

Abstract
Phosphonium lipocations were synthesized and evaluated for inhibition of the development of Plasmodium falciparum and Trypanosoma cruzi, etiological agents of malaria and Chagas disease, respectively. Optimal phthalimides and 1,4-naphthoquinone-based lipocations were active in vitro at mid-high nM concentrations against P. falciparum and low μM concentrations against T. cruzi.
Keywords: Malaria, Chagas, Antiparasitic drugs, Plasmodium, Trypanosoma
Malaria and American trypanosomiasis (Chagas disease) are vector-borne infections that cause hundreds of millions of illnesses and nearly a million deaths in the tropics each year.1 With malaria, clinical illness results when erythrocytes become infected with Plasmodium parasites. Effective antimalarial therapies must eliminate erythrocytic parasites while inflicting minimal host toxicity. Older antimalarial therapies such as chloroquine are seriously limited by drug resistance2. Newer agents include the artemisinins and the ubiquinone (CoQ) antagonist atovaquone (ATV). Artemisinins are rapidly active components of new combination regimens now widely used to treat malaria3, although resistance to artemisinins may be emerging4. ATV is co-formulated with proguanil (Malarone) for the treatment and prevention of malaria because of synergistic interaction of the two components and high recrudescence rates after monotherapy.5 ATV acts on the mitochondrion and demonstrates low toxicity due in part to evolutionary dissimilarities between plasmodial and mammalian mitochondria. However, for pharmaceuticals that require internalization by mitochondria, subcellular and therapeutic efficacy can be limited by the physiochemical properties of a compound.
Phosphonium cations are used extensively as molecular probes for studying mitochondrial function6 and have demonstrated utility as mediators of antioxidant7, antimicrobial8, and antineoplastic9 transport into mitochondria. Mitoquinone7a (MitoQ, Fig. 1), a synthetic analog of ubiquinone, is the most clinically advanced phosphonium cation, and it has progressed to phase II trials in the U.S. for the management of Parkinson’s disease, hepatitis C, and fatty liver disease. As a positively charged analog of the dietary antioxidant idebenone (Catena/Sovrima, Fig. 1), the enhanced therapeutic efficacy of mitoquinone is conferred by attachment of a triphenylphosphonium group to the decyl side chain. This modification guides the lipocation into energized mitochondria by electrostatic attraction and leads to increased drug concentrations in the mitochondrial matrix.7d
Figure 1.

Chemical structures of ubiquinone analogs atovaquone, idebenone, and mitoquinone.
A structural feature common to mitoquinone and other CoQ mimics (e.g. ATV) is a lipophilic side chain, which increases permeability in the inner phospholipid bilayers of mitochondria, where electron transport complexes are embedded. We hypothesized that if a phosphonium moiety were bound to the lipid substituent in antagonists of the Plasmodium and Trypanosoma electron transport chains, higher drug concentrations would be achieved inside the mitochondrion and lead to increased antiparasitic effects. In this initial report, we examine the effects on anti-parasitic activity of phosphonium group attachment to structural analogs of CoQ containing phthalimide and 1,4-naphthoquinone platforms.
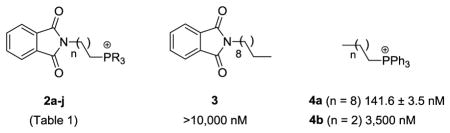
The initial molecules chosen for evaluation were derived from phthalimide, which has structural similarities to CoQ, and served as a simple platform to conduct an SAR study on the lipocation chain component. N-Alkylphthalimides 1a-c (Scheme 1) were prepared from dibromoalkanes of various chain lengths and then coupled with a tertiary phosphine under microwave10 or conventional heating conditions (methods A–C) to afford the phosphonium lipocations.
Scheme 1.

Synthesis of phthalimide-based phosphonium lipocations 2 (Table 1).
Minimum inhibitory concentrations (IC50) were determined for the phthalimide-based lipocations 2a-j against the chloroquine-resistant P. falciparum strain W2, using methods as previously described.11 The IC50s ranged from 134.0 nM to >3.5 μM (Table 1), compared to 66.9 nM for chloroquine (CQ). Analysis of structure-activity relationships revealed that antiplasmodial effects were conferred by the phosphonium moiety, and the presence or absence of the phthalimide group had minimal influence on activity. This conclusion was established by inclusion of phthalimide 3 and triphenylphosphonium cations 4 in the study. Conversely, substituents on the phosphonium moiety and the lipid chain length (n) had a moderate impact on activity. The P-substituent could be either alkyl (e.g. R = cyclohexyl, 2i) or aromatic (e.g. R = Ph, 2g), with only a moderate effect on the IC50 values. The trimethyl derivative 2a was an exception having much weaker activity compared to analogs of equal chain length (i.e. 2d-j). A similar detrimental effect was observed for compounds possessing shorter chain lengths, with the 4- and 6-carbon chain analogs, 2b and c respectively, demonstrating 4–15 fold reduced activity compared to the 10-carbon chain lipocation 2g.
Table 1.
Comparisons of IC50s for P. falciparum growth.
| |||||
|---|---|---|---|---|---|
| compd | n | R | IC50 (nM) | Log Da | MWb |
| 2a | 9 | Me | >10,000 | −0.08 | 362.5 |
| 2b | 3 | Ph | 3,500 | 0.01 | 464.5 |
| 2c | 5 | Ph | 1021 ± 93.2 | 1.14 | 492.6 |
| 2d | 9 | 4-PhF | 345.6 ± 15.3 | 2.13 | 602.6 |
| 2e | 9 | Bn | 288.5 ± 35.2 | 1.37 | 590.8 |
| 2f | 9 | n-Bu | 194.2 ± 3.8 | 3.68 | 488.7 |
| 2g | 9 | Ph | 172.7 ± 4.0 | 1.67 | 548.7 |
| 2h | 9 | 2-PhMe | 143.4 ± 7.0 | −0.75 | 590.8 |
| 2i | 9 | C6H11 | 140.6 ± 0.7 | 4.50 | 566.8 |
| 2j | 9 | 4-PhOMe | 134.0 ± 1.7 | 3.19 | 638.7 |
|
| |||||
| CQ | 66.93 ± 2.3 | 319.9 | |||
measured by 1-octanol–water partition at pH 7.4
molecular weights of lipocations 2 minus the counterion
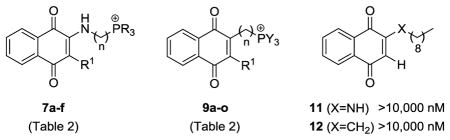
Although investigation of the phthalimide-based lipocations was beneficial in confirming activity and examining SAR, it was apparent that the phthalimide group was not serving as an antagonist of CoQ, as supported by similarities in the IC50 values of phosphonium salts 2f-j and 4a. Attention was turned to lipocations containing the 1,4-naphthoquinone platform which is found in ATV and many other known inhibitors of the Plasmodium cytochrome bc1 complex12. The analogs were synthesized from quinones 5 using 3 different approaches to install the lipocation chain (Scheme 2). The first method was by nucleophilic addition13 of straight-chain amino alcohols to 1,4-naphthoquinone (R = H), followed by mesylation of the corresponding alcohol, and generation of the phosphonium salts under microwave irradiation. The second approach introduced the cation by chloroalkylation14 of naphthoquinone 5 (R = Me) with formaldehyde and subsequent conversion to triphenylphosphonium salt 9f. The final method was by alkylation of naphthoquinones 5 (R = H, Me) via radical decarboxylation.15 The resulting bromides were then converted to the lipocations by methods A–C (Scheme 1), which often resulted in substantial amounts of reduced products (i.e. 1,4-naphthalenediols) being formed. When this occurred, re-oxidation to the quinone was accomplished with nitric oxide generated in situ from NaNO216 and subsequent counterion exchange to the mesylate salt form to ensure chemical conformity of the product.
Scheme 2.

Synthesis of 1,4-naphthoquinone-based phosphonium lipocations 7 and 9 (Table 2).
Susceptibility testing of the naphthoquinone-based lipocations against W2-strain P. falciparum revealed an SAR profile similar to that of the phthalimide series (Table 2). Antiplasmodial activity was conferred by the installation of a phosphonium-containing hydrocarbon chain, and a variety of P-substituents could be utilized without significant deviations in the IC50 values (i.e. 9i-m). The activities for many of the 10-carbon chain analogs were once again comparable to the lipocation control 4a (IC50 141.6 nM), suggesting that the inhibitory effects were due not to a specific mechanism (e.g. cytochrome bc1 inhibition), but rather to a non-specific action such as disruption of plasma membrane integrity. The two apparent exceptions were lipocations 9n (IC50 48.3 nM) and 9o17 (IC50 18.7 nM), which displayed 3–7 fold greater activity than control 4a. The 5- and 4-carbon chain analogs were also more active than their 10-carbon chain counterpart 9k (IC50 143.4 nM), a reversal in the SAR profile observed with the phthalimide series. In addition, a comparison of analog 9o to its 4-carbon chain control 4b and phthalimide 2b (IC50s 3.5 μM) established that the naphthoquinone platform had a role in achieving sub-50 nM activities.
Table 2.
Comparisons of IC50s for P. falciparum growth.
| ||||||
|---|---|---|---|---|---|---|
| compd | n | R | R1 | IC50 (nM) | Log Da | MWb |
| 7a | 1 | Ph | H | 519.9 ± 61.2 | −1.08 | 462.5 |
| 7b | 10 | 4-PhF | H | 292.0 ± 53.2 | 1.67 | 628.7 |
| 7c | 10 | Bn | H | 214.6 ± 0.5 | 2.80 | 616.8 |
| 7d | 10 | 4-PhOMe | H | 134.2 ± 10.5 | 4.37 | 664.8 |
| 7e | 10 | Ph | H | 113.9 ± 7.6 | 3.60 | 574.7 |
| 7f | 10 | C6H11 | H | 94.4 ± 35.2 | 3.41 | 592.9 |
|
| ||||||
| 9a | 10 | Bn | H | 1303.5 ± 13.4 | 5.32 | 601.8 |
| 9b | 10 | 4-PhF | H | 846.3 ± 0.7 | 1.66 | 613.7 |
| 9c | 10 | Ph | H | 259.2 ± 18.7 | 4.08 | 559.7 |
| 9d | 5 | Ph | H | 156.6 ± 6.5 | 1.00 | 489.6 |
|
| ||||||
| 9e | 5 | Me | Me | 955.6 ± 2.5 | 0.21 | 317.4 |
| 9f | 1 | Ph | Me | 543.4 ± 79.2 | 1.89 | 447.5 |
| 9g | 10 | Me | Me | 404.2 ± 16.7 | 1.95 | 373.5 |
| 9h | 10 | Bn | Me | 212.0 ± 27.2 | 6.64 | 615.8 |
| 9i | 10 | n-Bu | Me | 155.8 ± 9.0 | 5.10 | 513.8 |
| 9j | 10 | 2-PhMe | Me | 143.4 ± 7.0 | 3.58 | 615.8 |
| 9k | 10 | Ph | Me | 143.4 ± 6.3 | 3.97 | 573.7 |
| 9l | 10 | C6H11 | Me | 134.3 ± 1.8 | 3.64 | 591.9 |
| 9m | 10 | 4-PhOMe | Me | 130.9 ± 6.6 | 3.52 | 663.8 |
| 9n | 5 | Ph | Me | 48.3 ± 1.5 | 2.15 | 503.6 |
| 9o | 4 | Ph | Me | 18.7 ± 0.3 | 1.91 | 489.6 |
|
| ||||||
| ARTc | 7.31 ± 0.14 | 262.3 | ||||
| ATV | 0.28 ± 0.19 | 366.3 | ||||
measured by 1-octanol–water partition at pH 7.4
molecular weights of lipocations 7 and 9 minus the counterion
artemisinin
The naphthoquinone-derived lipocations were additionally evaluated as growth inhibitors against Trypanosoma cruzi, the etiological agent of Chagas disease. Like P. falciparum, T. cruzi has a complex life cycle which includes infective, non-replicating bloodstream trypomastigote forms and intracellular amastigotes that infect cardiac and other cells, leading to disease.18 The compounds were assessed for efficacy against Vero cell-infected T. cruzi amastigotes19. The IC50s for inhibition of parasite development ranged from 1.6 to 5.4 μM and variable degrees of Vero cell toxicity was observed (Table 3). For comparison, benznidazole was used as a positive control (IC50 2.1 μM) and was non-toxic at the concentrations indicated.
Table 3.
Comparisons of IC50s for T. cruzi growth and Vero cell toxicity.
| compd | n | R | T. cruzi IC50 (μM) | Vero cell toxicity (μM)a
|
||
|---|---|---|---|---|---|---|
| 25 | 12.5 | 6.25 | ||||
| 11 | 17.3 ± 7.1 | − | − | − | ||
| 12 | 9.3 ± 1.3 | − | − | − | ||
| 9n | 5 | Ph | 5.4 ± 0.1 | + | − | − |
| 9i | 10 | n-Bu | 4.0 ± 3.6 | + | + | − |
| 9h | 10 | Bn | 3.7 ± 1.8 | + | + | − |
| 9a | 10 | Bn | 3.0 ± 1.3 | − | − | − |
| 9o | 4 | Ph | 2.7 ± 1.0 | − | − | − |
| 9l | 10 | C6H11 | 2.4 ± 0.7 | + | + | − |
| 7e | 10 | Ph | 1.6 ± 0.5 | + | + | + |
| 9k | 10 | Ph | 1.6 ± 0.5 | + | + | + |
| BNZb | 2.1 ± 0.4 | − | − | − | ||
+ cytotoxicity observed; − cytotoxicity not evident
benznidazole
As with P. falciparum, the lipocation analogs were more effective antitrypanosomal agents than their uncharged naphthoquinone counterparts 11 and 12. Chain length and P-substituent type appeared to have little effect on the activity but, were influential on Vero cell cytotoxicity and may have resulted in the increased efficacy observed for lipocations 7e and 9k (IC50s 1.6 μM). The most potent compound not displaying toxicity at 25 μM was the 4-carbon chain analog 9o (IC50 2.7 μM) which also possessed the highest antiplasmodial activity at 18.7 nm. Additional testing of lipocation 9o further revealed therapeutic index values {TI = [IC50(Vero)]/[IC50(parasite)]} of 19.5 and 2,818 for T. cruzi and P. falciparum, respectively.
With these findings, a question to be addressed is whether pharmacological activity is conferred by charge-mediated accumulation of the lipocations inside the mitochondrion. As with mitochondrial-targeted modulators of similar design,7,20 subcellular internalization of the antagonists is thought to be governed by their physiochemical properties. It is hypothesized that the lipid character of the cations combined with electrostatic forces facilitates non-carrier-mediated transport through the cytoplasmic membranes of the host cell and parasite. Their lipophilic properties results from distribution of the cation charge across the molecules however, guiding movement of the inhibitors toward regions of negative charge within parasitized cells (Fig. 2).
Figure 2.

The antiparasitic effects of phosphonium lipocation 9o is believed to be due to electrostatic forces that increase subcellular (mitochondrial) concentrations of the inhibitor.
In the case of plasmodia, the inward cytoplasmic membrane potential (Δψp) of erythrocytes and P. falciparum are reportedly ≤−35 mV21 and −95 ± 2 mV22, respectively, which offers a rational basis for a Δψ-directed route into the parasite. Further, with the absence of mitochondria in uninfected erythrocytes, the Plasmodium mitochondrion would be the site of highest negative charge in an infected erythrocyte, and thereby be the primary location where the lipocations would be expected to accumulate. Conversely, mammalian cells usually possess hundreds of mitochondria also capable of taking up the lipocations, which may have led to the reduced efficacy observed against Vero cell-infected T. cruzi. Although host mitochondria will cause a decrease of lipocation concentrations in the parasite, T. cruzi amastigotes have only a single mitochondrion2. Lower drug concentrations would therefore likely be required to collapse the Δψm and confer lethal effects on the trypanosome compared to host cells.
In summary, phosphonium lipocations were found to be efficacious inhibitors of the development of cultured P. falciparum and T. cruzi. Preliminary SAR profiles were established for lipocations derived from phthalimide and 1,4-naphthoquinone platforms. In both series, antiparasitic activity was greater for compounds containing a phosphonium group. The P-substituent could be either aromatic or alkyl without significant deviations in the IC50s, while a lipid chain of 4-carbons appeared to be the optimal length for 1,4-naphthoquinone-based inhibitors. Current research efforts are focused on evaluating the antiparasitic activity of lipocations containing various platform types of known electron transport antagonists. The goal will be to identify compounds that are equipotent to other mitochondrion-acting agents (e.g. ATV) and demonstrate minimal cytotoxicity. Ideally, the lipocations will also have good distribution in tissues most susceptible to damage by the infection (erythrocytes for malaria parasites and cardiac and intestinal myocytes for T. cruzi) and cost-effective, orally active agents to merit their development as antiparasitic drugs.
Acknowledgments
Financial support was generously provided by endowed funds from the College of Pharmacy at The University of Georgia to T.E.L, the National Institutes of Health to R.D. (AI082542) and P.J.R. and the Doris Duke Charitable Foundation, with which P.J.R. is a Distinguished Clinical Scientist. Special thanks is also given to Dr. Jim Franklin for many insightful discussions and Dr. Dennis Phillips for mass spectroscopy analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Greenwood BM, Bojand K, Whitty CJM, Targett GA. Lancet. 2005;365:1487. doi: 10.1016/S0140-6736(05)66420-3. [DOI] [PubMed] [Google Scholar]; (b) Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. Nature. 2005;434:214. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rassi A, Jr, Rassi A, Marin-Neto JA. Lancet. 2010;375:1388. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]; (d) Urbina J, Docampo R. Trends Parasitol. 2003;19:495. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Petersen I, Eastman R, Lanzer M. FEBS Lett. 2011;585:1551. doi: 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 3.White NJ. Science. 2008;320:330. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 4.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. N Engl J Med. 2009;361:455. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canfield CJ, Pudney M, Gutteridge WE. Exp Parasitol. 1995;80:373. doi: 10.1006/expr.1995.1049. [DOI] [PubMed] [Google Scholar]
- 6.(a) Murphy MP. Biophys Acta, Bioenerg. 2008;1777:1028. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]; (b) Ross MF, Da Ros T, Blaikie FH, Prime TA, Porteous CM, Severina II, Skulachev VP, Kjaergaard HG, Smith RAJ, Murphy MP. Biochem J. 2006;400:199. doi: 10.1042/BJ20060919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ross MF, Kelso GF, Blaikie FH, James AM, Cochemé HM, Filipovska A, Da Ros T, Hurd TR, Smith RAJ, Murphy MP. Biochemistry (Moscow) 2005;70:222. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]; (d) Ono A, Miyauchi S, Demura M, Asakura T, Kamo N. Biochemistry. 1994;33:4312. doi: 10.1021/bi00180a027. [DOI] [PubMed] [Google Scholar]
- 7.(a) Smith RAJ, Murphy MP. Ann NY Acad Sci. 2010;1201:96. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]; (b) Murphy MP. Biochim Biophys Acta. 2008;1777:1028. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]; (c) Murphy MP, Smith RAJ. Annu Rev Pharmcol Toxicol. 2007;47:629. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]; (d) James MA, Cochemé HM, Smith RJ, Murphy MP. J Biol Chem. 2005;280:21295. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]; (e) Asin-Cayuela J, Manas AB, James AM, Smith RAJ, Murphy MP. FEBS Lett. 2004;571:9. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]; (f) Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RAJ, Murphy MP. J Biol Chem. 2001;276:4588. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 8.(a) Luque-Ortega JR, Reuther P, Rivas L, Dardonville C. J Med Chem. 2010;53:1788. doi: 10.1021/jm901677h. [DOI] [PubMed] [Google Scholar]; (b) Kanazawa A, Ikeda T, Endo T. Antimicrob Agents Chemother. 1994;38:945. doi: 10.1128/aac.38.5.945. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kanazawa A, Ikeda T, Endo T. J Appl Bacteriol. 1995;78:55. doi: 10.1111/j.1365-2672.1995.tb01673.x. [DOI] [PubMed] [Google Scholar]; (d) McAllister PR, Dotson MJ, Grim SO, Hillman GR. J Med Chem. 1980;23:862. doi: 10.1021/jm00182a010. [DOI] [PubMed] [Google Scholar]; (e) Levi-Schaffer F, Tarrab-Hazdai R, Meshulam H, Arnon R. Int J Immunopharmacol. 1984;6:619. doi: 10.1016/0192-0561(84)90073-0. [DOI] [PubMed] [Google Scholar]; (f) Kinnamon KE, Steck EA, Rane DS. J Med Chem. 1979;22:452. doi: 10.1021/jm00190a019. [DOI] [PubMed] [Google Scholar]; (g) Hanson WL, Chapman WL, Jr, Kinnamon KE. Int J Parasitol. 1977;7:443. doi: 10.1016/0020-7519(77)90004-2. [DOI] [PubMed] [Google Scholar]; (h) Kinnamon KE, Steck EA, Hanson WL, Chapman WL., Jr J Med Chem. 1977;20:741. doi: 10.1021/jm00216a001. [DOI] [PubMed] [Google Scholar]
- 9.(a) Millard M, Pathania D, Shabaik Y, Taheri L, Deng J, Neamati N. PLoS One. 2010;5:e13131. doi: 10.1371/journal.pone.0013131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Delikatny EJ, Cooper WA, Brammah S, Sathasivam N, Rideout DC. Cancer Res. 2002;62:1394. [PubMed] [Google Scholar]; (c) Cooper WA, Bartier WA, Rideout DC, Delikatny EJ. Magn Reson Med. 2001;45:1001. doi: 10.1002/mrm.1133. [DOI] [PubMed] [Google Scholar]; (d) Manetta A, Gamboa G, Nasseri A, Podnos YD, Emma D, Dorion G, Rawlings L, Carpenter PM, Bustamante A, Patel J, Rideout D. Gynecol Oncol. 1996;60:203. doi: 10.1006/gyno.1996.0026. [DOI] [PubMed] [Google Scholar]; (e) Rideout D, Bustamante A, Patel J. Int J Cancer. 1994;57:247. doi: 10.1002/ijc.2910570220. [DOI] [PubMed] [Google Scholar]; (f) Dubois RJ, Lin CC, Beisler JA. J Med Chem. 1978;21:303. doi: 10.1021/jm00201a016. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kiddle JJ. Tetrahedron Lett. 2000;41:1339. [Google Scholar]; (b) Cvengros J, Toma S, Marque S, Loupy A. Can J Chem. 2004;82:1365. [Google Scholar]
- 11.Coteron JM, Catterick D, Castro J, Chaparro MJ, Diaz B, Fernandez E, Ferrer S, Gamo FJ, Gordo M, Gut J, de las Heras L, Legac J, Marco M, Miguel J, Munoz V, Porras E, de la Rosa JC, Ruiz JR, Sandoval E, Ventosa P, Rosenthal PJ, Fiandor JM. J Med Chem. 2010;53:6129–52. doi: 10.1021/jm100556b. [DOI] [PubMed] [Google Scholar]
- 12.(a) Rodrigues T, Lopes F, Moreira R. Curr Med Chem. 2010;17:929. doi: 10.2174/092986710790820660. [DOI] [PubMed] [Google Scholar]; (b) Porter TH, Folkers K. Angew Chem Int Edit. 1974;13:559. doi: 10.1002/anie.197405591. [DOI] [PubMed] [Google Scholar]
- 13.Bittner S, Gorohovsky S, Paz-Tal O, Becker JY. Amino Acids. 2002;22:71. doi: 10.1007/s726-002-8202-3. [DOI] [PubMed] [Google Scholar]
- 14.Lipshutz BH, Kim SK, Mollard P, Stevens KL. Tetrahedron. 1998;54:1241. [Google Scholar]
- 15.Commandeur C, Chalumeau C, Dessolin J, Laguerre M. Eur J Org Chem. 2007:3045. [Google Scholar]
- 16.Bozell JJ, Hoberg JO, Dimmel DR. Tetrahedron Lett. 1998;39:2261. [Google Scholar]
- 17.(4-(3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)butyl)triphenyl-phosphonium methanesulfonate (9o): orange oil; TLC (SiO2) Rf 0.27 (9:1 DCM:MeOH); 1H NMR (500 MHz, CDCl3) δ 8.04-8.02 (m, 1H), 7.97-7.96 (m, 1H), 7.81-7.68 (m, 17H), 3.69-3.63 (m, 2H), 2.68-2.66 (m, 5H), 2.17 (s, 3H), 1.86-1.80 (m, 2H), 1.76-1.69 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 185.1, 184.9, 146.0, 144.4, 135.09, 135.06, 133.64, 133.56, 133.5, 133.4, 132.1, 131.9, 130.6, 130.5, 126.3, 126.2, 118.6, 117.9; 31P NMR (202 MHz, CDCl3) δ 24.9; ESI-HRMS calcd for C33H30O2P [M+] 489.1977, found 489.1982.
- 18.Docampo R. Curr Pharm Des. 2001;7:1157. doi: 10.2174/1381612013397546. [DOI] [PubMed] [Google Scholar]
- 19.Demoro B, Caruso F, Rossi M, Benítez D, Gonzalez M, Cerecetto H, Parajón-Costa B, Castiglioni J, Galizzi M, Docampo R, Otero L, Gambino D. J Inorg Biochem. 2010;104:1252. doi: 10.1016/j.jinorgbio.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frantz MC, Wipf P. Environ Mol Mutagen. 2010;51:462. doi: 10.1002/em.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikkelsen RB, Tanabe K, Wallach DHF. J Cell Biol. 1982;93:685. doi: 10.1083/jcb.93.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen RJ, Kirk K. J Biol Chem. 2004;279:11264. doi: 10.1074/jbc.M311110200. [DOI] [PubMed] [Google Scholar]
- 23.Fidalgo LM, Gille L. Pharm Res. 2011;28:2758. doi: 10.1007/s11095-011-0586-3. [DOI] [PubMed] [Google Scholar]