

Abstract
If smoking is a risk factor for Alzheimer’s disease (AD) but a smoker dies of another cause before developing or manifesting AD, smoking-related mortality may mask the relationship between smoking and AD. This phenomenon, referred to as competing risk, complicates efforts to model the effect of smoking on AD. Typical survival regression models assume that censorship from analysis is unrelated to an individual’s probability of developing AD (i.e., that censoring is noninformative). However, if individuals who die before developing AD are younger than those who survive long enough to develop AD, and if they include a higher percentage of smokers than nonsmokers, the incidence of AD will appear to be higher in older individuals and in nonsmokers. Further, age-specific mortality rates are higher in smokers because they die earlier than nonsmokers. Therefore, if we fail to take into account the competing risk of death when we estimate the effect of smoking on AD, we bias the results and are really only comparing the incidence of AD in nonsmokers with that in the healthiest smokers. In this study, we demonstrate that the effect of smoking on AD differs in models that are and are not adjusted for competing risks.
Keywords: Alzheimer disease, competing risks, elderly, mortality, smoking
INTRODUCTION
Epidemiological studies of risk and protective factors for dementia are often viewed with skepticism because of inconsistencies in results and conclusions.1,2 One risk relationship that has been particularly controversial is that observed between cigarette smoking and Alzheimer’s disease (AD). Several earlier studies (both cross-sectional and prospective) of older adults suggested that smoking might have a protective effect against developing incident AD.3-7 In contrast, more recent studies have reported that midlife smoking significantly increases the risk of developing incident AD in later life.8 This paradox parallels the phenomenon observed with mid-life versus late-life cholesterol, blood pressure, and body mass index.9 An intuitive explanation is that those who develop the outcome (AD) in late life are those who lived long enough to enter the age of risk for developing AD and to enter the study in question.10 Recruitment of an older sample is thus unavoidably biased in favor of survivors, and smokers who survive are likely healthier than smokers who die. This is called survival bias, and the impact of ignoring it has been discussed by others.11,12 Further, after entering the study, smokers are likely to die earlier (younger) than nonsmokers. If smoking is a risk factor for AD but a smoker dies of another cause before developing or manifesting AD, smoking-related mortality may mask the relationship between smoking and AD. This phenomenon is referred to as competing risk.
Statistical modeling of the effect of smoking on AD is complicated by the presence of competing risk due to death. The usual survival regression model assumes that an individual’s being “censored” from the cohort (due to death, loss to follow-up, or being still alive and free of dementia at the end of the study) is unrelated to his/her innate probability of developing incident AD, i.e., that censoring is non-informative. However, if individuals who die before developing AD are younger than those who develop AD, and if they include a higher percentage of smokers than nonsmokers, the incidence rates of AD will appear elevated in older individuals and among nonsmokers. In addition, even though overall mortality rates for smokers and nonsmokers might be comparable, age-specific mortality may be higher in smokers since they tend to die earlier than nonsmokers.13,14 Therefore, if we fail to take into account competing risk due to death when estimating the effect of smoking on AD, we may really only be comparing AD incidence between nonsmokers and the healthiest smokers. Merely censoring out individuals who die before developingAD might bias the results. Here, we demonstrate the variation in the estimated effect of smoking on incident AD (the “main event”) depending on whether or not the analytic model takes mortality (the “competing risk”) into account.
METHODS
Study population
We used data derived from a completed population-based project entitled the Monongahela Valley Independent Elders Survey (MoVIES), which investigated various aspects of normal and abnormal aging between 1987 and 2002 in a community in southwestern Pennsylvania. The study cohort was drawn from a rural, largely blue-collar community in the mid-Monongahela Valley. The original cohort of 1,681 individuals aged 65 years or older was assembled between 1987 and 1989 and was followed in biennial data collection “waves” until 2002, for a total of 6 waves. At wave 2, which is the baseline for the current analysis, the cohort included 1,342 participants. They were aged 67+ years with a mean age of 74.9±5.5 years, was approximately 60.7% female, was 97.4% white (reflecting the base population of the rural mid-Monongahela Valley), and had a median educational level of high school graduate. The analytic dataset for the present analyses includes 1,242 participants at wave 2 after excluding 92 individuals who had prevalent dementia with onset before wave 2 and 8 individuals with unknown dementia onset dates.
Predictor variables
Beginning with wave 2, current and lifetime smoking were determined by the participant’s response to the questions “Do you smoke cigarettes now?” and “Have you ever smoked cigarettes regularly?” Number of cigarettes smoked per day was not ascertained. Other baseline covariates included demographic information (age, sex, and education), potential confounders (alcohol, depression, number of prescription drugs use, lipid-lowering drug use, NSAID use, self-reported health status, number of alcoholic drinks consumed at a time), APOE ε4 carrier status (determined at wave 3), and self-reported disease history (collected at wave 3), determined by the participant’s response to the question “Has a doctor or nurse ever told you that you have (the condition of interest)?” Specifically here we included self-reported cerebrovascular disease (stroke or TIA) and cardiovascular disease (diabetes mellitus, myocardial infarction, hypertension, and high cholesterol). We examined the frequencies and proportions of these variables by age among smokers and nonsmokers.
Outcome variables
Incidence of Alzheimer’s disease (AD)
As reported previously, dementia was defined by DSM-III-R criteria and staged according to the Clinical Dementia Rating (CDR) scale,15,16 and its date of onset estimated based on all available data.17 For the present analyses, we treat a CDR value <0.5 as indicating the absence of dementia (dementia-free) and a CDR value ≥0.5 as indicating the presence of dementia. Between wave 2 and the study cutoff date on December 31, 2001 (follow-up duration: mean 8.2±3.9 years, median 10.0 years), we observed 275 incident cases of dementia with a CDR value ≥0.5. Among them, 202 cases were Probable or Possible AD; 73 cases were non-AD dementias; and one case was of indeterminate cause.18 The latter 74 cases were treated as censored in the survival analyses described below.
Mortality
Over the average 8.2 years of follow-up (10129.6 person-years of follow-up), there were 485 (39.1%) deaths. Mortality was the main source of attrition, as would be expected in an aging cohort (9%–15% between 2-year waves). As previously reported, given the stability of the region’s older population, we had complete ascertainment of mortality. Further, the mortality rates from MoVIES are comparable to U.S. national rates for whites, and reported mortality rates in AD and population attributable risk of mortality from AD.19,20
Choice of survival model
The outcome variable for these analyses was years from baseline assessment (wave 2) interview to the estimated onset date for incident AD. The main predicting variable was self-reported smoking status (never smoker vs. lifetime smoker) at wave 2. Lifetime smokers were defined as current smoking at wave 2 or ever having smoked regularly.
We first used log-log survival plot to assess the proportional hazards assumption for the main covariate. The lines for nonsmokers and smokers were not significantly deviated from parallel, indicating that the proportional hazards (PH) assumption was not violated and PH models were appropriate for this data set. We used Cox PH regression as the main model to estimate the effect of smoking on incident AD. The model stratified age at wave 2 into two categories: 65-74 and ≥75 years old.
Adjustment for competing risk due to death
Deaths that occur before dementia onset in individuals who would have developed dementia (had they lived long enough) have the effect of censoring the latent failure time to dementia. To account for the possible bias from competing risk due to death, we have - in theory - to eliminate these deaths. Since death cannot be eliminated in reality, a statistical solution is needed. These censored lifetimes can be completed statistically by estimating the latent failure times for dementia, based on data from the participants who did develop dementia during their lives.21,22
Both smokers and nonsmokers had high mortality rates, as expected in this aging cohort. The Cox PH model assumes that death is a source of noninformative censoring. This is problematic because factors that are positively correlated to mortality might also be positively correlated to developing incident AD. A commonly used regression model taking competing risks into account is the Fine and Gray (FG) proportional subdistribution hazards model.23 The FG model assumes that those who died without developing AD would neither have died of AD nor developed AD had they lived throughout the follow-up period. This is also problematic because our population was aged 65 years or older, an age group in which neither incident AD cases nor deaths from AD are rare.
To account for possible violation of the FG competing risks regression model, we performed a further modification to incorporate the probability of developing AD among those who died, had they survived the entire follow-up period. First, we constructed a propensity model to estimate this probability at the last observed follow-up for each individual, using a logistic regression model with covariates (smoking, age, sex, and education) that were potential risk factors for developing AD. In the propensity model, those who developed AD during the follow-up were treated as “events,” while those who remained alive and free of AD until the end of the study, were treated as “non-events.” Each individual (including those who died without developing AD) was then assigned a propensity score by calculating his/her estimated probability.
In the second step, the propensity scores for those who died without developing AD were used to reclassify them into main events (incident AD or death with high probability of having AD) or competing events (death with low probability of having AD), in order to correct for the bias due to informative censoring of death. Those who died were reclassified as having had main events (if their propensity scores were above a cutpoint) or as competing events if below the cutpoint. The cutpoint we chose for the reclassification is the median propensity score among those who developed AD (Figure 1). Sensitivity analyses showed that the median was the most conservative among the cutpoints that produced stable results (data not shown).
Figure 1.
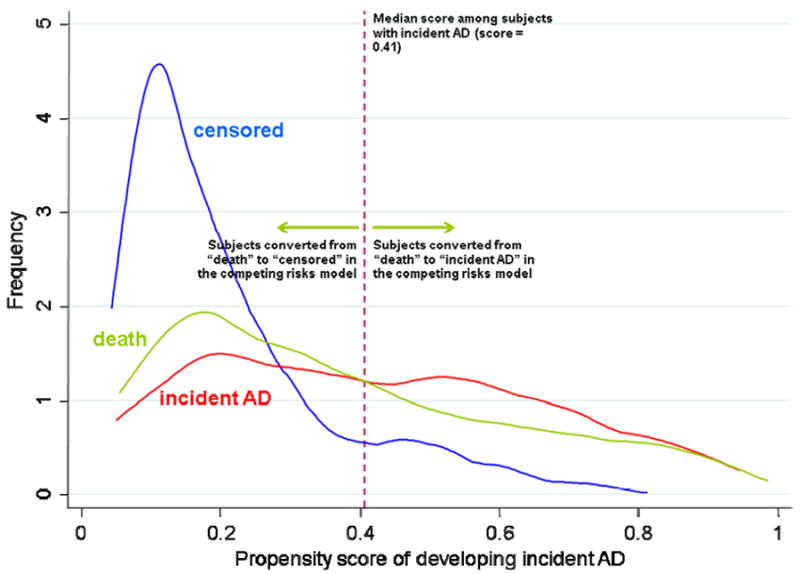
Distribution of propensity scores among those who developed AD, those who died before developing AD, and those survived the entire follow-up period without developing AD.
Fitted regression models
We fitted two types of age-stratified regression models to estimate the effect of smoking on the incidence of AD. First we fit Cox PH regression models, in which participants who survived and remained AD-free, as well as those who died without developing AD, were treated as censored. Next, we fit the modified FG competing risks regression models. Here, participants who remained alive and AD-free were again treated as censored, but those who died without developing AD were treated either as competing risks (if their propensity scores were above the cutpoint) or as censored (if their propensity scores were below the cutpoint). Lifetime smoking status was the main predictor variable in both types of models.
For both Cox and modified FG regressions, we fit three different models by sequentially adding more covariates:
Model 1: smoking and incident AD;
Model 2: smoking and incident AD adjusting for baseline and demographic covariates; and
Model 3: smoking and incident AD adjusting for baseline and demographic covariates, APOE ε4 allele, and vascular risk factors.
Because age was the main confounding variable, we fit these three models stratified by age (n = 751, 60.5% for age <75 years at baseline, and n = 491, 39.5% for age ≥75 years at baseline). Cox PH regression were applied for all 3 models without adjusting for competing risks, and hazard ratios (HR) were reported for the smoking effect. The modified FG regression models were applied for all 3 models adjusting for competing risks, and subdistribution hazard ratios (SHR) were reported for the smoking effect.
RESULTS
At study baseline (wave 2), smokers were significantly younger than never-smokers. The proportion of smokers was significantly higher in those aged <75 years (14.7%) than those aged >=75 years (7.7%) (p<0.001). Among younger elderly (aged 65-74 years), smokers were more likely to have depressive symptoms and more likely to consume alcohol. Among older elderly (age ≥75 years), smokers and nonsmokers were not significantly different (Table 1).
Table 1.
Individual characteristics by smoking status and by age at baseline (wave 2).
| All | 65-74 years of age | >=75 years of age | P value of >=75 vs. 65-74 smokers* | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Never smokers | Smokers | Never smokers | Smokers | Never smokers | Smokers | ||||||
| (n = 1094) | (n = 148) | P value* | (n = 641) | (n = 110) | P value* | (n = 453) | (n = 38) | P value* | |||
| Baseline covariates | |||||||||||
| Female | 668 (61.1%) | 91 (61.5%) | >0.99 | 379 (59.1%) | 66 (60.0%) | 0.92 | 289 (63.8%) | 25 (65.8%) | 0.86 | 0.57 | |
| Education ? high school | 674 (61.6%) | 86 (58.1%) | 0.42 | 457 (71.3%) | 66 (60.0%) | 0.02 | 217 (47.9%) | 20 (52.6%) | 0.62 | 0.45 | |
| Use 3 or more prescription drugs | 350 (32.0%) | 45 (30.4%) | 0.78 | 164 (25.6%) | 32 (29.1%) | 0.48 | 186 (41.1%) | 13 (34.2%) | 0.49 | 0.55 | |
| Lipid-lowering drug use | 32 (2.9%) | 1 (0.7%) | 0.17 | 26 (4.1%) | 1 (0.9%) | 0.16 | 6 (1.3%) | 0 (0.0%) | >0.99 | >0.99 | |
| NSAID use | 753 (68.8%) | 101 (68.2%) | 0.93 | 446 (69.6%) | 79 (71.8%) | 0.74 | 307 (67.8%) | 22 (57.9%) | 0.21 | 0.16 | |
| >5 depressive symptoms | 98 (9.0%) | 23 (15.9%) | 0.02 | 45 (7.1%) | 17 (15.6%) | 0.01 | 53 (11.8%) | 6 (16.7%) | 0.42 | >0.99 | |
| Self-reported health poor** | 19 (1.7%) | 7 (4.7%) | 0.03 | 7 (1.1%) | 4 (3.6%) | 0.06 | 12 (2.7%) | 3 (7.9%) | 0.10 | 0.37 | |
| # of alcoholic drinks at a time per day: | none | 507 (46.4%) | 56 (37.8%) | <0.001 | 265 (41.4%) | 38 (34.6%) | <0.001 | 242 (53.4%) | 18 (47.4%) | 0.07 | 0.18 |
| 1/day | 402 (36.8%) | 41 (27.7%) | 247 (38.6%) | 31 (28.2%) | 155 (34.2%) | 10 (26.3%) | >0.99 | ||||
| 2+/day | 184 (16.8%) | 51 (34.5%) | 128 (20.0%) | 41 (37.3%) | 56 (12.4%) | 10 (26.3%) | 0.24 | ||||
| APOE ε4 allele | 164/1488 (11.0%) | 23/190 (12.1%) | 0.63 | 120/956 (12.6%) | 18/160 (11.3%) | 0.70 | 44/532 (8.3%) | 5/30 (16.7%) | 0.17 | 0.37 | |
| Vascular risk factors | |||||||||||
| History of stroke | 30 (3.2%) | 2 (1.6%) | 0.57 | 12 (2.2%) | 1 (1.1%) | 0.70 | 18 (4.7%) | 1 (3.5%) | >0.99 | 0.45 | |
| History of MI | 123 (13.1%) | 10 (8.1%) | 0.15 | 67 (12.0%) | 9 (9.5%) | 0.60 | 56 (14.6%) | 1 (3.5%) | 0.16 | 0.45 | |
| History of diabetes | 122 (13.0%) | 8 (6.5%) | 0.04 | 66 (11.8%) | 5 (5.3%) | 0.07 | 56 (14.6%) | 3 (10.3%) | 0.78 | 0.42 | |
| History of hypertension | 436 (46.3%) | 47 (37.9%) | 0.08 | 254 (45.5%) | 33 (34.7%) | 0.06 | 182 (47.5%) | 14 (48.3%) | >0.99 | 0.43 | |
| History of high cholesterol | 275 (30.7%) | 27 (24.3%) | 0.19 | 188 (35.5%) | 21 (25.0%) | 0.06 | 87 (23.8%) | 6 (22.2%) | >0.99 | 0.81 | |
Based on Fisher’s exact tests.
Compared to fair/good/excellent.
Among 148 smokers, 12 (8.1%) developed AD and 64 (43.2%) died, during 1228.6 person-years of follow up. Among 1,094 never-smokers, 190 (17.4%) developed AD and 421 (38.5%) died, during 8901.1 person years of follow up. From these crude numbers, smoking seems to decrease the risk of AD and increase risk of mortality, relative to not smoking. Taking time into account, using the log-rank test for the Kaplan-Meier survival estimates, smokers developed AD later than nonsmokers (p=0.01) (online supplement Figure a,). While smokers died earlier than nonsmokers, the overall mortality rates were comparable (p=0.54) (online supplement Figure b; Figure 2).
Figure 2.
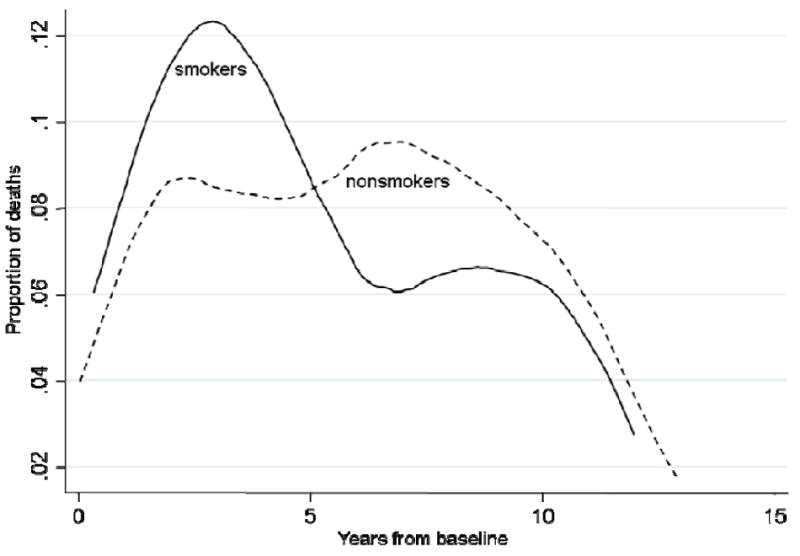
Proportion of death among smokers (solid line) and nonsmokers (dash line) who died during followup.
If smoking increases the risks of both incident dementia and mortality, we would expect different results from the models adjusting and not adjusting for competing risks. In the Cox PH model, our data show that smoking decreases the risk of incident dementia (hazard ratio = 0.47, 95% CI: 0.26-0.86, p = 0.01 from the Cox PH model) but increases the risk of mortality (hazard ratio = 1.09, 95% CI: 0.83-1.41, p = 0.54). Therefore, we may expect adjustment for competing risks to change the effect size, but not necessarily reverse the results.
Table 2 gives the results of estimated effect of smoking on incident AD with and without adjustment for competing risks. Without adjusting for any covariates (Model 1), the estimated HR for smoking suggested an apparent protective effect against incident AD. The key comparison here shows that the HR for smoking is similar between the Cox model (no adjustment for competing risks) and the modified FG model (with adjustment for competing risks). After adjusting for covariates (Models 2 and 3), the hazards for smokers and never smokers are more similar to each other when estimated from the competing risks model than from the Cox model.
Table 2.
Estimated effect of smoking on incident AD age <75 years (n=751) and for age ≥75 years (n=491) with and without adjustment for competing risks.
| Model | Age stratum | Cox model HR† (95% CI) | Competing risks model SHR† (95% CI) |
|---|---|---|---|
| Model 1: include smoking status only | 65-74 | 0.79 (0.39, 1.58) | 0.75 (0.37, 1.49) |
| ≥75 | 0.32 (0.10, 0.99) | 0.48 (0.26, 0.86) | |
| Model 2*: include smoking status, and other baseline covariates | 65-74 | 0.82 (0.41, 1.68) | 0.75 (0.37, 1.53) |
| ≥75 | 0.37 (0.17, 1.17) | 0.53 (0.29, 0.97) | |
| Model 3**: include smoking status, and other baseline covariates, and vascular risk factors | 65-74 | 0.88 (0.39, 2.00) | 0.87 (0.38, 1.99) |
| ≥75 | 0.20 (0.03, 1.45) | 0.34 (0.11, 1.05) |
Baseline covariates included age at baseline (wave 2), gender, education, use 3 or more prescription drugs, lipid-lowering drug use, NSAID use, >5 depressive symptoms, self-reported health, and number of drinks.
APOE ε4 carrier (determined at wave 3); vascular risk factors (measured at wave 3) included history of stroke, myocardial infarction, diabetes, hypertension, and high cholesterol.
HR: hazard ratio; SHR: subdistribution hazard ratio.
Examining the model results in the two age strata, we found that in the younger group (age 65-74), Cox models and modified FG competing risks models gave very similar HR estimates in all three models. In contrast, in the older group (age ≥75), Cox models show a much stronger “protective” effect for smoking than the modified FG competing risks model. This finding indicates that, only in the older group, smokers were more likely than nonsmokers to die with AD or develop AD if they had lived throughout the follow-up period.
Figure 3 depicts the adjusted rates of developing AD between smokers and nonsmokers derived from models 1-3 with and without adjusting for competing risks. It shows that adjusted rates estimated from Cox model and modified FG competing risks model are similar for participants aged 65-74 years old. The FG competing risks models gave higher estimates of probability of developing AD.
Figure 3.
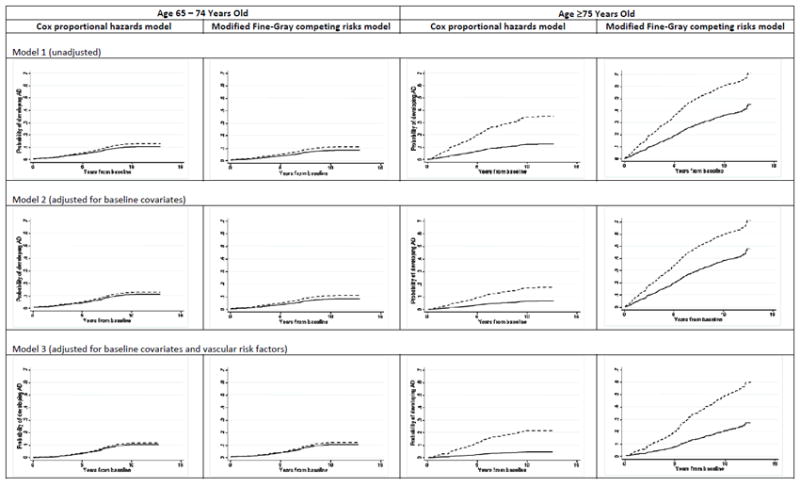
Unadjusted and adjusted cumulative incidence probabilities of developing AD for smokers (solid line) and nonsmokers (dash line) using Cox proportional hazards regression and modified FG competing risks regression models.
DISCUSSION
In this approximately 13-year cohort study of individuals aged 67 and older at the start of the follow-up period, lifetime smoking was associated with a lower hazard of incident AD. This association was statistically non-significant but consistent across all statistical models and both age groups. The focus of our analysis was to compare the results across models that did and did not take competing risks into account, i.e., the possibility that the apparent protective effect of smoking could be explained by smokers at risk for AD dying before developing dementia. The results of the adjusted and unadjusted models were very similar in participants aged 65-74 but quite different in those aged 75 years and older. These data suggest that, in older participants, competing risk from mortality is partly responsible for the decreased hazard of AD observed among smokers. Identifying and addressing competing risk can therefore help eliminate or reduce bias in the risk analysis.
When early observational studies showed negative associations between smoking history and Alzheimer’s disease, commentators appropriately suspected that the finding was likely an artifact of survival bias and/or competing risks.11,12,24-25 Yet, experimental research has consistently shown nicotine-induced short-term enhancement of attention and information processing.26-31 A recent review of molecular and cellular studies suggests that nicotinic acetylcholine receptor mediates protection against neurotoxicity induced by beta amyloid and glutamate.32 Perhaps these findings gained traction in part because were consistent with smokers’ anecdotal experience of improved alertness during and after smoking. However, the deleterious effects of smoking on the cardiovascular and pulmonary systems and its carcinogenic effects were sufficient to increase the risk: benefit ratio of smoking to unacceptable levels. In fact, the population-based Cardiovascular Health Study (CHS) found that second-hand smoking exposure increased the risk of dementia in individuals with subclinical (undiagnosed) cardiovascular disease; we lack data on second-hand smoking to replicate these results.33 In our analyses, self-reported vascular risk factors and vascular disease history did not alter the associations between smoking and AD, but the MoVIES study did not measure subclinical vascular disease. In a recent meta-analysis of studies published from 1984 to 2006, 18 case-control studies without tobacco industry affiliation yielded a nonsignificant pooled odds ratio (0.91, 95% CI: 0.75-1.10) of smoking effect on AD; 8 case-control studies with tobacco industry affiliation yielded a significant protective effect of smoking on AD (odds ratio = 0.86, 95% CI: 0.75-0.98); 14 cohort studies without tobacco industry affiliation showed that smoking had a significant increased relative risk of AD (1.45, 95% CI: 1.16-1.80); and 3 cohort studies with tobacco industry affiliation found that smoking had a nonsignificant pooled relative risk (0.60, 95% CI: 0.27-1.32) on AD.34
An important longitudinal study that followed participants from age 50 onwards found that midlife smoking increased risk of AD in late life.8 In contrast, most studies of the effects of various exposures on AD incidence involve cohorts enrolled after the age of 60, before which AD onset is relatively rare.35 Although this is a cost-effective approach with respect to case detection, a study sample restricted to older adults is by definition a survivor sample and lacks information on those who died before they could enter the study. Further non-random information is lost when individuals die during the course of the study without (before) developing AD. Here, we have demonstrated a statistical approach to adjusting for survival bias and competing risk. The fact that we did not find a large effect size for competing risk may reflect the 2-year followup with relatively low attrition rates in the MoVIES study; the probability is low that an individual without dementia at one assessment wave would develop dementia and die before the next assessment. However, it is possible that an individual with incipient AD could die of a smoking-related disease before clinically manifesting dementia.
We have shown that the magnitudes of the estimated effect of smoking on incident AD vary according to whether or not the analytic models take competing risks into account. We could under- or over-estimate the effect of exposure if those who experience the competing event (mortality) have higher (or lower) risk of developing the main event (AD) if the competing events had not occurred. If we simply treat death as censored in the analysis, we presume that the risk of death and the risk of developing AD are uncorrelated, and hence either underestimate or overestimate the effect of smoking on AD.
To judge whether a survival model should take competing risks into account when data involves competing risks, the simplest way is to ask whether the main event and the competing event are highly correlated. If the answer is yes, a model taking competing risks into account will be appropriate, whether or not the competing risk is found to have a significant impact. The Fine and Gray (FG) model is the most widely used competing risks regression model, but it assumes that, due to biological or other characteristics, those who experienced the competing event would have had zero chance of developing the main event during the entire follow-up period. This assumption is violated for the present study because individuals who died during follow-up still had the chance to develop AD before they died. Our modification of the FG model based on propensity scores adjusts for this possibility. It is worth noting that we used smoking status and demographic information (age, sex, and education) to predict the probability of developing AD in the propensity score model. If this model is misspecified, the results of the modified competing risks model might not be ideal although it should still perform better than the nonadjusted Cox model and the FG model.
In addition to competing risks, survival bias at the time of sample selection is a form of unavoidable selection bias that should also be taken into account in studies of older adults. The impact of selection bias on the estimation of the effect of the exposure has been described in detail by Hernán et al.11 and Kukull.12 Essentially, those who died directly or indirectly because of smoking, and those who already developed AD, cannot be included in the study sample. We can speculate that selective survival accounts for the lower proportion of smokers in the older compared to the younger subgroup of our cohort, and thus that survival bias might account for the larger size of the apparent protective effect of smoking against AD in the older subgroup. In general, when we recruit an elderly cohort, we will underestimate the negative effect of smoking on incident AD unless we can adjust for survival bias in the selected sample. To adjust for survival bias, we would need data of age-specific rates of mortality and AD incidence for both smokers and nonsmokers. We will also need age-specific information on proportions of smokers. Since such data cannot usually be obtained, there is a strong argument to be made for recruiting study cohorts at earlier ages and following them through the age of risk for diseases of later life. When such cohorts are not available, statistical approaches such as those described here can help provide relatively unbiased estimates of the risk of disease associated with chronic exposures.
Supplementary Material
Acknowledgments
Sources of funding support:
This study was supported in part by grants R01 AG07562, R01 AG023651, and K24 AG022035 from the National Institute on Aging, US Department of Health and Human Services.
Footnotes
Financial disclosure: None reported.
References
- 1.Ganguli M, Kukull WA. Lost in translation; epidemiology, risk, and Alzheimer’s disease. Archives of Neurology. 2010;67:107–111. doi: 10.1001/archneurol.2009.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson SC, Hauser SL. The challenge of publishing newsworthy epidemiology. Annals of Neurology. 2010;68:A8–A10. doi: 10.1002/ana.22131. [DOI] [PubMed] [Google Scholar]
- 3.Graves AB, Van Duijn CM, Chandra V, Fratiglioni L, Heyman A, Jorm AF. Alcohol and tobacco consumption as risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. International Journal of Epidemiololy. 1991;20(Suppl 2):S48–S57. doi: 10.1093/ije/20.supplement_2.s48. [DOI] [PubMed] [Google Scholar]
- 4.van Duijn C, Hofman A. Relation between nicotine intake and Alzheimer’s disease. British Medical Journal. 1991;302:1491–1494. doi: 10.1136/bmj.302.6791.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hebert LE, Scherr PA, Beckett LA, Funkenstein HH, Albert MS, Chown MJ, Evans DA. Relation of smoking and alcohol consumption to incident Alzheimer’s disease. American Journal of Epidemiology. 1992;135(4):347–355. doi: 10.1093/oxfordjournals.aje.a116296. [DOI] [PubMed] [Google Scholar]
- 6.Hillier V, Salib E. A case-control study of smoking and Alzheimer’s disease. International Journal of Geriatric Psychiatry. 1997;12(3):295–300. doi: 10.1002/(sici)1099-1166(199703)12:3<295::aid-gps476>3.3.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Tyas SL, Pederson LL, Koval JJ. Is smoking associated with the risk of developing Alzheimer’s disease? Results from three Canadian data sets. Annals of Epidemiology. 2000;10(7):409–416. doi: 10.1016/s1047-2797(00)00061-2. [DOI] [PubMed] [Google Scholar]
- 8.Rusanen M, Kivipelto M, Quesenberry CP, Zhou J, Whitmer RA. Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Archives of Inernal Medicine. 2011;171(4):333–229. doi: 10.1001/archinternmed.2010.393. [DOI] [PubMed] [Google Scholar]
- 9.Kloppenborg RP, van den Berg E, Kappelle LJ, Biessels GJ. Diabetes and other vascular risk factors for dementia: Which factor matters most? A systematic review. Europe Journal of Pharmacology. 2008;585:97–108. doi: 10.1016/j.ejphar.2008.02.049. [DOI] [PubMed] [Google Scholar]
- 10.Sabbagh MN, Tyas SL, Emery SC, Hansen LA, Alford MF, Reid RT, Tiraboschi P, Thal LJ. Smoking affects the phenotype of Alzheimer disease. Neurology. 2005;64(7):1301–1303. doi: 10.1212/01.WNL.0000156912.54593.65. [DOI] [PubMed] [Google Scholar]
- 11.Hernán MA, Alonso A, Logroscino G. Cigarrete smoking and dementia: potential selection bias in the elderly. Epidemiology. 2008;19:448–450. doi: 10.1097/EDE.0b013e31816bbe14. [DOI] [PubMed] [Google Scholar]
- 12.Kukull WA. The association between smoking and Alzheimer’s disease: effects of study design and bias. Biological Psychiatry. 2001;49:194–199. doi: 10.1016/s0006-3223(00)01077-5. [DOI] [PubMed] [Google Scholar]
- 13.Kim IS, Ohrr H, Jee SH, Kim H, Lee Y. Smoking and total mortality: Kangwha cohort study, 6-year follow-up. Yonsei Medical Journal. 1993;34(3):212–222. doi: 10.3349/ymj.1993.34.3.212. [DOI] [PubMed] [Google Scholar]
- 14.Gupta PC, Mehta HC. Cohort study of all-cause mortality among tobacco users in Mumbai, India. Bulletin of the World Health Organization. 2000;78(7):877–883. [PMC free article] [PubMed] [Google Scholar]
- 15.Ganguli M, Dodge HH, Chen P, Belle S, DeKosky ST. Ten-year incidence of dementia in a rural elderly US community population. The MoVIES Project. Neurology. 2000;54:1109–1116. doi: 10.1212/wnl.54.5.1109. [DOI] [PubMed] [Google Scholar]
- 16.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. British Journal of Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 17.Chen P, Ganguli M, Mulsant BH, DeKosky ST. The temporal relationship between depressive symptoms and dementia: a community-based prospective study. Archives of General Psychiatry. 1999;56(3):261–266. doi: 10.1001/archpsyc.56.3.261. [DOI] [PubMed] [Google Scholar]
- 18.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 19.Ganguli M, Dodge HH, Mulsant BH. Rates and predictors of mortality in an aging, rural, community-based cohort: the role of depression. Archieves of General Psychiatry. 2002;59(11):1046–1052. doi: 10.1001/archpsyc.59.11.1046. [DOI] [PubMed] [Google Scholar]
- 20.Ganguli M, Dodge HH, Shen C, Pandav RS, DeKosky ST. Alzheimer disease and mortality: a 15-year epidemiology study. Archieves of Neurology. 2005;62(5):779–784. doi: 10.1001/archneur.62.5.779. [DOI] [PubMed] [Google Scholar]
- 21.Grunkemeier GL, Jin R, Eijkemans MJC, Takkenberg JJM. Actual and actuarial probabilities of competing risks: apples and lemons. The Annals of Thoracic Surgery. 2007;83:1586–1592. doi: 10.1016/j.athoracsur.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 22.Freiberg M, Chang CH, Skanderson M, McGinnis K, Kuller LH, Kraemer KL, Rimland D, Goetz M, Butt AA, Rodriguez-Barradas MC, Gibert C, Leaf D, Brown ST, Samet J, Kazis L, Bryant K. Justice AC for the Veterans Aging Cohort Study. The risk of incident coronary heart disease among Veterans with and without HIV and hepatitis C. Circulation: Cardiovascular Quality and Outcomes. 2011;4(4):425–432. doi: 10.1161/CIRCOUTCOMES.110.957415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Journal of the American Statistical Association. 1999;94:496–509. [Google Scholar]
- 24.Riggs JE. The “protective” influence of cigarette smoking on Alzheimer’s and Parkinson’s diseases. Quagmire or opportunity for neuroepidemiology? Neurologic Clinics. 1996;14(2):353–358. doi: 10.1016/s0733-8619(05)70261-9. [DOI] [PubMed] [Google Scholar]
- 25.Debanne SM, Bielefeld RA, Cheruvu VK, Fritsch T, Rowland DY. Alzheimer’s disease and smoking: bias in cohort studies. Journal of Alzheimer’s Disease. 2007;11:313–321. doi: 10.3233/jad-2007-11308. [DOI] [PubMed] [Google Scholar]
- 26.Le Houezec J, Benowitz NL. Basic and clinical psychopharmacology of nicotine. Clinics in Chest Medicine. 1991;12(4):681–699. [PubMed] [Google Scholar]
- 27.Jones GMM, Sahakian BH, Levy R, Warburton DM, Gray JA. Effects of acute subcutaneous nicotine on attention, information processing and short-term memory in Alzheimer’s disease. Psychopharmacology. 1992;108:485–494. doi: 10.1007/BF02247426. [DOI] [PubMed] [Google Scholar]
- 28.Levin ED. Nicotinic systems and cognitive function. Psychopharmacology. 1992;108:417–431. doi: 10.1007/BF02247415. [DOI] [PubMed] [Google Scholar]
- 29.Rusted JM, Warburton DM. Facilitation of memory by post-trial administration of nicotine: evidence for an attentional explanation. Psychopharmacology. 1992;108:452– 455. doi: 10.1007/BF02247420. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence AD, Sahakian BJ. Alzheimer disease, attention, and the cholinergic system. Alzheimer Disease & Associated Disorders. 1995;9(Suppl 2):43–49. [PubMed] [Google Scholar]
- 31.Whitehouse P, Kalaria RN. Nicotinic receptors and neurodegenerative dementing diseases: basic research and clinical implications. Alzheimer Disease & Associated Disorders. 1995;9(Suppl 2):3–5. doi: 10.1097/00002093-199501002-00002. [DOI] [PubMed] [Google Scholar]
- 32.Kawamata J, Shimohama S. Stimulating nicotinic receptors trigger multiple pathways attenuating cytotoxicity in models of Alzheimer’s and Parkinson’s diseases. Journal of Alzheimer’s Disease. 2011;24:95–109. doi: 10.3233/JAD-2011-110173. [DOI] [PubMed] [Google Scholar]
- 33.Barnes DE, Height, Height TJ, Mehta KM, Carlson MC, Kuller LH, Targer IB. Second-hand smoke, vascular disease, and dementia incidence: findings from the Cardiovascular Health Cognition Study. American Journal of Epidemiology. 2010;171:292–302. doi: 10.1093/aje/kwp376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. Journal of Alzheimer’s Disease. 2010;19(2):465–480. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: the Baltimore longitudinal study of aging. Neurology. 2000;54(11):2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.