

Background: Deleting the acetyl-CoA carboxylase 2 gene (ACC2) in mice results in continuous fatty acid oxidation.
Results: Less fat accumulates in the liver, despite up-regulation of the de novo lipogenic pathway and dietary induction of fatty liver.
Conclusion: Continuous fatty acid oxidation results in a futile metabolic cycle.
Significance: ACC2-specific inhibitors could be a viable treatment against fatty liver disease.
Keywords: Fatty Acid Metabolism, Fatty Acid Oxidation, Fatty Acid Synthase, Gene Expression, Gene Knockout, Lipids, Lipid Droplet, Lipid Metabolism, Lipotoxicity, Liver Metabolism
Abstract
Hepatic fat accumulation resulting from increased de novo fatty acid synthesis leads to hepatic steatosis and hepatic insulin resistance. We have shown previously that acetyl-CoA carboxylase 2 (Acc2−/−) mutant mice, when fed a high-fat (HF) or high-fat, high-carbohydrate (HFHC) diet, are protected against diet-induced obesity and maintained whole body and hepatic insulin sensitivity. To determine the effect of an ACC2 deletion on hepatic fat metabolism, we studied the regulation of the enzymes involved in the lipogenic pathway under Western HFHC dietary and de novo lipogenic conditions. After completing the HFHC regimen, Acc2−/− mutant mice were found to have lower body weight, smaller epididymal fat pads, lower blood levels of nonesterified fatty acids and triglycerides, and higher hepatic cholesterol than wild-type mice. Significant up-regulation of lipogenic enzymes and an elevation in hepatic peroxisome proliferator-activated receptor-γ (PPAR-γ) protein were found in Acc2−/− mutant mice under de novo lipogenic conditions. The increase in lipogenic enzyme levels was accompanied by up-regulation of the transcription factors, sterol regulatory element-binding proteins 1 and 2, and carbohydrate response element-binding protein. In contrast, hepatic levels of the PPAR-γ and PPAR-α proteins were significantly lower in the Acc2−/− mutant mice fed an HFHC diet. When compared with wild-type mice fed the same diet, Acc2−/− mutant mice exhibited a similar level of AKT but with a significant increase in pAKT. Hence, deleting ACC2 ameliorates the metabolic syndrome and protects against fatty liver despite increased de novo lipogenesis and dietary conditions known to induce obesity and diabetes.
Introduction
The accumulation of excessive triglycerides (TGs)4 in animal tissues, including the liver, is strongly associated with insulin resistance, a major component of the metabolic syndrome (1, 2). The intracellular pooling of hepatic TGs results from increased uptake and esterification of exogenous fatty acids from dietary sources, increased de novo synthesis from acetyl-CoA, and/or a decrease in fatty acid oxidation (3, 4).
An unresolved question in this process is whether the isoenzymes of acetyl-CoA carboxylase, ACC1 and ACC2, play distinct and separate roles in the synthesis of fatty acids and the regulation of fatty acid oxidation, respectively. Another unresolved question is how these isoenzymes are interrelated. Both ACC1 and ACC2 are highly homologous and share the serine residues implicated in their inactivation by AMP kinase-mediated phosphorylation (5, 6). Such tight, coordinated regulation of the ACC1 and ACC2 activities would likely prevent simultaneous fatty acid oxidation and synthesis, resulting in a futile metabolic cycle.
A key to a viable cycle may be the possible compartmentalization of ACC1 and ACC2 and their malonyl-CoA products in the cytosol and mitochondria, respectively (7). ACC1-synthesized malonyl-CoA is the two-carbon donor in the synthesis of fatty acids by the fatty acid synthase (FAS); ACC2-synthesized malonyl-CoA is the regulator of mitochondrial fatty acid oxidation through its inhibition of carnitine palmitoyltransferase 1 (7, 8). Both ACC1 and ACC2 are expressed at significant levels in the liver (5, 6), making this organ unique in its ability to synthesize fatty acids (lipogenesis) in nourished animals and to oxidize them (ketogenesis) in starved animals. Under lipogenic conditions, such as when starved animals are fed a fat-free, high-carbohydrate (FFHC) diet, the hepatic levels of ACC and FAS are increased severalfold, resulting in increased de novo fatty acid synthesis (9, 10).
Under dietary conditions that promote obesity and insulin resistance in humans and other mammals, free fatty acids are elevated in the blood and enter the liver where they are esterified to form TGs instead of being oxidized. On the other hand, de novo fatty acid synthesis is markedly increased under these pathological conditions as a result of the activation of transcription factors, such as sterol regulatory element-binding protein 1 (SREBP-1) and peroxisome proliferator-activated receptor γ (PPAR-γ), in lipogenic tissues (11).
As we have shown previously, Acc2−/− mutant mice are protected against obesity and type 2 diabetes as induced by an HFHC diet (12). We subsequently showed that because of continuous fatty acid oxidation, Acc2−/− mutant mice fed an HF diet were protected against fat-induced peripheral and hepatic insulin resistance (13). The improvement in insulin-stimulated glucose metabolism resulted in part from enhanced insulin signaling in the liver, as evidenced by a significant increase in insulin-induced repression of hepatic glucose production (13). Herein, we report our findings from a study of the effects of a chronic ACC2 deletion on liver lipogenesis in Acc2−/− mutant mice under different dietary conditions.
EXPERIMENTAL PROCEDURES
Animals and Dietary Conditions
Male Acc2−/− mutant and wild-type mice (Sv/129) were housed under controlled environmental conditions (12-h light/dark cycle; 25 °C temperature) in the Animal Care Center at Baylor College of Medicine. Animal experiments were conducted in accordance with the NIH guidelines (32). Five mice, either all Acc2−/− mutants or all wild-type, were housed per cage and had free access to standard laboratory chow (Purina Mills, Richmond, IN). To study the effects of an HFHC diet (59% of calories derived from fat and 24% from carbohydrates; Bioserv, Frenchtown, NJ), 3–4-month-old Acc2−/− mutant mice were fed this diet for 2 months. To create lipogenic conditions, wild-type and Acc2−/− mutant mice between 4 and 5 months of age underwent a 48-h fast and were subsequently fed an FFHC diet for 48 h. Under fasting conditions, food was removed for 48 h, and the mice had access to water only.
Measurement of Blood Constituents
Whole blood was withdrawn from the tails of the Acc2−/− mutant and wild-type mice after a brief fasting period of 4–5 h, and the serum constituents were determined as described previously (7, 12). Serum levels of glucose, TGs, total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, and very low-density lipoprotein cholesterol were measured by technicians in the Comparative Pathology Laboratory at Baylor College of Medicine. Serum nonesterified fatty acids (NEFAs) were measured by using a NEFA C kit (Wako Chemicals, Richmond, VA).
Liver Analyses
All mice were sacrificed after completing their respective dietary regimens, and the livers and epididymal fat pads of the individual animals were weighed. All the livers were surgically sectioned. For each liver, some of the sections were stained with Oil Red O to visualize the TGs as previously described (8); the remaining sections were frozen in liquid nitrogen and kept at −80 °C for further analysis. The tissue concentrations of TGs and cholesterol were measured by using a Cholesterol E Kit (Wako Chemicals) and an Infinity Triglyceride Kit (Thermo Electron, Melbourne, Australia) as described previously (14).
Enzymatic Activities and Western Blot Analyses
The preparation of liver extracts for enzyme activities and Western blot analyses was carried out as described previously (15). A portion of the frozen liver was ground to powder in liquid nitrogen. The powder was suspended in 10 ml of PBS containing 0.1 mm phenylmethanesulfonyl fluoride, 5 mm benzamidine, and 5 mg/ml of a protease inhibitor mixture. The suspension was homogenized using a Polytron homogenizer (3 × 30 s at high speed) and was sonicated briefly to degrade the DNA. The extracts were clarified by centrifugation at 16,000 × g for 20 min. The concentrations of the proteins in the supernatant were measured, and the protein precipitate was subjected to Western blot analysis by using antibodies against FAS, ACC, p-ACC, ACL, PPAR-γ, PPAR-α, AMPK-α, pAMPK, AKT, pAKT, and SCD1. The intensities of specific bands of the proteins of interest were scanned and normalized against Ponceau staining for quantification. The FAS and ACC activities in the liver extracts were determined as described previously (15).
Real-time Quantitative PCR
Total RNA was isolated from liver tissues using an RNeasy Plus Micro kit (Qiagen). The cDNA was synthesized in a 20-μl volume containing 5 μg of RNA, 0.25 μg of random hexamer, 200 μm dNTPs, 10 mm DTT, 20 units of RNase inhibitor, and 0.5 μl of Superscript II enzyme (Invitrogen). Real-time quantitative PCR was performed with a DNA Engine Opticon 2 system by using DynamoTM HS SYBR Green qPCR (New England Biolabs), as described previously (15, 16). Specific primer sequences for the cDNAs of ACC1, FAS, mevalonate pyrophosphate decarboxylase, ATP citrate-lyase (ACL), stearoyl-CoA desaturase 1 (SCD1), 3-hydroxy-3-methylglutaryl-CoA reductase, SREBP-1, SREBP-2, carbohydrate response element-binding protein (ChREBP), PPAR-α, and PPAR-γ were described previously (15–17) and are available upon request. RNA samples were normalized for comparison by determining the 18 S levels with real-time quantitative PCR. The relative quantity of each gene was calculated by using the 2−ΔΔCT formula, in which ΔΔCT equals the difference between the CT values of mRNA from Acc2−/− mutant mice and wild-type cohorts (16–18).
Northern Blot Analysis
Total RNA (6–8 μg) was subjected to electrophoresis on a 1% agarose gel in the presence of formalin. The fractionated RNA was transferred to Hybond N filters (Amersham Biosciences), which were hybridized with 32P-labeled cDNA probes for PPAR-α and PPAR-γ.
RESULTS
Effects of Different Dietary Conditions on Blood Constituents
To determine the role ACC2 plays in the metabolic changes that occur in serum under different dietary conditions, we measured and compared the levels of these metabolites in 4–5-month-old male Acc2−/− mutant and control mice. When both groups of mice were fed normal chow after a brief fast (∼4–5 h), there was little difference in the total cholesterol levels between the groups; however, the serum levels of TGs and VLDL cholesterol were about 30% less than in wild-type controls (p < 0.05; Table 1). In addition, serum glucose levels in the Acc2−/− mutant mice were 10% lower than those in the wild-type mice (p = 0.1; Table 1). When both groups of mice were fed an HFHC diet, the Acc2−/− mutants had significantly lower serum glucose levels (p = 0.01; Table 1). In addition, the serum levels of TGs and VLDL cholesterol were about 40% lower in the Acc2−/− mutants (Table 1). Interestingly, however, the serum levels of total, HDL, and LDL cholesterols were about 30% higher in the Acc2−/− mutant mice (p < 0.05; Table 1).
TABLE 1.
Serum metabolite data from wild-type and Acc2−/− mutant mice under different dietary conditions
Blood was withdrawn from tails after a 5–6-h fasting period, and serum was separated and kept at 20 °C for further analysis of the blood constituents.
| Blood constituents | NDa |
FFHC |
HFHC |
Fast |
||||
|---|---|---|---|---|---|---|---|---|
| WT | Mut | WT | Mut | WT | Mut | WT | Mut | |
| Glu (mg/dl) | 205 ± 23 | 182 ± 9 | 151 ± 10 | 151 ± 9 | 233 ± 19 | 186 ± 13b | 74 ± 13 | 71 ± 11 |
| Cholesterol (mg/dl) | 145 ± 22 | 154 ± 22 | 92 ± 15 | 123 ± 9b | 204 ± 38 | 270 ± 26 | 177 ± 11 | 149 ± 10b |
| HDL cholesterol (mg/dl) | 131 ± 33 | 140 ± 22 | 83 ± 10 | 104 ± 6b | 186 ± 31 | 242 ± 35b | 165 ± 11 | 145 ± 10 |
| LDL cholesterol (mg/dl) | 14 ± 2 | 18 ± 3 | 12 ± 11 | 21 ± 4b | 17 ± 4 | 29 ± 7b | 12 ± 2 | 11 ± 3 |
| TG (mg/dl) | 157 ± 52 | 101 ± 15b | 91 ± 20 | 61 ± 4b | 137 ± 41 | 93 ± 15b | 160 ± 40 | 80 ± 6b |
| VLDL cholesterol (mg/dl) | 32 ± 11 | 20 ± 9b | 18 ± 4 | 12 ± 1b | 28 ± 8 | 18 ± 3b | 38 ± 12 | 16 ± 1b |
| NEFA (mEq/dl) | 3.1 ± 0.3 | 2.3 ± 0.2b | 1.1 ± 0.1 | 0.8 ± 0.1 | 2.1 ± 0.4 | 1.5 ± 0.2b | 1.7 ± 0.1 | 1.2 ± 0.1b |
a ND, normal diet; FFHC, fasted-refed; Fast, fasted for 48 h; HFHC, high-fat, high-carbohydrate diet; mut, mutant; NEFA: nonesterified fatty acids; VLDL, very low-density lipoprotein.
b Values are mean ± S.D. (n = 5), p < 0.05.
When both groups of mice fasted for 48 h, the serum levels of total cholesterol were 17% lower (p < 0.05; Table 1) and those of TGs were 50% lower (Table 1) in the Acc2−/− mutants. No significant differences were found in serum glucose and LDL cholesterol levels between both types of mice (Table 1).
To determine serum levels of the various metabolites under lipogenic conditions, both groups of mice underwent a 48-h fast and a 48-h feeding with an FFHC diet. This dietary regimen resulted in 30% lower serum levels of TGs and VLDL cholesterol (p < 0.05; Table 1) and 30, 25, and 75% higher serum levels of total, HDL, and LDL cholesterols, respectively, in the Acc2−/− mutants (p < 0.05; Table 1). The serum levels of glucose were similar in both groups of mice.
The presence of NEFAs in blood serum is an indicator of lipolytic activities and fat mobilization in adipose tissue and in fat-oxidizing tissues such as muscle. In addition, a high serum level of NEFAs has been linked to insulin resistance and indicates a slower fatty acid oxidation rate. When we measured the serum levels of these fatty acids in both groups of mice under different dietary conditions, we found that the levels of the NEFAs were lower in the Acc2−/− mutants no matter the dietary condition (Table 1): normal diet (p = 0.02), fasting and refeeding (p = 0.3), starvation (p = 0.006), and an HFHC diet (p = 0.04; Table 1). These results suggest that although white adipose tissues have higher lipolytic activity (16), NEFAs are lower in the blood, most likely because of increased delivery of free fatty acids to different tissues.
Reduced Body Weight and Smaller Epididymal Fat Pads and Livers in Acc2−/− Mutant Mice under Different Dietary Conditions
We have previously shown that Acc2−/− mutant mice are protected against obesity and diabetes even when fed HF or HFHC diets (12, 13). In this study, we determined the effects of different dietary conditions, including those characteristic of a Western HFHC diet, on the morphology of Acc2−/− mutant and wild-type mice. When fed a normal diet, the Acc2−/− mutant mice weighed about 25% less (p = 0.0005; Fig. 1A); and the livers and epididymal fat pads of the Acc2−/− mutant mice weighed about 20% less than those of the wild-type controls and were significantly lower when calculated per body weight (p = 0.001; Fig. 1, B and C). We also measured morphological changes after 48 h of starvation and after refeeding with an FFHC diet for another 48 h. Before fasting, the body weight of the Acc2−/− mutant mice was about 25% lower than that of the wild-type controls (Fig. 1D; p = 0.005). Although both groups lost about 14% of their body weight after the starvation period (Fig. 1D), this loss was not statistically significantly different (p = 0.15). By the end of the 48-h refeeding, the Acc2−/− mutants consumed about 22% more food than the wild-type controls (7.9 versus 6.2 g/mouse, respectively; data not shown). After the 48-h refeeding, both groups of mice regained about 90% of their initial body weight before starvation (28.88 ± 4.4 versus 32.16 ± 4.5 g/mouse and 21.4 ± 1.2 g versus 22.5 ± 1.9 g/mouse, respectively; Fig. 1D). The weights of the livers were similar for both groups of mice after the 48-h refeeding (1.02 ± 0.15 versus 0.96 ± 0.12 g; Fig. 1E), but, the epididymal fat pads of the Acc2−/− mutants weighed about 65% less (1.19 ± 0.31 versus 0.45 ± 0.09 g; p = 0.04; Fig. 1, E and F). These results suggest that the wild-type mice regained most of their white adipose tissue after being refed the FFHC diet, whereas the Acc2−/− mutants mice only partially regained this tissue.
FIGURE 1.

Body and tissue weights of Acc2−/− mutant and control mice under different feeding conditions. The weights of 4–5-month-old male mice (n = 5) in each different feeding group, as indicated at the right of each panel, were measured. ND, mice that were maintained on a normal diet for 4–5 mo; FRFD, mice that fasted for 48 h and then fed a fat-free, high-carbohydrate diet for an additional 48 h; HFHC, 3–4-month-old male mice that were fed a high-fat, high-carbohydrate diet for an additional 2 months; FAST, 4–5-month-old mice that fasted for 48 h. Before the mice were sacrificed, blood was collected from their tails after a 4–5-h fasting period to determine the blood constituents. At the end of each experiment, the mice were weighed and sacrificed, and the extracted tissues were weighed and kept at −80 °C for further analyses.
When both groups of mice were fed the HFHC diet, the Acc2−/− mutant mice weighed about 20% less before eating the diet and 42% less after eating the diet (Fig. 1G). On average, the Acc2−/− mutants gained about 7.5 g/mouse (range: 20.88 ± 0.98 to 28.3 ± 1.3 g/mouse), whereas the wild-type controls gained about 22 g/mouse (range: 26.34 ± 2.05 to 48.42 ± 5.3 g/mouse; Fig. 1G).
Under these feeding conditions, the livers of Acc2−/− mutants weighed about half the weight of the livers of the wild-type controls (0.88 ± 0.09 versus 1.75 ± 0.5 g; p = 0.018; Fig. 1H). However, when the ratio of liver weight to body weight was calculated for both groups, the ratio was similar (p = 0.14; Fig. 1I). On the other hand, the size of the epididymal fat pads of the Acc2−/− mutant mice was about 38% of that of the wild-type controls (p = 0.0005) and about 50% less when the ratio of adipose tissue weight to body weight was calculated (p = 0.001; Fig. 1I).
Under prolonged fasting (48 h), the wild-type controls lost an average of 2.9 ± 1.01 g of their body weight, and the Acc2−/− mutants lost an average of 1.6 ± 0.40 g (Fig. 1J). The livers of the Acc2−/− mutants weighed about 44% less (p = 0.003) and their epididymal fat pads weighed about 50% less than those of the wild-type controls (p = 0.02; Fig. 1K); when calculated per body weight, the weight of the epididymal fat pads of the Acc2−/− mutants was significantly less (p < 0.05; Fig. 1L).
Effects of ACC2 Deletion on Accumulation of Hepatic Lipids and Function of Lipogenic Enzymes After Normal Diet
In addition to its central role in glucose homeostasis, the liver is a metabolically active tissue in both fatty acid synthesis and oxidation. Disturbing the balance between the lipogenic pathway and fatty acid oxidation could result in fatty liver. We determined the activities and levels of lipogenic enzymes and the levels of TGs and cholesterols in the livers of Acc2−/− mutant and wild-type mice fed normal chow. The FAS and ACC activities in the liver extracts were not significantly different between the groups of mice (data not shown), a finding that affirms our previous discovery that ACC1 is the dominant isoform in the liver and the major contributor to ACC activity and its product malonyl-CoA (3). The protein levels of ACC1, FAS, and ACL were also similar, suggesting that the lipogenic pathway does not change under normal feeding conditions (Fig. 2, A and B). The level of hepatic TGs in the Acc2−/− mutants was lower (∼30%; p = 0.09; Fig. 2C), the level of hepatic cholesterol was about 23% lower in the wild-type controls (p = 0.008; Fig. 2D).
FIGURE 2.

Representative Western blots of lipogenic enzymes, and TG and cholesterol levels in liver extracts from control and Acc2−/− mutant mice fed a normal diet. In Western blot analyses, crude liver extracts (“Experimental Procedures”) were separated by 4–12% NuPAGE MES gels. A, 30-μg protein samples of liver extracts from control and Acc2−/− mutant mice were separated by gel electrophoresis. Antibodies against ACL, ACC1, and FAS were used to detect their respective levels with ECL. The bottom panel shows a representative Western blot after staining with Ponceau S to indicate equal loading. B, the respective bands in the blots in A were scanned and quantified. C and D, activities of ACC and FAS in liver crude extracts, respectively. E and F, triglyceride and cholesterol levels in liver extracts from the two groups of mice were determined as described under “Experimental Procedures.”
Effects of ACC2 Deletion on Accumulation of Hepatic Lipids and Lipogenic Enzymes after an HFHC Diet
Acc2−/− mutant mice are protected against obesity and diabetes when fed an HFHC diet, which induces these pathological conditions (12). To compare weight gains after extended feeding, we fed 5-week-old Acc2−/− mutant mice and wild-type controls an HFHC diet for 16 weeks. As shown in Fig. 3A, the Acc2−/− mutants gained significantly less weight during the feeding period. Afterward, the Acc2−/− mutants weighed 32.30 ± 0.56 g/mouse and the wild-type controls weighed 39.65 ± 1.06 g/mouse. To determine the combined effect of the ACC2 deletion and an HFHC diet and other dietary conditions, we fed 3–4-month-old Acc2−/− mutant mice an HFHC diet for 2 months. We found a significant decrease in the protein levels of the key enzymes in the lipogenic pathways as follows: 65% for ACC1, 30% for ACL, and 40% for FAS (Fig. 3, B and C). In liver extracts, ACC activity was about 30% lower (p = 0.019; Fig. 3D) and FAS activity was about 60% lower (p = 0.01; Fig. 3E) in Acc2−/− mutant mice. When the effects of the HFHC diet were compared with those of the normal diet (Fig. 2), hepatic TGs were elevated in both the wild-type and Acc2−/− mutants, with the level of TGs being statistically significantly lower in the Acc2−/− tissues (p = 0.001; Fig. 3F). Interestingly, although the level of hepatic cholesterol was higher in the liver extracts of the Acc2−/− mutants, this difference did not reach statistical significance (p = 0.09; Fig. 3G). Consistent with these low levels of hepatic TGs, staining of the liver tissues with Oil Red O showed that the wild-type livers were very fatty with large oil droplets, whereas the Acc2−/− mutant livers were less fatty and had small oil droplets (Fig. 3H).
FIGURE 3.

Body weights, representative Western blots, enzyme activities of ACC and FAS, and levels of TGs and cholesterol in samples of liver extracts from control and Acc2−/− mutant mice fed a high-fat, high-carbohydrate diet. A, body weights of 5-week-old male mice fed a high-fat, high-carbohydrate diet for 16 weeks (n = 20). B–H, represent data from 3–4-month-old male mice fed a high-fat, high-carbohydrate diet for 2 months. B and C, representative Western blots of liver extracts and the quantification of the respective bands as described for Fig. 2. For detection of ACC, we used ACC1 antibodies, as we described previously (3). D and E, enzyme activities of ACC and FAS, respectively. F and G, levels of TGs and cholesterol in liver extracts, as determined by enzymatic methods. H, Oil Red O staining of frozen liver tissues showing the accumulation of oil droplets in orange; the bar indicates 20 μm.
Increased Hepatic AKT Activity after an HFHC Diet
Under hyperinsulinemic/euglycemic conditions, hepatic glucose production is suppressed in Acc2−/− mutant mice, suggesting increased insulin sensitivity (13). Because AKT and its active form, pAKT, play an important role in insulin signaling, we decided to measure the levels of AKT and pAKT (Ser473) in the livers of 3–4-month-old male, Acc2−/− mutant and wild-type mice fed an HFHC diet for 2 months. The AKT level was very similar in both groups of mice, but the pAKT (Ser473) level was about 40 to 50% higher in the livers of the Acc2−/− mutants (p = 0.05; Fig. 4). These results suggest that insulin sensitivity was maintained in the Acc2−/− mutants, partly because of improved insulin signaling through AKT activation.
FIGURE 4.
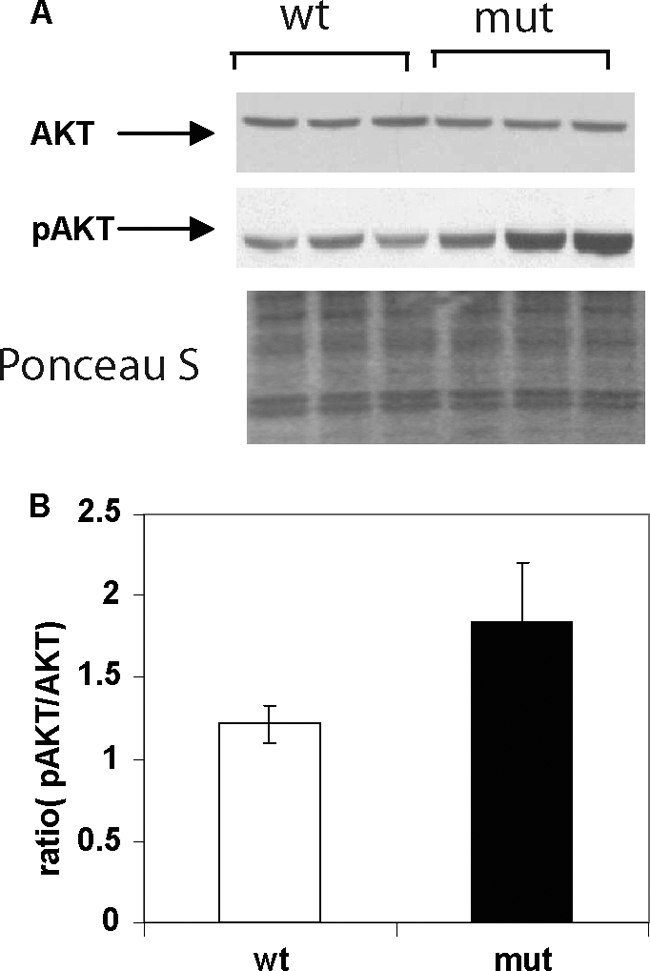
Representative Western blots of AKT and pAKT levels in samples from liver extracts of wild-type and Acc2−/− mutant mice fed a high-fat, high-carbohydrate diet. A, 30-μg protein samples from liver extracts of wild-type and Acc2−/− mutant mice were subjected to electrophoresis gel separation using 4–12% NuPAGE MES gels. Blots were probed with antibodies against AKT or pAKT and detected with ECL. The bottom panel shows a representative blot stained with Ponceau S as a control for equal loading. B, average ratio of the scanned values of pAKT and AKT.
Significantly Fewer Hepatic Lipids Accumulate with Fasting and Refeeding
To determine the effect of continuous fatty acid oxidation on hepatic lipogenesis as a consequence of the ACC2 deletion, we followed a standard dietary protocol: a 48-h fast followed by a 48-h feeding with an FFHC diet. Under these conditions, the activities and levels of the enzymes involved in the lipogenic pathway, ACC1, ACL, and FAS, were induced in the livers of both the wild-type and the Acc2−/− mutant mice (Fig. 5A), yielding a 2-fold increase in the ACC1 protein, a 1.8-fold increase in the ACL protein, and a 3.5-fold increase in the FAS protein in the Acc2−/− mutant livers (Fig. 5B).
FIGURE 5.

Representative Western blots and activities of lipogenic enzymes and lipid levels in samples from liver extracts of wild-type and Acc2−/− mutant mice under fasted-refed dietary conditions. A, 30-μg protein samples from liver extracts of wild-type and Acc2−/− mutant mice were subjected to separation by gel electrophoresis using 4–12% NuPAGE MES gels and probed with the antibodies indicated, as described in the legends to Figs. 2–4, and avidin-peroxidase to detect ACC. B, the relative abundance of lipogenic enzymes in A, determined by scanning the respective bands. C and D, ACC and FAS activities in liver crude extracts, respectively. E and F, hepatic levels of TGs and cholesterol determined by enzymatic methods.
Interestingly, despite the 2-fold increase in the level of ACC1 in the livers of Acc2−/− mutants, the ACC activity was similar in the livers of both the Acc2−/− mutant and the wild-type mice (p = 0.29; Fig. 5C). This similarity can be attributed to a parallel, 2-fold increase in phosphorylated ACC (pACC) that, in turn, led to reduction of these activities (Fig. 5A). These results also underscore the importance of the acute regulation of ACC by a phosphorylation/dephosphorylation mechanism, which results, respectively, in activation and inhibition of ACC activity. FAS activity, which is not known to be regulated at the post-translational level, was about 4-fold higher in the livers of the Acc2−/− mutant mice when compared with the wild-type controls (58.6 ± 7.3 and 14.9 ± 5.1 nmol/min/mg, respectively), which is consistent with the increase in FAS protein (Fig. 5, A–D). Unexpectedly, despite significant increases in the protein levels and activities of the key enzymes in the lipogenic pathway, the level of TGs in the Acc2−/− mutant mouse livers was about 65% lower than that in the wild-type livers (16.03 ± 5.75 versus 4.63 ± 1.29 mg/g of tissue, respectively; p = 0.017; Fig. 5 E). On the other hand, total cholesterol was about 20% higher in the Acc2−/− mutant livers than the wild-type livers (4.11 ± 0.23 versus 3.35 ± 0.53 mg/g, respectively; p = 0.05; Fig. 5F). These results suggest that newly synthesized fatty acids may be oxidized and therefore less likely to be esterified and converted to TGs, as was the case in the wild-type livers. On the other hand, increased hepatic cholesterol may suggest increased de novo cholesterol synthesis due to increased levels of acetyl-CoA in the Acc2−/− mutant mice, because cholesterol cannot be oxidized in the β-oxidation pathway.
Effects of ACC2 Deletion on Metabolic Changes after Prolonged Fasting
During prolonged fasting, the activities of lipogenic enzymes are significantly lower than during feeding, where the synthesis of fatty acids is limited and fatty acid oxidation increases to serve the energy demand. For this reason, we investigated the effect of the chronic ACC2 deletion on fatty acid enzymes and lipids. As expected, when the Acc2−/− mutant mice fasted for 48 h, the expression levels of ACC1 and ACL were not statistically significantly different from those of the wild-type mice (Fig. 6, A and B). In addition, the ACC activity tended to be higher in the Acc2−/− mutant livers than in wild-type livers (0.84 ± 0.1 versus 0.66 ± 0.05 nmol/min/mg; p = 0.1; Fig. 6C). Furthermore, FAS protein and FAS activity were about 30% higher in the Acc2−/− mutant livers than in the wild-type livers (2.42 ± 0.18 versus 1.82 ± 0.0.25 nmol/min/mg; p = 0.005; Fig. 6, B and D).
FIGURE 6.

Representative Western blots, activities of lipogenic enzymes, and TG and cholesterol levels in samples from liver extracts of control and Acc2−/− mutant mice after 48-h fasting. A, 30-μg protein samples from liver extracts of wild-type and Acc2−/− mutant mice were separated by gel electrophoresis using 4–12% NuPAGE MES gels and probed with the indicated antibodies, as described in the legend to Fig. 2. B, relative values of scanned bands shown in A. C and D, ACC and FAS activities, respectively, in liver extracts. E and F, levels of TGs and cholesterol, respectively.
The cholesterol profiles of both groups of mice were similar, as were the levels of hepatic TGs (∼65 mg/g; Fig. 6E). Although the hepatic total cholesterol level was slightly lower in the wild-type livers (11.82 ± 2.15 mg/g) than in the Acc2−/− mutant livers (14.52 ± 2.30 mg/g), this difference in elevation was not statistically significant (p = 0.13; Fig. 6F).
Up-regulation of Transcription Regulators of Fatty Acid Metabolism and Their Gene Targets
The lipogenic and oxidation pathways are under transcriptional regulation by the SREBPs. To determine the effects of the ACC2 deletion on these transcriptional regulators, we used the quantitative RT-PCR method to determine their mRNA levels. Under the lipogenic conditions of fasting and refeeding, which produce the greatest up-regulation of FAS, ACC1, and ACL, we found a significant 2.5-fold increase in the mRNA levels of SREBP-1 and SREBP-2 and about a 2-fold increase in the mRNA ChREBP level (Fig. 7A). Consistent with up-regulation of these transcription factors, we found that the levels of their target genes were also up-regulated significantly, being between 1.5- and 3.5-fold (Fig. 7B). These target genes included FAS, ACC1, ACL, and SCD1 (all of which are under the control of SREBP-1 and ChREBP) and MVD (mevalonate pyrophosphate decarboxylase) and HMGR (3-hydroxy-3-methyl-glutaryl-CoA reductase) (which are regulated by SREBP-2 and involved in cholesterol synthesis).
FIGURE 7.

Relative mRNA levels of transcription factors and target genes in liver. RNA was isolated from livers of both wild-type and Acc2−/− mutant mice (n = 5) and measured by real-time RT-PCR as described previously (17). The values were normalized to 18 S ribosomal RNA and are depicted as mean ± S.D. (*, p < 0.05).
Effect of ACC2 Deletion on Expression of PPARs
Under the fasting-refeeding dietary regimen, the level of PPAR-γ protein was about 2-fold higher in livers of Acc2−/− mutant mice than in those of the wild-type controls (Figs. 8, A and B), but there were no significant differences in the level of PPAR-α between the two groups (Figs. 8, A and C). Interestingly, the mRNA levels of both PPAR-γ and PPAR-α were similar in both groups of mice (Fig. 8, D and E), suggesting that up-regulation of PPAR-γ in the livers of the Acc2−/− mutants was at the translational level. When both groups of mice were fed the HFHC diet, the hepatic PPAR-γ level in the Acc2−/− mutants was about 30% lower than in the wild-type controls (Fig. 9, A and B).
FIGURE 8.

Levels of transcription factors PPAR-γ and PPAR-α in liver extracts of control and Acc2−/− mutant mice under fasted-refed (FRFD) conditions. A, representative Western blots for PPAR-γ and PPAR-α as indicated by arrows. Bottom panel shows a representative blot stained with Ponceau S as a control for equal loading. B and C, scanned values of PPAR-γ and PPAR-α, respectively. D, representative Northern blot of total RNA, which was electrophoresed on a 1% agarose gel in the presence of formalin and transferred to Hybond N filters. The filters were hybridized with 32P-labeled cDNA probes of PPAR-γ and PPAR-α. Bottom panel shows an ethidium bromide-stained agarose gel for control of equal loading; the ribosomal RNA 28 S and 18 S are indicated. E, scanned values of the PPAR-γ and PPAR-α bands. Data are expressed as mean ± S.D. (n = 5; *, p < 0.05).
FIGURE 9.

Levels of transcription factors PPAR-γ and PPAR-α in liver extracts of control and Acc2−/− mutant mice under high-fat, high-carbohydrate feeding conditions. A, representative Western blots of PPAR-γ and PPAR-α, and Ponceau S-stained filters as a control for equal loading. B and C, relative levels of PPAR-γ and PPAR-α as determined by scanning the respective bands. D, relative mRNA levels of PPAR-γ and PPAR-α transcripts determined by real-time quantitative PCR. Data are expressed as mean ± S.D. (n = 5; *, p < 0.05).
With the HFHC diet, we also unexpectedly found that the hepatic level of PPAR-α in the Acc2−/− mutant mice was 70% lower than that in the wild-type controls (Fig. 9, A and C). The down-regulation of PPAR-γ occurred at the transcriptional level, because the mRNA level, as measured by real-time PCR, was significantly lower (Fig. 9D). The mRNA level of PPAR-α did not change with the HFHC diet (Fig. 9D), suggesting that PPAR-α is down-regulated at the translational level.
DISCUSSION
We assessed the impact of a chronic ACC2 deletion, and its consequential unregulated mitochondrial fatty acid oxidation on hepatic lipid metabolism under different dietary conditions. Under all of the experimental dietary conditions used, including a normal chow diet, fasting and refeeding an FFHC diet, an HFHC diet only, and prolonged fasting, there were significant decreases in body weights, the weights of epidydimal fat pads, and hepatic levels of TGs in Acc2−/− mutant mice (Figs. 1–3 and 5). Interestingly, few changes occurred in the fat synthesis pathway under normal dietary conditions, which was reflected in similar levels of ACC, FAS, and ACL. Both ACC1 and ACC2 have little physiological impact during prolonged fasting because of their very low expression levels under this dietary condition. Consequently, and as we expected, we did not observe major differences in the lipogenic pathway between the Acc2−/− mutant and wild-type mice (Fig. 6). These results suggest that the ACC2 deletion alone did not cause chronic remodeling of the lipogenic pathway. However, under dietary conditions that induce fatty liver, Acc2−/− mutant mice were protected against hepatic fat accumulation, which involved different regulatory mechanisms (Fig. 10).
FIGURE 10.

Schematic proposed model for major metabolic pathways affected by ACC2 deletion in the livers of wild-type mice fed FFHC and HFHC diets. Master regulators of fat metabolic enzymes (SREBP1, SREBP2, ChREBP, and PPAR-γ) are induced under FFHC feeding conditions that result in up-regulation of key enzymes in fatty acid and cholesterol metabolism (ACL, ACC1, ACC2, FAS, mevalonate pyrophosphate decarboxylase (MVD), and 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR)). In livers of wild-type mice, where ACC2 is highly active, the malonyl-CoA produced blocks the activity of CPT1, thus inhibiting fatty acid oxidation and leading to excessive accumulation of excessive TGs and more fat (A). When ACC2 is deleted, there is a further increase in the transcriptional factors of fat synthesis, resulting in even more up-regulation of the lipogenic enzymes. However, the lack of CPT1 inhibition by ACC2-produced malonyl-CoA results in increased fatty acid oxidation and significant reduction in the accumulation of long-chain fatty acids and TGs and prevents fatty liver (B). Under HFHC feeding conditions, the livers of wild-type mice accumulate high levels of TGs as a result of exogenous fatty acids and de novo synthesis from the high carbohydrate in the diet (C). In livers of ACC2 mutant mice, fewer TGs accumulate due both to down-regulation of lipogenic enzymes and increased fatty acid oxidation, thus preventing excessive accumulation of TGs in these livers (D). The decrease in acylglycerides in livers of Acc2−/− mutant mice on a HFHC diet will result in an increase in pAKT that will enhance insulin sensitivity and increase glucose uptake (D). Thick arrows indicate up-regulation.
Eating an FFHC diet after prolonged fasting is known to induce the lipogenic pathway, including master transcription regulators such as SREBP1, SREBP2, and ChREBP (19). As a result, the livers of mice fed an FFHC diet become very fatty because of the high levels of TGs. Surprisingly, in the livers of Acc2−/− mutant mice, there was an even greater increase (2–3-fold) in the levels of SREBP1, SREBP2, and ChREBP that led to similar increases in the transcription levels of the genes (ACC, ACL, and FAS) controlled by these transcription factors (Figs. 1–3, 5, 7, and 10). Despite significant up-regulation of the de novo lipogenic pathway in Acc2−/− mutant livers, however, there were significantly lower hepatic levels of TGs. These results suggest that newly synthesized fatty acids were oxidized instead of esterified and accumulated as TGs, a futile metabolic cycle. The lower levels of intracellular fatty acids in Acc2−/− mutant livers may signal the cells that there is a need for newly synthesized fatty acids, which will result in up-regulation of the lipogenic pathway.
Another intriguing finding from our study is the effect of different diets on the regulation of PPARs in the livers of wild-type and Acc2−/− mutant mice. PPAR-γ, which is known to be moderately expressed in mammalian livers (20), was induced more than 2-fold in the livers of Acc2−/− mutant mice under lipogenic conditions (Fig. 8). These results support our previous findings that PPAR-γ is partially responsible for the up-regulation of hepatic glucose and lipid metabolism in the livers of Acc2−/− mutant mice (12, 13, 16, 17). Additional support comes from a study by Way et al. (21) in which diabetic rats were treated with PPAR-γ agonists, which resulted in increased lipogenic gene expression in addition to up-regulation of glucose transporter 2 (GLUT2) in the rat livers.
On the other hand, we found no change in PPAR-α expression with an FFHC diet, suggesting that the increased mitochondrial fatty acid oxidation in the livers of the Acc2−/− mutant mice did not occur because of up-regulation of the mitochondrial fatty acid oxidation genes controlled by PPAR-α. Interestingly, with an HFHC diet, we observed a significant reduction in PPAR-γ protein and mRNA and a similar decrease in PPAR-α protein. The down-regulation of PPAR-γ in the livers of Acc2−/− mutant mice is consistent with the significant decrease in the levels of enzymes involved in the lipogenic pathway, specifically ACC1, FAS, and ACL (Fig. 3). However, despite down-regulation of PPAR-α, fatty acid oxidation was still higher in tissues of the Acc2−/− mutant mice than those of the wild-type mice, a finding consistent with our earlier reports (12, 13, 16, 17). For this reason, we suggest that in the absence of the inhibitory effect of mitochondrial malonyl-CoA, fatty acid oxidation will increase despite significant down-regulation of PPAR-α. Moreover, it is conceivable that down-regulation of PPAR-α in the Acc2−/− mutant mice livers is an adaptive physiological mechanism that prevents excessive levels of fatty acid oxidation that may harm the liver.
Excessive accumulation of TGs causes fatty liver and is linked to the development of insulin resistance. Liver-synthesized fatty acids and exogenous NEFAs will either be oxidized or esterified to produce TGs. The size of the lipid droplets is controlled by the net result of lipid degradation and synthesis, which involves lipases and enzymes that catalyze the synthesis of TGs (22, 23). Based on our current findings, we propose that hepatic free fatty acids are continuously oxidized in Acc2−/− mutant mice, making these fatty acids less likely to be esterified and stored as TGs, ultimately leading to the formation of smaller lipid droplets.
Fat accumulation in animal tissues, including the liver, is associated with insulin resistance (24). Our previous studies of Acc2−/− mutant mice have not demonstrated insulin resistance, even after prolonged HFHC diet feeding (12) or an HF diet that was associated with insulin-stimulated glucose uptake and reduced lipid accumulation (13). The results of our present study are consistent with these findings; even when the Acc2−/− mutant mice were fed an HFHC diet for 4 months, they maintained insulin sensitivity (Fig. 4). Insulin sensitivity was most likely preserved because of an increase in pAKT, a key player in insulin signaling.
Other investigators have developed strains of Acc2 knock-out mice in which the knock-out is mediated by Cre recombinase (25, 26). Hoehn et al. (25) found a metabolic phenotype that was similar to our null Acc2 knock-out mouse, but did not observe a similar effect of their Acc2 knock-out on adiposity. Olson et al. (26) did not find a significant reduction in total malonyl-CoA content in their Acc2 knock-out mice, even in muscle tissues where ACC2 is the dominant and main isoform, and observed no change in fatty acid oxidation. Their results are surprising, not only because they contradict our findings and those of Hoehn et al. (25), but also because they are inconsistent with reports that showed significant lowering of malonyl-CoA by selective inhibitors against ACC2 (2, 27). As for the discrepancies between our findings and those of Hoehn et al. (25) despite the similar biochemical phenotypes of our Acc2 knock-out mice, these can be attributed to several factors. 1) Differences in the genetic background of the ES cells and transgenic mice. In our studies, we used littermates both of mixed genetic background (129/SvEv/C57BL/6J) and inbred genetic background (129/SvEv). In addition, the ES cells we used are of 129/SvEv origin. Hoehn et al. (25) used Bruce4 ES cells in mice with a C57BL/6 genetic background. It is noteworthy that the C57BL/6J mice showed heterogeneous phenotypes, that is, lean normoglycemic, lean diabetic, and obese diabetic, when fed HF diets (28, 29). 2) Differences in the methods used to achieve the ACC2 deletion. Hohen et al. (25) used Cre recombinase to mediate the deletion. Side effects related to Cre recombinase have been reported, such as impairments of pancreatic β-cell function, which could further complicate the effect of the ACC2 deletion on mouse physiology when using the Cre recombinase system (30, 31). 3) Differences in the dietary regimens used in the feeding experiments and the duration of the feedings.
In summary, we have shown that the ACC2 deletion protects against fatty liver, despite increased de novo lipogenesis and a diet that induces obesity, fatty liver, and diabetes. The adaptation of the liver to these metabolic conditions involves different mechanisms, such as down-regulation of the lipogenic pathway, as occurs with an HFHC diet, or an increase in fatty acid oxidation and synthesis, as occurs with fasting and refeeding an FFHC diet. Moreover, it is noteworthy that the metabolic changes that occurred with a normal diet were minor, suggesting that there are no major chronic physiological changes in the livers of Acc2−/− mutant mice under normal dietary conditions. These findings suggest that ACC2 could be a useful target for ameliorating the metabolic syndrome.
Acknowledgments
We acknowledge the importance of the contribution of Rubin Bressler, M.D., now deceased, to the collaborative development of the scientific thought underlying this work. We also thank Parichher Kordari and Assia Bentebibel for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-63115, the Hefni Technical Training Foundation, the Medallion Foundation (to S. J. W.), and the McLean Foundation Pilot Project (to L. A.).
- TG
- triglycerides
- ACC
- acetyl-CoA carboxylase
- ACL
- ATP citrate-lyase
- ChREBP
- carbohydrate responsive element-binding protein
- FAS
- fatty acid synthase
- FFHC
- fat-free, high-carbohydrate
- HFHC
- high-fat, high-carbohydrate
- NEFAs
- nonesterified fatty acids
- PPAR
- peroxisome proliferator-activated receptor
- SCD1
- stearoyl-CoA desaturase 1
- SREBP
- sterol regulatory element-binding protein
- VLDL
- very low-density lipoprotein.
REFERENCES
- 1. Browning J. D., Horton J. D. (2004) Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheung O., Sanyal A. J. (2010) Recent advances in nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 26, 202–208 [DOI] [PubMed] [Google Scholar]
- 3. Shulman G. I. (2000) Cellular mechanisms of insulin resistance. J. Clin. Invest. 106, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sul H. S., Wang D. (1998) Nutritional and hormonal regulation of enzymes in fat synthesis. Studies of fatty acid synthase and mitochondrial glycerol-3-phosphate acyltransferase gene transcription. Annu. Rev. Nutr. 18, 331–351 [DOI] [PubMed] [Google Scholar]
- 5. Abu-Elheiga L., Almarza-Ortega D. B., Baldini A., Wakil S. J. (1997) Human acetyl-CoA carboxylase 2. Molecular cloning, characterization, chromosomal mapping, and evidence for two isoforms. J. Biol. Chem. 272, 10669–10677 [DOI] [PubMed] [Google Scholar]
- 6. Ha J., Lee J. K., Kim K. S., Witters L. A., Kim K. H. (1996) Cloning of human acetyl-CoA carboxylase-β and its unique features. Proc. Natl. Acad. Sci. U.S.A. 93, 11466–11470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abu-Elheiga L., Matzuk M. M., Kordari P., Oh W., Shaikenov T., Gu Z., Wakil S. J. (2005) Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl. Acad. Sci. U.S.A. 102, 12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abu-Elheiga L., Matzuk M. M., Abo-Hashema K. A., Wakil S. J. (2001) Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291, 2613–2616 [DOI] [PubMed] [Google Scholar]
- 9. Delzenne N. M., Hernaux N. A., Taper H. S. (1997) A new model of acute liver stenosis induced in rats by fasting followed by refeading a high carbohydrate-free diet. Biochemical and morphological analysis. J. Hepatol. 4, 880–885 [DOI] [PubMed] [Google Scholar]
- 10. Winder W. W., MacLean P. S., Lucas J. C., Fernley J. E., Trumble G. E. (1995) Effect of fasting and refeeding on acetyl-CoA carboxylase in hindlimb muscle. J. Appl. Physiol. 2, 578–582 [DOI] [PubMed] [Google Scholar]
- 11. Jump D. B., Botolin D., Wang Y., Xu J., Christian B., Demeure O. (2005) Fatty acid regulation of hepatic gene transcription. J. Nutr. 135, 2503–2506 [DOI] [PubMed] [Google Scholar]
- 12. Abu-Elheiga L., Oh W., Kordari P., Wakil S. J. (2003) Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. U.S.A. 100, 10207–10212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi C. S., Savage D. B., Abu-Elheiga L., Liu Z. X., Kim S., Kulkarni A., Distefano A., Hwang Y. J., Reznick R. M., Codella R., Zhang D., Cline G. W., Wakil S. J., Shulman G. I. (2007) Continuous fat oxidation in acetyl-CoA carboxylase 2 knock-out mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc. Natl. Acad. Sci. U.S.A. 104, 16480–16485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandler C. E., Wilder D. E., Pettini J. L., Savoy Y. E., Petras S. F., Chang G., Vincent J., Harwood H. J., Jr. (2003) CP-346086, an MTP inhibitor that lowers plasma cholesterol and triglycerides in experimental animals and in humans. J. Lipid Res. 44, 1887–1901 [DOI] [PubMed] [Google Scholar]
- 15. Mao J., DeMayo F. J., Li H., Abu-Elheiga L., Gu Z., Shaikenov T. E., Kordari P., Chirala S. S., Heird W. C., Wakil S. J. (2006) Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8552–8557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oh W., Abu-Elheiga L., Kordari P., Gu Z., Shaikenov T., Chirala S. S., Wakil S. J. (2005) Glucose and fat metabolism in adipose tissue of acetyl-CoA carboxylase 2 knock-out mice. Proc. Natl. Acad. Sci. U.S.A. 102, 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Essop M. F., Camp H. S., Choi C. S., Sharma S., Fryer R. M., Reinhart G. A., Guthrie P. H., Bentebibel A., Gu Z., Shulman G. I., Taegtmeyer H., Wakil S. J., Abu-Elheiga L. (2008) Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am. J. Physiol. Heart Circ. Physiol. 295, H256–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCT)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 19. Horton J. D., Bashmakov Y., Shimomura I., Shimano H. (1998) Regulation of sterol regulatory element-binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. U.S.A. 95, 5987–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H. I., Ahn Y. H. (2004) Role of peroxisome proliferator-activated receptor-γ in the glucose-sensing apparatus of liver and β-cells. Diabetes 53, S60-S65 [DOI] [PubMed] [Google Scholar]
- 21. Way J. M., Harrington W. W., Brown K. K., Gottschalk W. K., Sundseth S. S., Mansfield T. A., Ramachandran R. K., Willson T. M., Kliewer S. A. (2001) Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor γ activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology 142, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 22. Walther T. C., Farese R. V., Jr. (2009) The life of lipid droplets. Biochim. Biophys. Acta 1791, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wiggins D., Gibbons G. F. (1992) The lipolysis/esterification cycle of hepatic triacylglycerol. Its role in the secretion of very low-density lipoprotein and its response to hormones and sulfonylureas. Biochem. J. 284, 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petersen K. F., Shulman G. I. (2006) Etiology of insulin resistance. Am. J. Med. 119, S10–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoehn K. L., Turner N., Swarbrick M. M., Wilks D., Preston E., Phua Y., Joshi H., Furler S. M., Larance M., Hegarty B. D., Leslie S. J., Pickford R., Hoy A. J., Kraegen E. W., James D. E., Cooney G. J. (2010) Acute or chronic up-regulation of mitochondrial fatty acid oxidation has no net effect on whole body energy expenditure or adiposity. Cell. Metab. 11, 70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olson D. P., Pulinilkunnil T., Cline G. W., Shulman G. I., Lowell B. B. (2010) Gene knock-out of Acc2 has little effect on body weight, fat mass, or food intake. Proc. Natl. Acad. Sci. U.S.A. 107, 7598–7603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gu Y. G., Weitzberg M., Clark R. F., Xu X., Li Q., Zhang T., Hansen T. M., Liu G., Xin Z., Wang X., Wang R., McNally T., Zinker B. A., Frevert E. U., Camp H. S., Beutel B. A., Sham H. L. (2006) Synthesis and structure-activity relationships of N-{3-[2-(4-alkoxyphenoxy-5-yl]-1-methylprop-2-ynyl}carboxy derivatives as selective acetyl-CoA carboxylase 2 inhibitors. J. Med. Chem. 49, 3770–3773 [DOI] [PubMed] [Google Scholar]
- 28. Surwit R. S., Kuhn C. M., Cochrane C., McCubbin J. A., Feinglos M. N. (1988) Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37, 1163–1167 [DOI] [PubMed] [Google Scholar]
- 29. Burcelin R., Crivelli V., Dacosta A., Roy-Tirelli A., Thorens B. (2002) Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am. J. Physiol. Endocrinol. Metab. 282, E834–842 [DOI] [PubMed] [Google Scholar]
- 30. Lee J. Y., Ristow M., Lin X., White M. F., Magnuson M. A., Hennighausen L. (2006) RIP-Cre revisited, evidence for impairments of pancreatic β-cell function. J. Biol. Chem. 281, 2649–2653 [DOI] [PubMed] [Google Scholar]
- 31. Silver D. P., Livingston D. M. (2001) Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell 8, 233–243 [DOI] [PubMed] [Google Scholar]
- 32. National Institues of Health (1996) Guide for the Care and Use of Laboratory Animals, Publication number 85-23, National Academy Press, Washington, D.C [Google Scholar]