

Abstract
Understanding the molecular sequence of events that culminate in multiple abnormalities in brains from patients that died with Alzheimer’s Disease (AD) will help to reveal the mechanisms of the disease and identify upstream events as therapeutic targets. The activity of the mitochondrial α-ketoglutarate dehydrogenase complex (KGDHC) in homogenates from autopsy brain declines with AD. Experimental reductions in KGDHC in mouse models of AD promote plaque and tangle formation, the hallmark pathologies of AD. We hypothesize that deficits in KGDHC also lead to the abnormalities in endoplasmic reticulum (ER) calcium stores and cytosolic calcium following K+ -depolarization that occur in cells from AD patients and transgenic models of AD. The activity of the mitochondrial enzyme KGDHC was diminished acutely (minutes), long term (days) or chronically (weeks). Acute inhibition of KGDHC produced effects on calcium opposite to those in AD, while the chronic or long term inhibition of KGDHC mimicked the AD-related changes in calcium. Divergent changes in proteins released from the mitochondria that effect ER calcium channels may underlie the selective cellular consequences of acute versus longer term inhibition of KGDHC. The results suggest that the mitochondrial abnormalities in AD can be upstream of those in calcium.
Keywords: Calcium, Endoplasmic reticulum, Mitochondria, Ketoglutarate dehydrogenase
1. INTRODUCTION
Multiple changes including abnormalities in glucose and calcium homeostasis are apparent in autopsy brains and cells from patients that died with Alzheimer’s Disease (AD). An understanding of the pathological order of events that lead to AD is important for understanding the disease process and for drug development. Although in the genetic forms of AD the cause is known, in the vast majority of cases of AD, the causative event is unknown. The experiments described in this manuscript test whether altered mitochondrial function can lead to the calcium changes that occur in cells from AD patients.
1.1 Glucose metabolism in AD
A decline in glucose metabolism is an early and invariant change in AD that predicts who will get AD and who will progress from mild cognitive impairment to AD (Jack CR Jr et al, 2010; Mosconi et al, 2008). Reduced activities in brain homgenates of key mitochondrial tricarboxylic acid (TCA) cycle enzymes, whose decline is highly correlated to the clinical dementia rating of the patients before they died (Bubber et al, 2005), may underlie the AD-related reductions in brain metabolism. The decline in the TCA cycle α-ketoglutarate dehydrogenase complex (KGDHC) may be particularly important (Gibson et al, 2005; Gibson et al, 2000; Gibson et al, 1988,Chinopoulos & Adam-Vizi, 2006; Tretter & Adam-Vizi, 2000). Reductions in KGDHC can be directly related to the pathophysiology. Reductions in KGDHC activity promote plaque and tangle formation which suggests that the mitochondrial changes can be an upstream event (Dumont et al, 2009; Karuppagounder et al, 2009). The focus of this paper is to determine whether a reduction in KGDHC can also lead to the abnormalities in calcium regulation that accompany AD.
1.2. Calcium regulation in AD
Calcium regulation is known to be altered with aging and AD (Berridge, 2010; Bezprozvanny & Mattson, 2008; Gibson & Peterson, 1987; Peterson et al, 1985; Stutzmann, 2007; Supnet & Bezprozvanny, 2010). Calcium uptake is diminished in fibroblasts (Peterson et al, 1985) and lymphocytes (Gibson et al, 1987) from patients with AD. Furthermore, inositol trisphosphate (IP3) sensitive calcium stores in the endoplasmic reticulum (ER) are exaggerated in fibroblasts from AD patients (Ito et al, 1994), in fibroblasts and neurons cultured from mice bearing presenilin-1 (PS-1) mutations (Leissring et al, 2000), and in hippocampus and cortical neurons from 3XTg AD mice (Stutzmann et al, 2004; Stutzmann et al, 2006). Two possible mechanisms have been proposed to underlie the exaggerated ER calcium stores in cells bearing PS-1 mutations. Presenilins control the ER calcium leak channels, and PS-1 mutations that cause AD block these channels (Nelson et al, 2007; Tu et al, 2006). The second possibility is that mutations in presenilin interact with the inositol 1,4,5-trisphosphate receptor (InsP3R) Ca2+ release channel and exert profound stimulatory effects on its gating activity in response to saturating and suboptimal levels of InsP3. These interactions result in exaggerated cellular Ca2+ signaling in response to agonist stimulation (Cheung et al, 2008, Cheung et al., 2010). ER calcium stores are also altered in fibroblasts from AD patients without presenilin mutations (i.e., the vast majority of patients). In these patients, the cause of the changes in calcium regulation is unknown.
AD-causing mutations also alter cytosolic free calcium ([Ca2+]i) (Supnet & Bezprozvanny, 2010). PS-1 mutations enhance synaptosomal [Ca2+]i following exposure to depolarizing agents, Aβ or mitochondrial toxins (Begley et al, 1999a). Diminishing the calcium release from ER completely abrogates the enhanced mitochondrial dysfunction in synaptosomes from PS-1 mutant mice (Begley et al, 1999a).
1.3. Linkage of mitochondria and endoplasmic reticulum
Mitochondria and ER are structurally and functionally coupled (Giorgi et al, 2009; Szabadkai & Rizzuto, 2007; Walter & Hajnóczky, 2005). The mitochondrial associated membranes that bridge the mitochondria and ER contain presenilins (Area-Gomez et al, 2009). Calcium released from the ER selectively increases mitochondrial calcium (Rizzuto et al, 1998), and consequentially alters mitochondrial function (Szabadkai et al, 2006) including apoptosis (Giorgi et al, 2009), KGDHC activity and ROS production (Lawlis & Roche, 1981; Tretter & Adam-Vizi, 2004; Tretter & Adam-Vizi, 2005) and enhanced ATP generation (McCormack & Denton, 1979; Walter & Hajnoczky, 2005). Furthermore, oxidants that inactivate KGDHC also elevates ER calcium stores (Huang et al, 2005). Whether elevated ER calcium is directly caused by diminished KGDHC is unknown.
Few studies have examined the consequences of diminished mitochondrial function on ER calcium regulation. The current experiments tested whether diminishing KGDHC alters ER calcium levels in cultured neurons from embryonic or adult mice as well as in a mouse neuroblastoma cell line. KGDHC was inhibited acutely with a specific inhibitor, long term with adenovirus expressing shRNA to the E1k subunit of KGDHC and chronically by using neurons cultured from transgenic mice with diminished KGDHC activity.
2. Materials and methods
2.1. Cell culture
N2a neuroblastoma cells (American Type Culture Collection, Manassas; VA) were maintained at 37°C in a humidified incubator under 5% CO2 and 95% air in the complete medium (DMEM supplemented with 10% fetal bovine serum) (Invitrogen; Carlsbad, CA). Cells were trypsinized when they reached 80% confluence and were seeded into 24-well plates (1×104 cells/well), 6-well plates (4×104 cells/well) or into Delta TPG dishes (Bioptechs, Butler, PA) (2×104 cells/well) for in-situ KGDHC activity assay, adenovirus infection and calcium measurements, respectively.
Cultured neurons were prepared from C57BL/6 mice or from E2k+/- mice (Yang et al, 2009). E2k [dihydrolipoyl succinyltransferase (DLST)] is the second subunit of KGDHC. The Institutional Animal Care and Use Committee of Weill Cornell Medical College approved all procedures with the animals. Neurons from embryos were prepared from the cerebral cortices of E15.5 of C57BL/6 mice (Charles River, Wilmington MA) (Brewer & Torricelli, 2007) and E2k+/- mice as described in detail previously (Huang et al, 2002). Neurons were seeded onto poly-D-Lysine (Sigma; St. Louis, MO) coated 24-well plates (2 × 105 cells /well) and poly-D-Lysine coated Delta TPG dishes (4 × 105 cells /dish) for in-situ KGDHC activity assay and calcium measurements, respectively. Two hours after incubation at 37°C in a humidified incubator with 5% CO2, the medium was replaced with neurobasal/B27 medium containing 0.5 mM glutamine and 25 μM glutamate (Invitrogen; Carlsbad, CA). The medium was changed to neurobasal/B27 medium without glutamate after 4 days and the neurons were cultured for a total of 10 days before treatments.
Neurons from adults were prepared from the cerebral cortices of C57BL/6 mice (Charles River, Wilmington MA) and E2k+/- mice (Yang et al, 2009) at 6 weeks of age as described previously (Brewer & Torricelli, 2007). Cells were plated onto poly-D-Lysine coated Delta TPG dishes at a seeding density of 4 × 105 cells /dish and incubated at 37°C in a humidified incubator with 5% CO2. Two hours after plating, the medium was replaced with neurobasal A/B27 containing 0.5 mM glutamine, 1 μg/ml Gentamycin and 5 ng/ml mouse FGF2 (Invitrogen; Carlsbad, CA). Medium was refreshed every four to five days and cultured for 10 days before treatments.
2.2. KGDHC activity measurements
KGDHC catalyzes the following reaction: α-ketoglutarate + NAD+ + CoA → succinyl CoA + CO2 + NADH. Two well established assays were used to measure KGDHC activities. Both methods use the specific substrate α-ketoglutarate (α-KG) to distinguish NADH production from KGDHC to that from other enzymes (i.e. substrate specificity). One method assays activity in cell lysates whereas the other assesses the activity in slightly permeabilized cells in which the mitochondria are intact (an in situ assay).
KGDHC activity assay in cell lysates
N2a cells in 6-well plates at 24 hr and 48 hr post-infection were washed twice with Dulbesso’s Phosphate Buffered Saline (D-PBS) and lysed with 250 μl of cell lysate buffer (50 mM Tris-HCl pH 7.2, 0.4% Triton X-100, 0.2 mM EGTA, 50 μM Leupeptin and 1 mM DTT). KGDHC activities were assayed immediately as described previously (Gibson et al, 1988).
In situ KGDHC activity assay
The reaction for in situ activity staining is basically the same as for the lysis method except that the reducing equivalents are coupled to nitroblue tetrazolium (NBT). The reduced dye forms crystals which accumulate in proportion to the reaction rate. In situ KGDHC activity of N2a cells or cortical neurons from embryos in 24-well plates after treatment was assayed as described previously (Park et al, 2000; Shi et al, 2005).
2.3. Inhibition of KGDHC
Acute inhibition of KGDHC by carboxyethyl succinyl phosphonate (CESP)
N2a cells, neurons from embryos and neurons from adult mice seeded in 24-well plates or poly-D-Lysine coated Delta TPG dishes were treated with varying concentrations of CESP [0, 10, 50 and 100 μM or diethyl succinyl phosphonate (DESP)] at room temperature for one hr in a balanced salt solution (BSS): (140 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 5 mM glucose, 10 mM HEPES, and 2.5 mM CaCl2, pH 7.4). Cells in 24-well plates were used for in-situ KGDHC assay. Cells in Delta TPG dishes were used for calcium measurements.
Long term inhibition of KGDHC by adenovirus mediated shRNA to KGDHC
The stability of the succinylphosphonates in water is unknown so long term inhibition was modeled with adeno-virus’s. To knockdown E1k gene expression, eight shRNA targeting E1k were designed, constructed and tested (Qiu et al, 2008). The shRNA with the strongest knockdown activity (shR-E1-1) was constructed into an adenoviral vector under the control of U6 promoter (DUAL-U6-EGFP). shR-E1-1 is composed of a sense strand (GATGAGAAGATCTTGCACATGAA), a loop (TTCAAGAGA), an antisense strand (TTGATGTGCAAGATCTTCTCATC) and a transcription termination (TTTTT) sequences. A scrambled shRNA (shR-Scr) that does not target any specific gene was used as a control. The sequence of the shR-Scr is GACACGCGACTTGTACCACTTCAAGAGAGTGGTACAAGTCGCGTGTCTTTTTT. The adenovirus expressing shRNA for E1k (Ad-E1k-shRNA) or scrambled shRNA (Ad-Sc-shRNA) were made in collaboration with Vectorbiolabs (Philadelphia, PA).
N2a neuroblastoma cells and neurons from embryos seeded in 6-well plates or poly-D-Lysine coated Delta TPG dishes were infected at the time of seeding with Ad-E1k-shRNA or Ad-Sc-shRNA (MOI = 100). MOI is the Multiplicity of Infection = ratio of infectious virus particles to cells. They were then incubated for 24 or 48 hr. The effect of E1k shRNA on mRNA and protein levels of E1k and KGDHC activity was assessed with N2a cells by real-time RT-PCR, Western blotting and KGDHC activity assay (see sections below). Calcium measurements were assessed with both N2a cells and neurons from embryos (see sections below).
Chronic inhibition of KGDHC
Neurons from embryos and adults were harvested from E2k+/- mice (Yang et al, 2009) and wild type mice and seeded onto poly-D-Lysine coated Delta TPG dishes using methods described above. Although a more precise comparions with the long term experiments would be to use E1k mice, they are not available. The activity of KGDHC in the E2k+/- mice is reduced approximately the same in the E2k+/- mice as in the neurons treated with adenovirus to reduce E1k.
2.4. Subcellular fractionation
N2a cells were seeded at 6 × 104 cells/cm2 on 10-cm dishes and maintained for 2 days prior to the experiment. On the day of experiment, the medium was aspirated, and cells were washed once with PBS. Cells were treated with CESP (acute inhibition of KGDHC) for 1 hr or infected with adenovirus expressing E1k shRNA (long term inhibition of KGDHC) for 48 hr. After treatment, mitochondrial and cytosolic fractions were isolated by published methods (Huang et al, 2003b; Yang et al, 1997).
2.5. SDS-PAGE and Western blotting
A 5X SDS-sample loading dye [50% glycerol, 250 mM Tris (pH 6.8), 10 mM EDTA, 10% SDS and 0.04% bromophenol blue] was added to N2a cell lysates to achieve 1X final concentration. Electrophoresis was done with a 4-20% Tris-Glycine gel (Invitrogen; Carlsbad, CA) using 90 volts for three hr. Western blotting was performed as described in a previous study (Shi et al., 2005, Shi et al., 2011) with different antibodies [E1k (1:1000 dilution, generated in collaboration with Rockland Immunochemicals Inc. Gilbertsville, PA); cytochrome c (1:1000 dilution, BD Biosciences, San Jose, CA); actin (1:5000 dilution, Sigma, St. Louis, MO)].
2.6 Real-time RT-PCR
Total RNA was isolated from N2a cells 24 hr and 48 hr post-infection by an RNeasy Plus Micro kit (Qiagen; Valencia, CA). First strand cDNA was synthesized from the isolated total RNA. Real-time PCR of E1k was performed using an Applied Biosystems 7500 Fast Real-Time PCR system with pre-designed Taqman® gene expression assays (Applied Biosystems; Foster City, CA) by a published method (Shi & Gibson 2011).
2.7. Measures of cytosolic free calcium ([Ca2+]i) in response to K+-depolarization
Cells in Delta TPG dishes were loaded with 2 μM Fura-2 AM (Invitrogen; Carlsbad, CA) in BSS for one hr at room temperature and rinsed twice with BSS. [Ca2+]i of cells in 2 ml BSS was monitored on the stage of an inverted Olympus IX70 microscope at room temperature with a Delta Scan System from PTI (Photon Technology International, Lawrenceville, NJ). Excitation wavelengths were alternated between 350 and 378 nm (band pass 4 nm) and emission was monitored at 510 nm with a Hamamatsu C2400 SIT camera at 5 s intervals. Basal [Ca2+]i was measured for one min. KCl (final concentration of 50 mM) was added to induce the influx of Ca2+ and the signal was measured for another 5 min. Each value was the average of 32 images taken within 5 s. Standard images of Fura-2 solutions with minimum and maximum [Ca2+]i were taken at the end of each day’s experiment to calculate the intracellular calcium concentrations (Huang et al, 1991).
2.8. Measures of bradykinin or caffeine releasable calcium stores (BRCS, CRCS)
Cells were loaded with 2 μM Fura-2 AM in BSS for one hr at room temperature and rinsed twice with Ca2+-free BSS (140 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 5 mM glucose, 10 mM HEPES, 0.1 mM CaCl2, 1 mM EGTA, pH 7.4) (Gibson et al, 2002). Then, 2 ml of Ca2+-free BSS was added to each dish and [Ca2+]i was monitored with the Delta Scan System described in Section 2.7. Basal [Ca2+]i was measured for one min. Bradykinin (200 nM) or caffeine (25 mM) (Sigma; St. Louis, MO) was added to release ER calcium and the signal was measured for another 5 min. Each value was the average of 32 images taken within 5 s. Standard images of Fura-2 solutions with minimum and maximum [Ca2+]i were taken at the end of each day’s experiment to calculate the intracellular calcium concentrations (Huang et al, 2004; Huang et al, 2005).
2.9. Statistical comparisons
Statistical differences for multiple comparisons were determined by ANOVA followed by Student Newman-Keuls. Individual comparisons were done with a Student’s t-test. Every experiment was done at least twice in triplicate which provided a minimum of n=6. The “n” is included with each experiment.
3. RESULTS
3.1. Acute, long term and chronic inhibition of KGDHC
Succinylphosphonate and its cell permeable analogue carboxyethyl succinyl phosphonate (CESP) are potent and selective inhibitors of KGDHC (Bunik et al, 2005). CESP produced acute inhibition of KGDHC. On the other hand, the diethyl ester derivative of succinylphosphonate (DESP) does not inhibit KGDHC. Thus, similar to using scrambled shRNA as control for adenovirus E1k shRNA experiments, DESP provides a perfect control for non-specific effects of CESP (Bunik et al, 2005). CESP caused a concentration-dependent inhibition of in situ KGDHC activity. CESP (50 μM) inhibited KGDHC activity by 60% in N2a cells (Figure 1A) and 35% in primary cultured cortical neurons (Figure 1B). DESP had no effect (Data not shown).
Figure 1. Acute inhibition of KGDHC activity with CESP in N2a neuroblastoma (A) and neurons cultured from embryos (B).

Cells on 24-well plates were incubated with KGDHC reaction mixture containing CESP (0, 10, 50 and 100 μM) for one hr at room temperature and in situ KGDHC was measured as described in Methods. The CESP structural analogue DESP had no effect on KGDHC (Bunik et al, 2005) (data not shown). Panel A is data from N2a cells (2 × 104 cells/well). Panel B has results from neurons cultured from embryos (2× 105 cells/well). Values (percent of control) are the means ± S.E.M. (n=8~10). *** Denotes significantly different (p<0.005) from control.
Adenovirus expressing shRNA to the E1k subunit of KGDHC (Ad-E1k-shRNA) produced a long term inhibition of KGDHC in N2a cells (i.e., 24 to 48 hours) (Figure 2). Adenovirus expressing scrambled shRNA (Ad-Sc-shRNA) was used as control. The infection efficiency of the Ad-E1k-shRNA was 70%, while that of the Ad-Sc-shRNA was 86%. E1k-shRNA diminished the mRNA level of E1k by more than 80% at 24 and 48 hour post-infection as compared to the Ad-Sc-shRNA infected cells (Figure 2A). Activity levels of KGDHC were diminished by 15% and 30% at 24 and 48 hour post-infection, respectively (Figure 2B). The percent (44%) of neurons infected was lower than N2a cells (70%). Thus, measuring overall activity does not reflect activity in the infected neurons. KGDHC activity was diminished by 15.7% in the in situ assay. If we divide this by the percent infected, the estimated inhibition is 35.7%. The calcium response was only measured in the infected cells. This percent inhibition is nearly identical to the inhibition in N2a cells.
Figure 2. Both mRNA (A) and activity levels (B) show long term inhibition of KGDHC.

Long term inhibition of KGDHC by adenovirus expressing shRNA to E1k in N2a cells alters the levels of E1k mRNA and activity of KGDHC. N2a cells were seeded at a density of 4 × 104 cells/well in 6-well plates and infected with100 MOI of Ad-E1k-shRNA or Ad-Sc-shRNA. The cells were then cultured for indicated number of hours. The infection efficiency was 70% to 86% for 24 hr of infection and 80% to 90% for 48 hr of infection. Values are the means ± S.E.M. of changes relative to the control group (Ad-Sc-shRNA). The mRNA level of E1k was measured by relative quantitative real-time PCR in triplicate and normalized with beta-2 microglobulin (b2m) (n=15). KGDHC activity was normalized with total protein (n=15). ** Denotes significantly different from control group (p<0.05). *** Denotes significantly different from control group (p<0.005).
The use of neurons cultured from either embryos or adult mice with reduced activity of KGDHC allowed us to study the effect of chronic inhibition of KGDHC. The activity of KGDHC in brain of E2k+/- mouse is diminished by about one half as compared to that of the wild-type mouse (E2k+/+) (Yang et al, 2009)
3.2. The response of cytosolic free calcium ([Ca2+]i) to K+-depolarization or to the release of endoplasmic reticulum (ER) calcium was determined in three cell types
Two aspects of calcium regulation were assessed to test the consequences of inhibition of KGDHC on calcium regulation in three different cell types (Figure 3). Both aspects of calcium regulation were assessed by the measurement of [Ca2+]i. The first measure was the response of [Ca2+]i to potassium depolarization (50 mM KCl). The responses of N2a cells (Figure 3a), neurons from embryos (Figure 3b) and neurons from adults (Figure 3c) were similar, but the magnitude and pattern of the responses differed. The second measure of calcium regulation was the stores of calcium in the endoplasmic reticulum (ER). The stores were released with bradykinin (or caffeine in adult neurons) and the increase in [Ca2+]i was determined. The responses of N2a cells to bradykinin (Figure 3d) was similar to that of neurons from embryos to bradykinin (Figure 3e), or neurons from adults to caffeine (Figure 3f), but the magnitude and pattern of the responses differed.
Figure 3. Changes in [Ca2+]i in response to K+ depolarization (a-c) or the release of ER calcium stores (d-f).

Calcium homeostasis was evaluated in multiple cell types by assessing the responses of cytosolic free calcium to depolarizing concentrations of K+ (50 mM) in N2a (a), neurons from embryonic mice (b), and neurons from adult mice (c). The release of ER calcium by either bradykinin (200 nM) or caffeine (25 mM) was determined in N2a cells (d), neurons from embryonic mice (e), and neurons from adult mice (f). The gray areas represent the calcium integration between 60 sec to 360 sec above basal. The values represent an average response of 187 to 355 neurons. The error bars represent the standard errors. In most cases they were too small to see.
3.3. The effects of reduced KGDHC activity on the response of [Ca2+]i to K+-depolarization
The responses of K+-induced changes in [Ca2+]i to acute, long term or chronic inhibition of KGDHC were determined in multiple cell types (Figure 4). In N2a cells, acute inhibition of KGDHC caused a dose-dependent reduction in the response of [Ca2+]i to K+ depolarization (top panel, Figure 4A). For example, 100 μM CESP reduced the response of [Ca2+]i to K+ depolarization by 75%. The structural analogue DESP, which has no effect on KGDHC, did not affect the calcium response. Acute inhibition of KGDHC did not alter the mitochondrial membrane potential in N2a cells. N2a cells were loaded with TMRM (20 nM) and the mitochondrial membrane potential was determined with the Delta scan system (Zhang et al., 2001). Data from two independent experiments (n=144~277 cells) did not reveal any significant differences between control (100.0 ± 3.3%) and 10 μM CESP (112.2 ± 3.9%) or 100 μM CESP (108.0 ± 3.7%). In neurons cultured from embryos, acute inhibition of KGDHC did not significantly alter the response of [Ca2+]i to K+-depolarization (middle panel, Figure 4A). In neurons cultured from adults, acute inhibition of KGDHC reduced the response of [Ca2+]i to K+-depolarization by 44% (bottom panel, Figure 4A). Thus, acute inhibition of KGDHC generally depresses the response of [Ca2+]i to K+-depolarization.
Figure 4. Selective effects of acute (A), long term (B) or chronic inhibition (C) of KGDHC on K+-induced changes in [Ca2+]i.

The effects of inhibiting KGDHC acutely (CESP) (N2a cells or neurons from embryos or adults), long term (shRNA) (N2a cells or neurons from embryos) or chronically (neurons from embryos or adult E2k+/- mice) on depolarization induced changes in calcium were determined. The vertical axis represents the integrated value for the peak calcium response. Values are means ± S.E.M. The number of cells measured is represented by “n”. **Denotes significantly different from control group (p<0.05). *** Denotes significantly different from control group (p<0.005).
Long term and chronic inhibition of KGDHC produced a very different effect than acute inhibition. Long term inhibition of KGDHC in N2a cells for 24 or 48 hours increased the calcium response to K+ depolarization by 28% and 43%, respectively (top panel, Figure 4B). Similarly, long term inhibition of KGDHC in neurons from embryos for 24 or 48 hours increased the calcium response following K+ depolarization by 37% and 41%, respectively (bottom panel, Figure 4B). Chronic inhibition of KGDHC in neurons cultured from either embryos or adults exaggerated the calcium response to potassium depolarization by 34% and 74%, respectively (Figure 4C). Although the reductions in KGDHC activity in the acute, long term and chronic conditions were approximately equivalent, the consequences on the calcium responses to depolarizing concentrations of K+ were very different.
3.4. The effects of reduced KGDHC on response of [Ca2+]i to the release of endoplasmic reticulum (ER) Ca2+
Acute, long term or chronic inhibition of KGDHC selectively altered the bradykinin or caffeine releasable calcium stores (BRCS or CRCS, respectively) from the ER (Figure 5). Acute inhibition of KGDHC with CESP decreased BRCS by 10% in N2a cells (top panel, Figure 5A) and by 71% in neurons cultured from embryos (middle panel, Figure 5A). CESP reduced the CRCS by 36% in neurons cultured from adults (bottom panel, Figure 5A). However, both long term and chronic inhibition of KGDHC altered ER calcium stores in a different manner. Long term inhibition in N2a cells increased BRCS by 39% after 48 hr infection (top panel, Figure 5B). The BRCS was increased by 11% and 29%, respectively, after 24 hr and 48 hr inhibition in primary neurons (bottom panel, Figure 5B). The increases in BRCS and CRCS were even more striking following chronic inhibition of KGDHC in neurons from embryos (114%) or adults (35%) (Figure 5C). Thus, acute inhibition of KGDHC produced a very different response than chronic or long term inhibition.
Figure 5. Selective effects of acute (A), long term (B) or chronic inhibition (C) of KGDHC on the bradykinin- or caffeine- induced release of ER calcium stores.
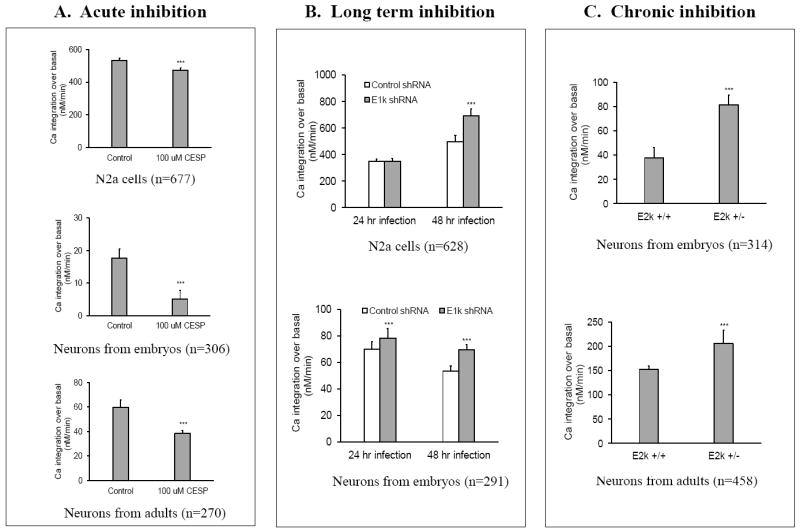
The effects of inhibiting KGDHC acutely (CESP) (N2a cells or neurons from embryos or adults),), long term (shRNA) (N2a cells or neurons from embryos) or chronically (neurons from embryos or adult E2k+/- mice) on the releasable ER calcium were determined. The vertical axis represents the integrated value for the peak calcium response. Values are means ± S.E.M. The number of cells measured is represented by “n”. ***Denotes significantly different from control group (p<0.005).
3.5 Release of cytochrome C from mitochondria upon acute inhibition of KGDHC
Cytochrome C is released from mitochondria following multiple mild metabolic insults and provides a marker of alterations in the protein signaling that is regulated by the mitochondria (Fiskum and Polster, 2004). Whether treatment of cells with CESP (acute inhibition of KGDHC) or infection with Ad-E1k-shRNA (long term inhibition of KGDHC) alters the release of cytochrome C from mitochondria into the cytosol was tested in cells. Mitochondrial and cytosolic fractions were separated from cells and were subjected to SDS-gel electrophoresis and Western blotting. Acute inhibition of KGDHC by CESP increased the release of cytochrome c from mitochondria to the cytosol by 63% and 80% as compared to the control based in two independent experiments (Figure 6). The increased release of cytochrome c is consistent with the reduced cytochrome c in mitochondrial fraction from CESP treated cells (Figure 6). In contrast, cytochrome c was not detectable in cytosol in cells infected with Ad-E1k-shRNA (i.e., long term inhibition of KGDHC, Figure 6).
Figure 6. Acute inhibition of KGDHC induces the release of cytochrome c from mitochondria to cytosol in N2a cell.
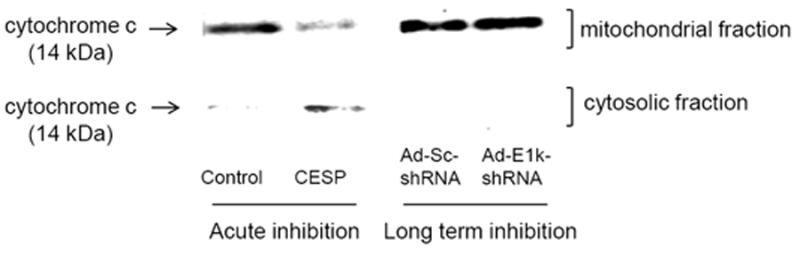
N2a cells were treated with CESP (0 and 50 μM) at RT for 1 hr, or infected with Ad-E1k-shRNA, or Ad-Sc-shRNA with 100 MOI at 37°C for 48 hr, cytochrome c protein levels were determined in mitochondrial and cytosolic fractions. A representative blot shows the release of cytochrome c to cytosol upon acute inhibition of KGDHC, but not in cells subjected to long term inhibition of KGDHC. A replication experiment gave qualitatively similar results.
4. DISCUSSION
An exaggerated calcium response to potassium depolarization or release of calcium from the ER would be expected to disrupt normal neuronal function and predispose to neuron death. These changes are well documented in fibroblasts from AD patients as well as neurons and fibroblasts from cells and mice bearing PS-1 mutations. Whether diminishing the mitochondrial TCA cycle enzyme complex KGDHC that is reduced in AD brains can initiate the abnormalities in cellular calcium regulation has not been assessed previously.
4.1 Long term/chronic inhibition of KGDHC leads to changes that mimic the exaggerated [Ca2+]i following K+-depolarization in cells from AD and PS-1 mutant mice
Elevated resting calcium levels and exaggerated calcium in response to stimulation including KCl depolarization occur in neurons from transgenic mice bearing presenilin mutations that cause AD. Although the calcium response in brain slices induced by a train of action potentials are not very different in brain slices from PS-1 mutant mice (Stutzmann et al, 2004), several studies demonstrate that PS-1 mutations lead to exaggerated response to depolarization and increased resting calcium. An exaggerated elevation in calcium following depolarization occurs in the cytosol in hippocampal neurons harvested from mice bearing PS1 M146V1 mutations (Guo et al, 1999) or PS1[A246E] mutations (Schneider et al, 2001). Synaptosomes prepared from this same strain also exhibit exaggerated elevations of [Ca2+]i following exposure to the same depolarizing KCl concentration as in the current paper, as well as to amyloid β-peptide, or a mitochondrial toxin (Begley et al, 1999b; Mattson & Chan, 2003). The stronger synaptic potentiation of the CA3 to CA1 projection demonstrates the physiological significance of the enhanced calcium response (Barrow et al, 2000).
Long term and chronic inhibition of KGDHC mimicked the exaggerated calcium levels following K+-depolarization observed in cells from patients with AD and in mutant mice, while acute inhibition of KGDHC produced the opposite response. Thus, the results are consistent with the suggestion that long term or chronic inhibition of KGDHC leads to changes in calcium regulation that are similar to those in neurons of mice that bear PS-1 mutations. The lack of change in the mitochondrial membrane potential suggests the effect is not likely bioenergetic, but measures of redox potentials including NADH/NAD ratios, glutathione and ATP would be required to conclusively make that conclusion (Jones and Brewer, 2010; Panihar et al., 2008). In agreement with our in vivo and in vitro studies, a recent modeling paper shows that ATP production is maintained with KGDHC deficiency through a shift in metabolism (Smith and Robinson, 2011)
Nor did studies with Rhod-2 reveal any changes in mitochondrial buffering capacity (data not shown). Thus, the inhibition of KGDHC may impair the calcium buffering capacity of the ER, which in turn leads to exaggerated [Ca2+]i. The dys-regulated calcium levels would sensitize the neurons to age-related metabolic and oxidative stress, which would promote neurodegeneration.
4.2. Long term/chronic inhibition of KGDHC leads to changes that mimic the exaggerated ER calcium store in cells from AD and PS-1 mutant mice
Exaggerated release of calcium from the ER in response to activation of IP3 receptors occurs in cells from AD patients and neurons from transgenic mice bearing presenilin mutations that cause AD. The exaggerated ER calcium stores in fibroblasts from AD patients are particularly well replicated in patients bearing PS-1 mutations (Ito et al, 1994), and in cells from patients with “non-genetic” forms of AD (Huang et al, 1994). ER calcium stores are also exaggerated in fibroblasts and cultured neurons from mice bearing PS-1mutations (Leissring et al, 2000). Moreover, caffeine-gated stores are exaggerated in hippocampus and cortical neurons from 3XTg AD mice (Stutzmann et al, 2004; Stutzmann et al, 2006). Although the molecular basis of the changes in calcium in patients bearing PS-1 mutations has been partially elucidated (see INTRODUCTION), the underlying mechanism in familial AD caused by other mutations and sporadic AD patients is entirely unknown. The current results suggest the changes could be secondary to abnormalities in the mitochondrial TCA cycle since long term and chronic inhibition of KGDHC exaggerated the ER calcium store. Increases in [Ca2+]i can alter synaptic activity by activation of calcineurin and calpains (Rao et al, 2008), elevate production of amyloidogenic fragments of APP and diminish memory. For example, strong data suggests that as a result of the specific interaction of PS1-M146L with the InsP3R stimulates amyloid beta processing, an important feature of AD pathology (Cheung et al., 2008).
4.3. Selective interaction of proteins with ER calcium channels may underlie the differential calcium responses
The duration of inhibition of KGDHC selectively alters ER calcium regulation and this is not associated with a change in bioenergetics large enough to alter the mitochondrial membrane potential. Neither CESP in the current studies, nor α-keto-β-methylvalerate (KMV) at concentrations that inhibited KGDHC similarly to the current studies (Huang et al, 2003b), diminish the mitochondrial membrane potential. Alteration in NADH or glutathione redox potentials or ROS production may be important and mild changes in these are not necessarily reflected in the mitochondrial membrane potential (Panihar et al. 2008; Jones and Brewer, 2010). Partial inhibition of KGDHC with CESP, in cerebellar granule neurons from E2k+/- mice as well as in HEK cells caused a redistribution of metabolism through the GABA shunt, but neither cell death nor evidence of energetic failure (Santos et al, 2006; Shi et al, 2009). Concentrations of KMV that do not alter the mitochondrial membrane potential cause release of cytochrome C from the mitochondria and activate caspases (Huang et al, 2003a). Caspase activation also occurs in AD (Gastard et al., 2003). Thus, inhibition of KGDHC alters mitochondrial-dependent protein signaling without altering the mitochondrial membrane potential.
Alterations in the interaction of proteins that shuttle between mitochondria and ER to regulate calcium channels in the ER provide a plausible explanation for the different response following acute, long term or chronic inhibition of KGDHC. The release of calcium through the IP3 sensitive calcium channels in the ER is modulated by more than 50 different proteins that can regulate the IP3 receptor including caspase 3, Bcl-2 family members and cytochrome C (see reviews Decuypere et al, 2011a; Decuypere et al, 2011b). Many of these same proteins associate with the mitochondria and the release of these proteins from the mitochondria affect ER calcium dynamics (Boehning et al, 2003; Boehning et al, 2004). The results demonstrate that acute inhibition of KGDHC increases cytosolic cytochrome C release, while long term inhibition does not. Cytochrome C can interact directly with the IP3 receptor. One nM cytochrome C blocks calcium-dependent inhibition of InsP receptor function (Boehning et al, 2003; Boehning et al, 2004). Stimulation of IP3 receptor by cytochrome c releases Ca2+ from ER. This would result in lower Ca2+ in ER, which is consistent with our findings that BRCS are reduced upon acute inhibition of KGDHC. In addition to a direct interaction of cytochrome C, the processes initiated by cytochrome C may play a critical role in the regulation of calcium release from the ER. Release of cytochrome C can activate caspase 3 and the IP3 receptor is a substrate for caspase 3. Following cleavage, the remaining channel domain is no longer IP3-sensitive (i.e., no longer sensitive to bradykinin) (Assefa et al, 2004; Hirota et al, 1999). Such a scenario could also underlie the decline in releasable ER calcium with acute inhibition of KGDHC. Many proteins that modulate the ER calcium stores likely change with KGDHC deficiency. Thus, the change in cytochrome C in this paper provides a “proof of principle” for much more complicated alterations. Modifications in the location of these other proteins may provide full explanation of the changes that we observe following chronic or long term inhibition of KGDHC.
4.4. Altered gene expression may also contribute to the differential response of ER calcium
The difference in the response of ER calcium in the acute, chronic and long term models could also be related to altered gene expression in the chronic and long term models. In mouse brain in vivo, chronic deficiency of KGDHC increases the expression of another mitochondrial enzyme, malate dehydrogenase (our unpublished data). Furthermore, chronic inhibition of KGDHC modifies the response of malate dehydrogenase to oxidative stress (Shi et al, 2008). Mitochondrial metabolic inhibitors activate a mitochondrion-to-nucleus stress signaling network that leads to alterations in gene expression that then affect a wide variety of cellular processes (Amuthan et al, 2001; Biswas et al, 1999). These processes include altered expression of the sarcoplasmic reticular ryanodine receptor-1, another major ER calcium release channel, and potentiate the Ca2+ release in response to caffeine (Amuthan et al, 2001; Biswas et al, 1999) (i.e., an AD like effect). Altered gene expression can alter the ratios of the Bcl-2 family. Expression of anti-apoptotic Bcl-2 family members decreases expression of IP3R-related ER Ca2+ release channels, leads to a reduction in the Ca2+ release in response to IP3. Bcl-XL-over expressing cells exhibit lower IP3-mediated Ca2+ release capacity (Li et al, 2002). These changes appear to be pathophysiologically important. A reduction in calcium in the ER decreases the probability of Ca2+ dependent apoptosis (Giorgi et al, 2009).
The results show that impairing a mitochondrial TCA cycle enzyme can lead to changes that mimic the abnormalities in cells from AD patients and mice bearing PS-1 mutations. Thus, correcting the mitochondrial deficit may also overcome abnormalities in calcium homeostasis.
Acknowledgments
We thank Olin Anderson (Department of Chemistry and Biochemistry, Eastern Washington University, Cheney, WA 99004) for his help in preparing the CESP. Grants PP-AG14930 and the Burke Medical Research Institute.
Footnotes
Conflicts. None of the authors have any financial conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amuthan G, Biswas G, Zhang S-Y, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. The EMBO journal. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175:1810–1816. doi: 10.2353/ajpath.2009.090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assefa Z, Bultynck G, Szlufcik K, Nadif Kasri N, Vermassen E, Goris J, Missiaen L, Callewaert G, Parys JB, De Smedt H. Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J Biol Chem. 2004;279:43227–43236. doi: 10.1074/jbc.M403872200. [DOI] [PubMed] [Google Scholar]
- Barrow PA, Empson RM, Gladwell SJ, Anderson CM, Killick R, Yu X, Jefferys JGR, Duff K. Functional Phenotype in Transgenic Mice Expressing Mutant Human Presenilin-1. Neurobiology of Disease. 2000;7:119–126. doi: 10.1006/nbdi.1999.0276. [DOI] [PubMed] [Google Scholar]
- Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999a;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 2010;459:441–449. doi: 10.1007/s00424-009-0736-1. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. The EMBO journal. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Snyder SH. Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle. 2004;3:252–254. [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nature protocols. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Annals of neurology. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Bunik VI, Denton TT, Xu H, Thompson CM, Cooper AJ, Gibson GE. Phosphonate analogues of alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry. 2005;44:10552–10561. doi: 10.1021/bi0503100. [DOI] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. The FEBS journal. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta. 2011a;1813:1003–1013. doi: 10.1016/j.bbamcr.2010.11.023. [DOI] [PubMed] [Google Scholar]
- Decuypere JP, Monaco G, Missiaen L, De Smedt H, Parys JB, Bultynck G. IP(3) Receptors, Mitochondria, and Ca Signaling: Implications for Aging. J Aging Res. 2011b;2011:920178. doi: 10.4061/2011/920178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Ho DJ, Calingasan NY, Xu H, Gibson G, Beal MF. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic Biol Med. 2009;47:1019–1027. doi: 10.1016/j.freeradbiomed.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer’s disease. Annals of Neurology. 2003;54:393–398. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, Mohs RC, Sheu RK, Blass JP. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Annals of neurology. 2000;48:297–303. [PubMed] [Google Scholar]
- Gibson GE, Nielsen P, Sherman KA, Blass JP. Diminished mitogen-induced calcium uptake by lymphocytes from Alzheimer patients. Biol Psychiatry. 1987;22:1079–1086. doi: 10.1016/0006-3223(87)90050-3. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiol Aging. 1987;8:329– 343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Archives of neurology. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Zhang H, Xu H, Park LC, Jeitner TM. Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme. Biochim Biophys Acta. 2002;1586:177–189. doi: 10.1016/s0925-4439(01)00091-6. [DOI] [PubMed] [Google Scholar]
- Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol. 2009;41:1817–1827. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Hirota J, Furuichi T, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J Biol Chem. 1999;274:34433–34437. doi: 10.1074/jbc.274.48.34433. [DOI] [PubMed] [Google Scholar]
- Huang HM, Chen HL, Gibson GE. Thiamine and oxidants interact to modify cellular calcium stores. Neurochemical research. 2010;35:2107–2116. doi: 10.1007/s11064-010-0242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HM, Chen HL, Xu H, Gibson GE. Modification of endoplasmic reticulum Ca2+ stores by select oxidants produces changes reminiscent of those in cells from patients with Alzheimer disease. Free Radic Biol Med. 2005;39:979–989. doi: 10.1016/j.freeradbiomed.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Huang HM, Martins R, Gandy SAM, Etcheberrigaray R, Ito E, Alkon DL, Blass J, Gibson G. Use of Cultured Fibroblasts in Elucidating the Pathophysiology and Diagnosis of Alzheimer’s Diseasea. Annals of the New York Academy of Sciences. 1994;747:225–244. doi: 10.1111/j.1749-6632.1994.tb44412.x. [DOI] [PubMed] [Google Scholar]
- Huang HM, Ou HC, Xu H, Chen HL, Fowler C, Gibson GE. Inhibition of alpha-ketoglutarate dehydrogenase complex promotes cytochrome c release from mitochondria, caspase-3 activation, and necrotic cell death. J Neurosci Res. 2003b;74:309–317. doi: 10.1002/jnr.10756. [DOI] [PubMed] [Google Scholar]
- Huang HM, Shen CC, Ou HC, Yu JY, Chen HL, Kuo JS, Hsieh SJ. Neuroprotective MK801 is associated with nitric oxide synthase during hypoxia/reoxygenation in rat cortical cell cultures. J Cell Biochem. 2002;84:367–376. doi: 10.1002/jcb.10022. [DOI] [PubMed] [Google Scholar]
- Huang HM, Toral-Barza L, Gibson G. Cytosolic free calcium and ATP in synaptosomes after ischemia. Life sciences. 1991;48:1439–1445. doi: 10.1016/0024-3205(91)90180-j. [DOI] [PubMed] [Google Scholar]
- Huang HM, Zhang H, Ou HC, Chen HL, Gibson GE. alpha-keto-beta-methyl-n-valeric acid diminishes reactive oxygen species and alters endoplasmic reticulum Ca (2+) stores. Free Radic Biol Med. 2004;37:1779–1789. doi: 10.1016/j.freeradbiomed.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Xu H, Shi Q, Chen LH, Pedrini S, Pechman D, Baker H, Beal MF, Gandy SE, Gibson GE. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol Aging. 2009;30:1587–1600. doi: 10.1016/j.neurobiolaging.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlis VB, Roche TE. Regulation of bovine kidney alpha-ketoglutarate dehydrogenase complex by calcium ion and adenine nucleotides. Effects on S0.5 for alpha-ketoglutarate. Biochemistry. 1981;20:2512–2518. doi: 10.1021/bi00512a023. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. The Journal of Cell Biology. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB. Bcl-XL affects Ca2+ homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proceedings of the National Academy of Sciences. 2002;99:9830–9835. doi: 10.1073/pnas.152571899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging. 2008;29:676–692. doi: 10.1016/j.neurobiolaging.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Kunz EA, Brewer GJ. Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res. 2008;86:2339–2352. doi: 10.1002/jnr.21679. [DOI] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Sheu KF, Gibson GE. Quantitative alpha-ketoglutarate dehydrogenase activity staining in brain sections and in cultured cells. Anal Biochem. 2000;277:86–93. doi: 10.1006/abio.1999.4359. [DOI] [PubMed] [Google Scholar]
- Peterson C, Gibson GE, Blass JP. Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N Engl J Med. 1985;312:1063–1065. doi: 10.1056/NEJM198504183121618. [DOI] [PubMed] [Google Scholar]
- Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- Qiu L, Wang H, Xia X, Zhou H, Xu Z. A construct with fluorescent indicators for conditional expression of miRNA. BMC Biotechnol. 2008;8:77. doi: 10.1186/1472-6750-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Peterhoff CM, Yang D-S, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, et al. Marked Calpastatin (CAST) Depletion in Alzheimer’s Disease Accelerates Cytoskeleton Disruption and Neurodegeneration: Neuroprotection by CAST Overexpression. The Journal of Neuroscience. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Santos SS, Gibson GE, Cooper AJL, Denton TT, Thompson CM, Bunik VI, Alves PM, Sonnewald U. Inhibitors of the α-ketoglutarate dehydrogenase complex alter [1-13C]glucose and [U-13C]glutamate metabolism in cerebellar granule neurons. Journal of Neuroscience Research. 2006;83:450–458. doi: 10.1002/jnr.20749. [DOI] [PubMed] [Google Scholar]
- Schneider I, Reversé D, Dewachter I, Ris L, Caluwaerts N, Kuipéri C, Gilis M, Geerts H, Kretzschmar H, Godaux E, et al. Mutant Presenilins Disturb Neuronal Calcium Homeostasis in the Brain of Transgenic Mice, Decreasing the Threshold for Excitotoxicity and Facilitating Long-term Potentiation. Journal of Biological Chemistry. 2001;276:11539–11544. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- Shi Q, Chen HL, Xu H, Gibson GE. Reduction in the E2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity. J Biol Chem. 2005;280:10888–10896. doi: 10.1074/jbc.M409064200. [DOI] [PubMed] [Google Scholar]
- Shi Q, Gibson GE. Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. J Neurochem. 2011;118:440–448. doi: 10.1111/j.1471-4159.2011.07333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Risa Ø, Sonnewald U, Gibson GE. Mild reduction in the activity of the α-ketoglutarate dehydrogenase complex elevates GABA shunt and glycolysis. Journal of Neurochemistry. 2009;109:214–221. doi: 10.1111/j.1471-4159.2009.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Xu H, Kleinman WA, Gibson GE. Novel functions of the alpha-ketoglutarate dehydrogenase complex may mediate diverse oxidant-induced changes in mitochondrial enzymes associated with Alzheimer’s disease. Biochim Biophys Acta. 2008;1782:229–238. doi: 10.1016/j.bbadis.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Xu H, Yu H, Zhang N, Ye Y, Estevez AG, Deng H, Gibson GE. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J Biol Chem. 2011;286:17640–17648. doi: 10.1074/jbc.M110.203018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Robinson A. A metabolic model of the mitochondrion and its use in modelling diseases of the tricarboxylic acid cycle. BMC Systems Biology. 2011;5(1):102. doi: 10.1186/1752-0509-5-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE. The Pathogenesis of Alzheimers Disease—Is It a Lifelong “Calciumopathy”? The Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supnet C, Bezprozvanny I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J Alzheimers Dis. 2010;20(Suppl 2):S487–498. doi: 10.3233/JAD-2010-100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Rizzuto R. Mitochondrial-endoplasmic reticulum interactions. In: Gibson GE, Dienel GA, editors. Handbook of Neurochemistry and Molecular Neurobiology. Brain Energetics. Integration of Molecular and Cellular Processes. Springer; 2007. pp. 617–640. [Google Scholar]
- Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta. 2006;1763:442–449. doi: 10.1016/j.bbamcr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philosophical transactions of the Royal Society of London. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Hajnóczky G. Mitochondria and Endoplasmic Reticulum: The Lethal Interorganelle Cross-Talk. Journal of Bioenergetics and Biomembranes. 2005;37:191–206. doi: 10.1007/s10863-005-6600-x. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yang L, Shi Q, Ho DJ, Starkov AA, Wille EJ, Xu H, Chen HL, Zhang S, Stack CM, Calingasan NY, et al. Mice deficient in dihydrolipoyl succinyl transferase show increased vulnerability to mitochondrial toxins. Neurobiology of disease. 2009;36:320–330. doi: 10.1016/j.nbd.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Huang HM, Carson RC, Mahmood J, Thomas HM, Gibson GE. Assessment of membrane potentials of mitochondrial populations in living cells. Anal Biochem. 2001;298:170–180. doi: 10.1006/abio.2001.5348. [DOI] [PubMed] [Google Scholar]