding interaction and the likely necessity of this aspartyl group over the course of evolution. It is likely no accident that this conserved aspartyl group is positioned potentially to interact with both the sulfur moiety of the [2Fe-2S] cluster as well as a hydrogen bond donor on NADPH.
Our findings led us to propose an additional dimension to our understanding of the physiological role of [2Fe-2S] cluster transfer in mNT and how the protein behaves in patients with type II diabetes (25). We propose that NADPH acts as a regulator of mNT [2Fe-2S] cluster transfer, whereby it not only prevents transfer of the [2Fe-2S] cluster to acceptor proteins or into the mitochondrial matrix, but also accelerates loss of the [2Fe-2S] cluster. The latter may be a means of down-regulating the protein under normal reducing conditions (Fig. 7). Our model proposes that under normal cytosolic conditions, i.e. reducing environment (36), mNT is incapable of cluster transfer and accumulation of Fe in the mitochondria is abrogated. If NADPH levels are dramatically decreased, such as under conditions of oxidative stress, NADPH is no longer available to prevent [2Fe-2S] cluster transfer to apo-acceptor proteins, nor is it able to down-regulate the protein. In this case iron accumulation occurs in the mitochondria, which is often observed in patients with type II diabetes (37, 38), and such a phenomenon is believed to damage cells, especially those that produce insulin (39–41). TZDs may act therapeutically by imitating the normal [2Fe-2S] cluster transfer inhibitory effects of NADPH. This view is supported by the fact that both NADPH and the TZDs cause an alkaline shift in pKox through either direct or indirect interactions with cluster-binding residue His-87 (24). A complete understanding of the role of NADPH association with members of the CGDSH family still remains to be understood. Specifically, whether NADPH binding shows similar effects on the CDGSH family member, Miner1, whose up-regulation leads to greater longevity in mice (42) but whose missplicing of the Miner1 gene, CISD2, leads to a rare but serious human disease known as Wolfram syndrome 2 (43). Determining whether NADPH inhibits cluster transfer in Miner1 is of high priority, as is determining whether NADPH can disrupt or enhance binding of Miner1 to antiautophagic/antiapoptotic proteins in the Bcl-2 family (44).
FIGURE 7.

Model for NADPH regulation of mNT stability and cluster transfer properties. Under nominal conditions, NADPH inhibits [2Fe-2S] cluster transfer from mNT to an apo-acceptor protein and also causes accelerated loss of the [2Fe-2S] cluster in mNT. This provides two different means to regulate the activity of the protein under normal cellular conditions. However, when the cell experiences oxidative stress, mNT [2Fe-2S] clusters become oxidized and thus capable of [2Fe-2S] cluster transfer. As oxidative stress also leads to a decrease in NADPH levels (19), the inhibitory effect of NADPH on cluster transfer is alleviated and transfer can proceed.
Here, we show [2Fe-2S] cluster transfer is inhibited by NADPH at physiological concentrations of the coenzyme, indicating that NADPH, previously shown to interact with the protein and accelerate loss of the [2Fe-2S] cluster, also affects a functional property of the protein: [2Fe-2S] cluster transfer to apo-acceptor proteins. We here propose a likely physiological role for the mNT-NADPH interaction, which is to prevent cluster transfer regardless of the [2Fe2S] oxidization state and to down-regulate the protein under normal reducing conditions.
Acknowledgments
We thank Lindsey Handley and Maria Luca for assistance in decay and redox measurements and Charles Wang for assistance in cluster transfer experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants GM41637 (to M. L. P.), GM54038 (to P. A. J.), and DK54441 (to P. A. J.). This work was also supported by Heme and Blood Proteins Training Grant 5T32DK007233-34 (to J. A. Z.) and Israel Science Foundation Grant 863/09 (to R. N.).
- FeS
- iron-sulfur
- a-Fd
- apo-ferredoxin
- BisTris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- Eacid
- redox potential at low pH
- mNT
- mitoNEET
- pKox
- pH at which titrating redox species is half-protonated
- TZD
- thiazolidinedione.
REFERENCES
- 1. Johnson D. C., Dean D. R., Smith A. D., Johnson M. K. (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74, 247–281 [DOI] [PubMed] [Google Scholar]
- 2. Lill R., Mühlenhoff U. (2008) Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700 [DOI] [PubMed] [Google Scholar]
- 3. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 4. Rouault T. A., Tong W. H. (2008) Iron-sulfur cluster biogenesis and human disease. Trends Genet. 24, 398–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adinolfi S., Iannuzzi C., Prischi F., Pastore C., Iametti S., Martin S. R., Bonomi F., Pastore A. (2009) Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 16, 390–396 [DOI] [PubMed] [Google Scholar]
- 6. Napier I., Ponka P., Richardson D. R. (2005) Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105, 1867–1874 [DOI] [PubMed] [Google Scholar]
- 7. Camaschella C., Campanella A., De Falco L., Boschetto L., Merlini R., Silvestri L., Levi S., Iolascon A. (2007) The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 8. Sohn Y. S., Breuer W., Munnich A., Cabantchik Z. I. (2008) Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood 111, 1690–1699 [DOI] [PubMed] [Google Scholar]
- 9. Hausmann A., Samans B., Lill R., Mühlenhoff U. (2008) Cellular and mitochondrial remodeling upon defects in iron-sulfur protein biogenesis. J. Biol. Chem. 283, 8318–8330 [DOI] [PubMed] [Google Scholar]
- 10. Hentze M. W., Muckenthaler M. U., Andrews N. C. (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117, 285–297 [DOI] [PubMed] [Google Scholar]
- 11. Hansen T. M., Nagley P. (2003) AIF: a multifunctional cog in the life and death machine. Sci. STKE 2003, PE31. [DOI] [PubMed] [Google Scholar]
- 12. Cusimano E. M., Knight A. R., Slusser J. G., Clancy R. L., Pierce J. D. (2009) Mitochondria: the hemi of the cell. Adv. Emerg. Nurs. J. 31, 54–62 [DOI] [PubMed] [Google Scholar]
- 13. Abdul-Ghani M. A., DeFronzo R. A. (2008) Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Curr. Diab. Rep. 8, 173–178 [DOI] [PubMed] [Google Scholar]
- 14. Lowell B. B., Shulman G. I. (2005) Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387 [DOI] [PubMed] [Google Scholar]
- 15. Saxena R., de Bakker P. I., Singer K., Mootha V., Burtt N., Hirschhorn J. N., Gaudet D., Isomaa B., Daly M. J., Groop L., Ardlie K. G., Altshuler D. (2006) Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am. J. Hum. Genet. 79, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Colca J. R., McDonald W. G., Waldon D. J., Leone J. W., Lull J. M., Bannow C. A., Lund E. T., Mathews W. R. (2004) Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am. J. Physiol. Endocrinol. Metab. 286, E252–260 [DOI] [PubMed] [Google Scholar]
- 17. Colca J. R., Kletzien R. F. (2006) What has prevented the expansion of insulin sensitisers? Expert Opin. Investig. Drugs 15, 205–210 [DOI] [PubMed] [Google Scholar]
- 18. Hofmann C. A., Colca J. R. (1992) New oral thiazolidinedione antidiabetic agents act as insulin sensitizers. Diabetes Care 8, 1075–1078 [DOI] [PubMed] [Google Scholar]
- 19. Pollak N., Dölle C., Ziegler M. (2007) The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem. J. 402, 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paddock M. L., Wiley S. E., Axelrod H. L., Cohen A. E., Roy M., Abresch E. C., Capraro D., Murphy A. N., Nechushtai R., Dixon J. E., Jennings P. A. (2007) MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. U.S.A. 104, 14342–14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin J., Zhou T., Ye K., Wang J. (2007) Crystal structure of human mitoNEET reveals distinct groups of iron sulfur proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 14640–14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou X., Liu R., Ross S., Smart E. J., Zhu H., Gong W. (2007) Crystallographic studies of human MitoNEET. J. Biol. Chem. 282, 33242–33246 [DOI] [PubMed] [Google Scholar]
- 23. Wiley S. E., Paddock M. L., Abresch E. C., Gross L., van der Geer P., Nechushtai R., Murphy A. N., Jennings P. A., Dixon J. E. (2007) The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster. J. Biol. Chem. 282, 23745–23749 [DOI] [PubMed] [Google Scholar]
- 24. Bak D. W., Zuris J. A., Paddock M. L., Jennings P. A., Elliott S. J. (2009) Redox characterization of the FeS protein MitoNEET and impact of thiazolidinedione drug binding. Biochemistry 48, 10193–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zuris J. A., Harir Y., Conlan A. R., Shvartsman M., Michaeli D., Tamir S., Paddock M. L., Onuchic J. N., Mittler R., Cabantchik Z. I., Jennings P. A., Nechushtai R. (2011) Facile transfer of [2Fe-2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proc. Natl. Acad. Sci. U.S.A. 108, 13047–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou T., Lin J., Feng Y., Wang J. (2010) Binding of reduced nicotinamide adenine dinucleotide phosphate destabilizes the iron−sulfur clusters of human mitoNEET. Biochemistry 49, 9604–9612 [DOI] [PubMed] [Google Scholar]
- 27. Wu S. P., Cowan J. A. (2003) Iron-sulfur cluster biosynthesis: a comparative kinetic analysis of native and Cys-substituted ISA-mediated [2Fe-2S]2+ cluster transfer to an apoferredoxin target. Biochemistry 42, 5784–5791 [DOI] [PubMed] [Google Scholar]
- 28. Dutton P. L. (1978) Redox potentiometry: determination of midpoint potentials of oxidation-reduction components of biological electron-transfer systems. Methods Enzymol. 54, 411–435 [DOI] [PubMed] [Google Scholar]
- 29. Zuris J. A., Halim D. A., Conlan A. R., Abresch E. C., Nechushtai R., Paddock M. L., Jennings P. A. (2010) Engineering the redox potential over a wide range within a new class of FeS proteins. J. Am. Chem. Soc. 132, 13120–13122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Albe K. R., Butler M. H., Wright B. E. (1990) Cellular concentrations of enzymes and their substrates. J. Theor. Biol. 143, 163–195 [DOI] [PubMed] [Google Scholar]
- 31. Andersen K. B., von Meyenburg K. (1977) Charges of nicotinamide adenine nucleotides and adenylate energy charge as regulatory parameters of the metabolism in Escherichia coli. J. Biol. Chem. 252, 4151–4156 [PubMed] [Google Scholar]
- 32. Dicus M. M., Conlan A., Nechushtai R., Jennings P. A., Paddock M. L., Britt R. D., Stoll S. (2010) Binding of histidine in the (Cys)3(His)1-coordinated [2Fe-2S] cluster of human mitoNEET. J. Am. Chem. Soc. 132, 2037–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baxter E. L., Jennings P. A., Onuchic J. N. (2011) Interdomain communication revealed in the diabetes drug target mitoNEET. Proc. Natl. Acad. Sci. U.S.A. 108, 5266–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tirrell T. F., Paddock M. L., Conlan A. R., Smoll E. J., Jr., Nechushtai R., Jennings P. A., Kim J. E. (2009) Resonance Raman studies of the (His)(Cys)3 2Fe-2S cluster of MitoNEET: comparison to the (Cys)4 mutant and implications of the effects of pH on the labile metal center. Biochemistry 48, 4747–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conlan A. R., Paddock M. L., Homer C., Axelrod H. L., Cohen A. E., Abresch E. C., Zuris J. A., Nechushtai R., Jennings P. A. (2011) Mutation of the His ligand in mitoNEET stabilizes the 2Fe-2S cluster despite conformational heterogeneity in the ligand environment. Acta Crystallogr. D Biol. Crystallogr. 67, 516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinovich G. G., Cherenkevich S. N., Sauer H. (2005) Intracellular redox state: towards quantitative description. Eur. Biophys. J. 34, 937–942 [DOI] [PubMed] [Google Scholar]
- 37. Hernandez C., Genesca J., Ignasi Esteban J., Garcia L., Simo R. (2000) Relationship between iron stores and diabetes mellitus in patients infected by hepatitis C virus: a case-control study. Med. Clin. 115, 21–22 [DOI] [PubMed] [Google Scholar]
- 38. Jiang R., Manson J. E., Meigs J. B., Ma J., Rifai N., Hu F. B. (2004) Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA 291, 711–717 [DOI] [PubMed] [Google Scholar]
- 39. Opara E. C. (2004) Role of oxidative stress in the etiology of type 2 diabetes and the effect of antioxidant supplementation on glycemic control. J. Investig. Med. 52, 19–23 [DOI] [PubMed] [Google Scholar]
- 40. Valko M., Morris H., Cronin M. T. (2005) Metals, toxicity and oxidative stress. Curr. Med. Chem. 12, 1161–1208 [DOI] [PubMed] [Google Scholar]
- 41. Wolff S. P. (1993) Diabetes mellitus and free radicals: free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br. Med. Bull. 49, 642–652 [DOI] [PubMed] [Google Scholar]
- 42. Chen Y. F., Wu C. Y., Kirby R., Kao C. H., Tsai T. F. (2010) A role for the CISD2 gene in lifespan control and human disease. Ann. N.Y. Acad. Sci. 1201, 58–64 [DOI] [PubMed] [Google Scholar]
- 43. Amr S., Heisey C., Zhang M., Xia X. J., Shows K. H., Ajlouni K., Pandya A., Satin L. S., El-Shanti H., Shiang R. (2007) A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 81, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang N. C., Nguyen M., Germain M., Shore G. C. (2010) Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 29, 606–618 [DOI] [PMC free article] [PubMed] [Google Scholar]