

Abstract
Herein we describe a two-step protocol to prepare m-tert-alkylbenzenes. The appropriate 3° benzylic alcohols are activated with SOCl2 or concentrated HCl, and then treated with trimethylaluminum, affording the desired products in 68–97% yields (22 examples). This reaction sequence is successful in the presence of a variety of functional groups, including acid-sensitive and Lewis-basic groups. In addition to t-Bu groups, 1,1-dimethylpropyl and 1-ethyl-1-methylpropyl groups can also be installed using this method.
Introduction
Due to intrinsic substituent directing effects, compounds bearing tert-alkyl groups on benzene rings meta- to ortho, para-directing substituents are challenging to prepare. In the course of our efforts to develop malaria mosquito-selective acetylcholinesterase inhibitors,1 we required such compounds, which we term “Friedel-Crafts-restricted” tert-alkyl benzenes. Traditional strategies to prepare these compounds use “temporary” ortho- or para-hydroxy or amino groups to direct Friedel-Crafts2 or other electrophilic aromatic substitution reactions;3,4 subsequent removal of the temporary directing group then unveils the desired meta-substitution pattern. We sought a more direct route that would benefit from the large number of commercially available meta-substituted benzoic acids and acetophenones. One approach that appeared especially promising was Reetz’s conversion of aryl ketone 1a to the corresponding tert-alkyl benzene 2a by treatment with Me2TiCl2 (Scheme 1).5
Scheme 1.

Published Reetz strategies5,6 to convert aromatic ketones to tert-alkyl–substituted aromatics
Two years earlier Reetz disclosed a potentially more general strategy:6 addition of EtLi to 1b, isolation of the lithium alkoxide 3b, and final treatment with ZnMe2/TiCl4 afforded 4b, which incorporated two different alkyl groups to give a 1,1-dimethylpropyl substituent. These remarkable transformations are worthy of wider application. We envisioned a related strategy starting from 3° benzylic alcohols: in situ activation of the alcohol followed by treatment with the appropriate methyl organometallic (M-Me) would afford aromatics featuring various tertalkyl groups (Scheme 2).
Scheme 2.

Planned transformation of 3° benzylic alcohols (6, 7, 8) to tert-alkyl benzenes (2, 4, 5), and precedent from Makriyannis (9)7.
Although ZnMe2 is known to react with 3° alkyl and benzylic chlorides,8 AlMe3 is 20-fold cheaper on a molar basis. Successful use of this reagent (40–51% yield) was demonstrated by Makriyannis in two examples, one of which (9)7 is shown in Scheme 2. Shishido also applied this method to the preparation of CF3- containing t-Bu isosteres from the corresponding 3° benzylic chlorides.9 These literature precedents encouraged us to investigate the substrate scope of this transformation, and determine if much milder conditions could be employed.
Results and Discussion
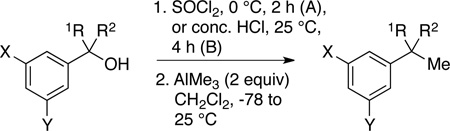
Standard synthetic methods were employed to prepare a range of 3° benzylic alcohols from commercially available acetophenones and carboxylic acids. These compounds (6b-l, 7b–d, f,h,i and 8c,d,f–h) varied in the identity of the meta-substituents (X and Y), and carbinol alkyl groups (R1 and R2, Table 1).
Table 1.
Transformation of 3° benzylic alcohols to tert-alkyl benzenes.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | cpd | X | Y | R1 | R2 | product | Activation Method |
Yield % |
| 1 | 6b | OMe | OMe | Me | Me | 2b | A | 88 |
| 2 | 6c | OMe | H | Me | Me | 2c | A | 93 |
| 3 | 6d | OH | H | Me | Me | 2d | A | 83 |
| 4 | 6e | Me | Me | Me | Me | 2e | A | 85 |
| 5 | 6f | Me | H | Me | Me | 2f | A | 85 |
| 6 | 6g | Ph | H | Me | Me | 2g | A | 70 |
| 7 | 6h | Cl | H | Me | Me | 2h | A | 93 |
| 8 | 6i | Br | H | Me | Me | 2i | A | 81 |
| 9 | 6j | OTBS | H | Me | Me | 2j | A | 93 |
| 10 | 6k | NHC(O)Ph | H | Me | Me | 2k | A | 97b |
| 11 | 6l | NHC(O)Me | H | Me | Me | 2l | A | 89 |
| 12 | 7b | OMe | OMe | Et | Me | 4b | A | 68 |
| 13 | 7c | OMe | H | Et | Me | 4c | A | 94 |
| 14 | 7d | OH | H | Et | Me | 4d | A | 70 |
| 15 | 7f | Me | H | Et | Me | 4f | A | 94 |
| 16 | 7h | Cl | H | Et | Me | 4h | A | 78a (87:13) |
| 17 | 4h | B | 95 | |||||
| 18 | 7i | Br | H | Et | Me | 4i | A | 72a (85:15) |
| 19 | 4i | B | 95 | |||||
| 20 | 8c | OMe | H | Et | Et | 5c | A | 97 |
| 21 | 8d | OH | H | Et | Et | 5d | A | 92 |
| 22 | 8f | Me | H | Et | Et | 5f | A | 80 |
| 23 | 8g | Ph | H | Et | Et | 5g | A | 71 |
| 24 | 8h | Cl | H | Et | Et | 5h | A | 90a (56:44) |
| 25 | 5h | B | 95a (96:4) | |||||
Weight recovery of a mixture of the intended methylated and unwanted elimination product (12h, 12i, and 13h, respectively) following chromatography; the mole ratio (1H NMR) is given in parentheses.
Methylation was slow at room temperature; reaction was carried out in 1,2-dichloroethane at 60 °C.
Reactions of dimethylaryl carbinols 6 were explored first. Each 3° alcohol was activated by treatment with neat SOCl2 (2.5 equiv, 0 °C, Activation Method A);10 after two hours, the residual SOCl2 was removed in vacuo at 0 °C. The residue (whose chemical identity is discussed below) was dissolved in CH2Cl2 and cooled to −78 °C, at which point AlMe3 (2 equiv) was added. After warming to room temperature overnight, the reaction was cooled to 0 °C and quenched with 1 M HCl. Aqueous workup and chromatographic purification yielded t-butylbenzenes 2b-l in 68–97% yield (Table 1, entries 1–11). Thus the reaction sequence tolerates a variety of meta-substituents (OMe, OH, Me, Ph, Cl, and Br), including acid sensitive groups (TBS-protected phenol, 6j) and amides (6k, 6l). The acid sensitivity of the TBS ether in 2j was confirmed by its quantitative conversion to 2d following exposure (1 h) to 1M HCl in MeOH. Note that moderately strong Lewis base functionalities (OH, OMe, OTBS, NHC(O)R) are compatible with this protocol. With regard to reaction temperature, most reactions of the activated alcohols with AlMe3 were found to proceed at or below room temperature, in contrast to the high temperature conditions reported for conversion of 9 (Scheme 2, 115 °C).7 Only in one case was it necessary to perform the reaction at elevated temperature (60 °C, amido-functionalized substrates 6k). Based on our reactions with similar substrates 6b and 7b, it seems likely that the conversion of 9 would also proceed well at room temperature.
Reaction of ethylmethylaryl carbinols 7 was nearly as successful as that of dimethylaryl carbinols 6. As seen in Table 1, entries 12–15, moderate to excellent yields of the desired 1,1-dimethylpropyl derivatives 4 were obtained. Unfortunately, in the case of m-Cl and m-Br carbinols 7h and 7i, the desired products 4h and 4i were contaminated with 13–15 mol% of the chromatographically inseparable elimination products (12h and 12i, entries 16 and 18 respectively).

To assess whether this product mixture arose in part from some difficulty in the alcohol activation step, 3° benzylic chlorides 14h and 14i were prepared by treating the corresponding carbinols with concentrated HCl, followed by extractive workup (Activation Method B). Reaction of 14h and 14i with AlMe3 at room temperature afforded the desired products 4h and 4i in 95% yield with no trace of the elimination products 12h and 12i (Table 1, entries 17, 19). Finally, reactions of diethylaryl carbinols 8 (Table 1, entries 20–23) gave moderate to excellent yields of 1-ethyl-1-methylpropylbenzenes 5. Elimination was again seen as a competing side reaction in the m-Cl substrate 8h, giving 44 mol% 13h (Table 1, Entry 24). Suspecting that some deficiency in the alcohol activation was again responsible, the 3° benzylic chloride 15h was prepared. As we had seen for reaction of 14h and 14i, reaction of 15h with AlMe3 gave an excellent yield (93% overall) with only a trace (4%) of the elimination product 13h (Table 1, Entry 25). Our analysis of the chloride intermediate 15h suggests that the small amount of elimination observed occurred prior to addition of AlMe3.
Given the divergent methylation results seen for 7h, 7i and 8h using activation method A (treatment with SOCl2) and activation method B (treatment with conc. HCl), we investigated the chemical identity of the SOCl2 activation products of two substrates. Treatment of 6b with SOCl2 at 0 °C for 2 hours, followed by rapid concentration in vacuo at 0 °C gave a product that was identical by 1H and 13C NMR spectroscopy to 3° benzylic chloride 16b formed by treatment of 6b with concentrated HCl. Presumably 6b quickly reacts with SOCl2 to form the chlorosulfite intermediate 17b, which then ionizes to a 3° benzylic carbocation/chlorosulfite ion pair 18b, ejects SO2, and recombines to form chloride 16b (Scheme 3).11,12
Scheme 3.

Rapid conversion of 6b to 16b on treatment with SOCl2 at 0 °C and possible mechanism.
However, treatment of 8h under the same conditions did not lead to chloride 15h, but rather to an intermediate we tentatively identified as the chlorosulfite 19h. This compound was nearly identical by 1H NMR spectroscopy (CDCl3) to starting alcohol 8h, but was much less polar on TLC, with an Rf very similar to that of 3° benzylic chloride 15h. To confirm our conclusion we carried out an NMR study of the reaction of 8h with SOCl2 (5 equiv) in CD2Cl2 (Scheme 4).
Scheme 4.

Course of the reaction of 8h with SOCl2; NMR chemical shifts were measured in CD2Cl2.
After 2 h at 0 °C, the NMR sample tube was allowed to reach room temperature and 1H and 13C NMR spectra of the reaction were taken at 1, 2, 5, and 24 h. After 1 h at room temperature, some conversion of 19h to the chloride 15h and elimination product 13h was evident. After 5 h at room temperature, only 17% of the chlorosulfite 19h remained, giving predominantly chloride 15h (52%) and elimination product 13h (31%). At 24 h all of the chlorosulfite 19h had been converted to chloride 15h and elimination product 13h. Olah has previously noted the tendency of tertiary chlorosulfites to eliminate.13
Why would the chlorosulfite 19h be slower than 17b in its conversion to the chloride? We note that based on σm values, the m-Cl substituent (σm = 0.37) is more electron-withdrawing than the m-OMe substitutent (σm = 0.10).14 If one m-Cl substituent is more electron-withdrawing than two m-OMe groups, the ionization of the chlorosulfite 19h would be slower than ionization of 17b. Thus it seems likely that the SOCl2 activation of m-Cl and m-Br ethylmethylaryl carbinols 7h and 7i (Br σm = 0.37)14 also progressed only to the chlorosulfite stage after 2 h at 0 °C. The inferior results for these substrates (and 8h) using the SOCl2 activation method A could be rationalized if 3° benzylic chlorosulfites are more prone to elimination on treatment with AlMe3 than are the corresponding 3° benzylic chlorides. As a final control experiment, alcohol 7h (without any prior activation) was treated with AlMe3 under the standard conditions. As expected, 7h was recovered and no methylated product 4h or elimination product 12h was formed.
To further assess the scope of this methylation protocol we examined two substrate classes expected to be problematic. First, if benzylic carbocation intermediates are formed during both the chlorination and methylation steps, then pyridyldialkylcarbinols might prove challenging substrates, due to the strongly electron-withdrawing nature of the pyridine ring. Hammett σ values for 2-, 3-, and 4-pyridyl rings are reported as 0.71, 0.55 and 0.94, respectively,15 and we chose to explore reactions of 2-pyridyl tertiary carbinol 20a and 3-pyridyl tertiary carbinol 20b.

As expected, methylation of these substrates was difficult and required elevated temperatures. Activation of 3-pyridyl substrate 20b using method A, followed by treatment with AlMe3 at 80 °C in 1,2-dichloromethane, gave the desired methylated product 21b, but contaminated with 21 mol% of chromatographically inseparable elimination product 22b. Application of this protocol to 20a was less successful, giving 21a and the chromatographically inseparable elimination product 22a in a 9:91 ratio. Activation of 20a and 20b by conversion to the corresponding mesylates9 was also explored, but did not provide improved outcomes. Thus neither pyridin-2-yl nor pyridin-3-yl dialkylcarbinols (e.g. 20a, b) qualify as good substrates for this reaction. Second, very strong electron-releasing substitutents might be problematic, because they could facilitate self-reaction of the benzylic chloride intermediate. Since hydroxy, alkoxy, silyloxy and amido substituents were well-tolerated (Table 1, entries 1–3, 9–14, 20–21), we explored dimethylamino-substituted aryldimethyl carbinol 6m. Regardless of the alcohol activation method employed, and conditions used for reaction with AlMe3, weight recoveries were very low (<20% of theoretical yield of 2m). 1H NMR spectroscopy of the reaction mixture suggested that oligomerization of the activated alcohol had occurred.
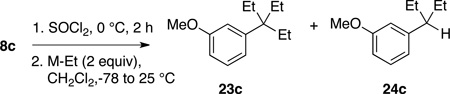
The protocol described above allows installation of tert-alkyl groups containing at least one methyl group. Installation of a 1,1-diethylpropyl (i.e. triethylcarbinyl) substituent would necessitate use of an ethyl organometallic. To attempt such a transformation, diethylaryl carbinol 8c was activated with SOCl2 and treated with various ethyl organometallics. Carbinol 8c was chosen for the high yield observed in its SOCl2-activation/methylation to 5c (Table 1, entry 17). Use of AlEt3 in the standard protocol did give the intended product 23c, but contaminated with the chromatographically inseparable 24c (Table 2, entry 1).
Table 2.
Attempted synthesis of 1,1-diethylpropyl-substituted benzene 23c
 | |||
|---|---|---|---|
| entry | M-Et | 23c:24ca | Weight recovery (%)b |
| 1 | AlEt3 | 77:23 | 94 |
| 2 | ClAlEt2 | 9:91 | 72 |
| 3 | BEt3 | no reaction | NAc |
| 4 | ZnEt2 | 46:54 | 96 |
Measured by 1H NMR spectroscopy.
Based on the stoichiometry indicated by 1H NMR spectroscopy.
Recovered 3° benzylic chloride-derived from 8c.
It seemed likely that 24c arose from β-hydride delivery, as Miller suggested in his studies of the reaction of AlEt3 with aliphatic & benzylic halides.16 We thus investigated other ethyl organometallics. Interestingly, reaction with ClAlEt2 gave a 9:91 mixture in favor of the hydride addition product 24c (Table 2, entry 2). Reaction with BEt3 did not proceed: neither 23c or 24c was detected, and the 3° benzylic chloride derived from 8c was recovered. Finally, use of ZnEt2 in the protocol gave a nearly 1:1 mixture of 23c and 24c. Thus an effective protocol to install the 1,1-diethylpropyl group by ethylation of a diethylaryl carbinol remains elusive.
To conclude our study we explored reaction of tertiary alcohol 25, bearing a tethered benzene ring, that was designed to probe the mechanism of the reaction of tertiary chlorides with AlMe3 (Scheme 5).
Scheme 5.

Methylation of 3° aliphatic alcohol 25.
In Miller’s studies of the reaction of AlEt3 with 1°, 2° and 3° and benzylic chlorides, it was proposed that the observed product mixtures and relative rates were consistent with the formation of ion pair intermediates.16 In this paradigm, the 3° chloride derived from 25 would react with AlMe3 to form ion pair intermediate 27, which could undergo two fates: methyl transfer to 26, or intramolecular Friedel-Crafts alkylation leading to 28. In the event, 26 was isolated in 74% yield, and Friedel-Crafts product 28 was not detected. Thus ion pair 27 appears to have a very short lifetime.
Conclusion
In closing, based on the work of Reetz,5,6 Makriyannis,7 and Shishido,9 we developed a mild two-step method to place tert-alkyl groups on aromatic rings meta- to ortho, para-directing groups. Tertiary benzylic alcohols are activated by treatment with SOCl2 (Method A) or concentrated HCl (Method B), and in most cases were found to react quickly with AlMe3 at or below room temperature. In addition to t-butyl, the 1,1-dimethylpropyl (ethyldimethylcarbinyl) and 1-ethyl-1-methylpropyl (diethylmethylcarbinyl) substitutents were successfully installed. Eleven different aromatic substitution patterns were examined, giving a total of 22 examples in 68–97% yields (Table 1). For substrates bearing moderate electron-withdrawing groups (e.g. m-Cl or m-Br), activation Method B was found to give the best results. However, elimination remained a persistent problem for strongly electron deficient substrates (e.g. pyridin-2-yl and pyridin-3-yl substrates, 20a,b). Substrates bearing very strong electron donors (e.g. NMe2, 6m) also proved problematic.
Experimental Section
General Methods
High resolution mass spectra (ESI and APCI) were recorded on a time of flight LC/MS instrument. Due to their demonstrated lability, characterization of 3° benzylic chlorides or chlorosulfites by HRMS or elemental analysis was not attempted. 1H NMR spectra of known tert-alkyl benzenes (i.e. those not listed below) synthesized in this work are provided in the Supporting Information to document purity. Known dimethylaryl carbinols 6b–i, l, m, and 20a,b were prepared in 89–97% yield by the addition of lithium trimethylmagnesate to the corresponding acetophenones in THF.17 Known ethylmethylaryl carbinols 7c, 7d, 7f, 7h, and 7i were prepared in 87–98% yields by Zn2+-catalyzed addition of EtMgCl to the corresponding acetophenones;18 ethylmethylaryl carbinol 7b19 was prepared in 75 % yield by addition of EtMgCl to 3,5-dimethoxyacetophenone. Known diethylaryl carbinols 8c, 8d, 8f and 8h, and diethyl carbinol 25 were prepared in 86–94% overall yield by conversion of the corresponding benzoic acids to the methyl ester, followed by reaction with EtMgBr (2.5 equiv) in THF.20 In all cases analytical data of synthesized known compounds matched that of the literature.
General Procedure for SOCl2 activation (Method A)/methylation of tertiary benzylic carbinols
1-(tert-butyl)-3-methoxybenzene (2c).21
A dry Schlenk flask (50 mL) equipped with a rubber septum and a magnetic stirbar was charged with 2-(3-methoxyphenyl)propan-2-ol (6c, 400 mg, 2.41 mmol), placed in an ice bath, and purged with nitrogen. Thionyl chloride (438 mg, 6.02 mmol) was added via syringe and the reaction was allowed to stir at 0 °C for 2 hours; in some cases up to 1 mL of CH2Cl2 was added to improve mixing. Volatiles were then removed at 0 °C under reduced pressure. The residue was diluted with dichloromethane (8 mL) and cooled to −78 °C in a dry-ice/acetone bath. Trimethylaluminum (2.0 M in hexanes, 2.4 mL, 4.8 mmol) was injected into the flask and allowed to stir for three hours at −78 °C, and then stirred overnight at room temperature. The reaction was then cooled to 0 °C and cautiously quenched by the addition of HCl (1 M, 10 mL). Following extraction with dichloromethane (3 × 25 mL) the organic layers were combined, washed with brine, dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified on silica gel (5 : 1 hexane : ethyl acetate) to yield a colorless oil (368 mg, 93% yield). 1H and 13C NMR spectral data matched those reported in the literature.
N-(3-(tert-butyl)phenyl)benzamide (2k)
This compound was prepared from N-(3-(2-hydroxypropan-2-yl)phenyl)benzamide (6k, 17.4 mg, 0.068 mmol) using the procedure described above for 2c, with minor modification: following activation via Method A, reaction with AlMe3 was conducted at 60 °C, using 1,2-dichloroethane as solvent. Following aqueous workup the residue was purified on silica gel (5 : 1 hexane : ethyl acetate) to yield 2k as a white solid (16.7 mg, 97% yield); 1H NMR (400 MHz, CDCl3) δ 7.90-7.87 (m, 3H), 7.63 (s, 1H), 7.54-7.45 (m, 4H), 7.32-7.25 (m, 1H), 7.28-7.18 (m, 1H), 1.34 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 165.73, 152.30, 137.65, 135.11, 131.75, 128.75, 126.99, 121.67, 117.51, 117.37, 34.69, 31.29; HRMS (APCI): 254.1539 calcd for C17H19NO [M+H]+ found 254.1535 (−1.56 ppm).
1-Methoxy-3-(tert-pentyl)benzene (4c)
This compound was prepared from 7c (199 mg, 1.10 mmol) using the Method A procedure for 2c, providing 185 mg of 4c (94% yield) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.23 (t, J = 8.0 Hz, 1H), 6.96 – 6.91 (m, 1H), 6.89 (t, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.0, 2.0 Hz, 1H), 3.81 (s, 3H), 1.63 (q, J = 7.4 Hz, 2H), 1.27 (s, 6H), 0.68 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 159.5, 151.5, 129.0, 118.71, 118.70, 112.9, 109.9, 55.3, 38.1, 37.0, 28.6, 9.3; HRMS (APCI): 179.1430 calcd for C12H19O [M+H]+ found 179.1429 (−0.79 ppm).
General Procedure for HCl activation (Method B)/methylation of tertiary benzylic carbinols
Chloro-3-(tert-pentyl)benzene (4h)
A flask was charged with 2-(3-chlorophenyl)butan-2-ol (7h, 202 mg, 1.09 mmol), to which was added concentrated hydrochloric acid (1.5 mL). The mixture was stirred at room temperature for 3 h prior to being extracted with CH2Cl2 (3 × 3 mL). The combined extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the corresponding chloride 14h (containing 2 mol% elimination product 12h) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.56 (t, J = 2.0 Hz, 1H), 7.44 (dt, J = 7.7, 1.8 Hz, 1H), 7.26–7.32 (overlapped, 2H), 2.13–2.24 (m, 2H), 1.95 (s, 3H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 147.1, 134.1, 129.4, 127.5, 126.5, 124.3, 73.7, 39.4, 31.2, 9.7. To a solution of the chloride, prepared from the above step, in dichloromethane (7 mL) at −78 °C was added trimethylaluminum in hexanes (2.0 M in hexanes, 1.4 mL, 2.8 mmol), and the reaction mixture was stirred at −78 °C for 3 h, and allowed to warm to room temperature overnight. The reaction was quenched cautiously at 0 °C with HCl (1 M) and extracted with dichloromethane (20 mL × 3). The combined extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated to provide methylated product 4h as a colorless oil (185 mg, 93% for two steps; contains 2 mol% elimination product 12h): 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 2.0 Hz, 1H), 7.27-7.22 (overlapped, 2H), 7.16 (dt, J = 7.1, 2.0 Hz, 1H), 1.65 (q, J = 7.4 Hz, 2H), 1.29 (s, 6H), 0.70 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.7, 134.0, 129.2, 126.4, 125.5, 124.2, 38.1, 36.8, 28.3, 9.1; HRMS (EI): calcd for C11H15Cl 182.0862; found 182.0860 (−0.2 mmu, −1.3 ppm).
Bromo-3-(tert-pentyl)benzene (4i)
This compound (129 mg, oil) was prepared in 95% yield from alcohol 7i (145 mg, 0.63 mmol) by the Method B procedure described above for 4h. Spectral data for chloride 14i (containing 2 mol% elimination 12i); 1H NMR (500 MHz, CDCl3) δ 7.70 (t, J = 2.0 Hz, 1H), 7.48 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.43 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 2.12–2.23 (m, 2H), 1.94 (s, 3H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 147.3, 130.4, 129.7, 129.3, 124.8, 122.4, 73.6, 39.4, 31.2, 9.7. Spectral data for 4i : 1H NMR (500 MHz, CDCl3) δ 7.47 (t, J = 1.9 Hz, 1H), 7.32 (ddd, J = 7.8, 1.9, 1.0 Hz, 1H), 7.26 (ddd, J = 7.8, 1.9, 1.0 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 1.64 (q, J = 7.5 Hz, 2H), 1.28 (s, 6H), 0.70 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.1, 129.5, 129.3, 128.4, 124.7, 122.4, 38.1, 36.8, 28.3, 9.1;
Elemental analysis: calcd for C11H15Br 58.17 %C, 6.66 %H; found 58.41 %C, 6.87 %H.
1-Methoxy-3-(3-methylpentan-3-yl)benzene (5c)
This compound was prepared from 8c (260 mg, 1.34 mmol) using the Method A procedure for 2c, providing 5c (249 mg, 95% yield) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.23 (t, J = 8.0 Hz, 1H), 6.91 – 6.86 (m, 1H), 6.84 (t, J = 2.2 Hz, 1H), 6.71 (dd, J = 8.0, 2.2 Hz, 1H), 3.81 (s, 3H), 1.72 (dq, J = 14.9, 7.5 Hz, 2H), 1.54 (dq, J = 14.9, 7.5 Hz, 2H), 1.22 (s, 3H), 0.67 (t, J = 7.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 159.5, 149.7, 128.8, 119.4, 113.6, 109.7, 55.2, 41.5, 35.4, 22.9, 8.9; HRMS (APCI): 193.1587 calcd for C13H21O [M+H]+ found 193.1581 (−3.23 ppm).
3-(3-Methylpentan-3-yl)phenol (5d)
This compound was prepared from 8d (102 mg, 0.566 mmol), using the Method A procedure for 2c, providing 5d (93 mg, 92%) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.16 (t, J = 8.0 Hz, 1H), 6.86 (ddd, J = 8.0, 1.7, 0.9 Hz, 1H), 6.81 – 6.72 (m, 1H), 6.64 (ddd, J = 8.0, 1.7, 0.9 Hz, 1H), 4.64 (s, 1H), 1.74 - 1.66 (m, 2H), 1.58 – 1.48 (m, 2H), 1.21 (s, 3H), 0.67 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 155.3, 150.2, 129.1, 119.5, 114.0, 112.2, 41.4, 35.4, 22.8, 8.8; HRMS (APCI): 177.1285 calcd for C12H17O [M-H]− found 177.1278 (−4.03 ppm).
1-Methyl-3-(3-methylpentan-3-yl)benzene (5f)
This compound was prepared from 8f (333 mg, 1.87 mmol) using the Method A procedure for 2c, providing 5f (263 mg, 80%) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.22 – 7.15 (m, 1H), 7.11 – 7.05 (m, 2H), 6.98 (d, J = 7.3 Hz, 1H), 2.35 (s, 3H), 1.73 (dq, J = 14.8, 7.5 Hz, 2H), 1.55 (dq, J = 14.8, 7.5 Hz, 2H), 1.23 (s, 3H), 0.67 (t, J = 7.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 147.8, 137.3, 127.8, 127.5, 126.0, 123.8, 41.24, 35.3, 23.0, 21.9, 8.9; HRMS (APCI): 370.3468 calcd for C26H44N [2M+NH4]+ found 370.3496 (7.35 ppm).
3-(3-methylpentan-3-yl)-1,1'-biphenyl (5g)
This compound was prepared from 8g (253 mg, 1.05 mmol) using the Method A procedure for 2c, providing 5g (170 mg, 71%) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.67–7.69 (m, 2H), 7.59 (t, 1H, J = 1.7 Hz), 7.36–7.52 (m, 5H), 7.34–7.36 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 148.3, 142.1, 140.9, 128.8, 128.4, 127.4, 127.2, 125.83, 125.80, 124.3, 41.5, 35.4, 23.1; HRMS (APCI) calcd for C18H22 [M]+ 238.1722; found 238.1716 (−2.52 ppm).
3-(1-Chloro-3-(3-methylpentan-3-yl)benzene (5h)
This compound (28 mg, oil) was prepared in 93% yield from alcohol 8h (30 mg, 0.15 mmol) via the corresponding chloride using the Method B procedure as described for 4h. Spectral data for chloride 15h (contains 3 mol% elimination product 13h) is given below in the section describing NMR analysis of the reaction of 8h with SOCl2. Spectral data for 5h (contains 3 mol% elimination product 13h): 1H NMR (500 MHz, CDCl3) δ 7.27 (dd, J = 7.8, 2.2 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.15–7.18 (overlapped, 2H), 1.69–1.76 (m, 2H), 1.53–1.60 (m, 2H), 1.24 (s, 3H), 0.68 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 150.1, 134.0, 129.1, 127.0, 125.4, 124.9, 41.5, 35.2, 22.7, 8.7; Elemental analysis: calcd for C12H17Cl 73.27 %C, 8.71 %H; found 73.17 %C, 8.69 %H.
2-(3-((tert-Butyldimethylsilyl)oxy)phenyl)propan-2-ol (6j)
This compound (710 mg) was prepared as a colorless oil in 89% yield by the addition of lithium trimethylmagnesate17 to 1-(3-((tert-butyldimethylsilyl)oxy)phenyl) ethanone (750 mg, 3 mmol) in THF, using the procedure outlined below for 6k; 1H NMR (500 MHz, CDCl3) δ 7.20 (t, J = 8.0 Hz, 1H), 7.07 (ddd, J = 8.0, 1.8, 1.0 Hz, 1H), 6.99 (t, J = 2.1 Hz, 1H), 6.73 (ddd, J = 8.0, 2.5, 1.0 Hz, 1H), 1.57 (s, 6H), 1.00 (s, 9H), 0.22 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 155.8, 151.2, 129.3, 118.4, 117.4, 116.7, 72.6, 31.9, 25.9, 18.9, −4.2; HRMS (ESI): 249.1675 calcd for C15H25OSi [M-H2O+H]+ found 249.1655 (−5.52ppm).
N-(3-(2-hydroxypropan-2-yl)phenyl)benzamide (6k)
Under Nitrogen, a dry 50 mL Schlenk flask equipped with a magnetic stirbar was charged with 15 ml dry THF and then placed in an ice bath. Methylmagnesium chloride (3M in ether, 0.84 ml, 2.51 mmol) and methyl lithium (3M in ether, 0.84 ml, 2.51 mmol) were added. After stirring at 0 °C for 30 minutes, a solution of N-(3-acetylphenyl)benzamide (200 mg, 0.84 mmol) in 10 mL dry THF was added. After 1 hour the ice bath was removed and the reaction was allowed to stir at room temperature overnight. The reaction was cooled to 0 °C and quenched with saturated NaHCO3 solution and extracted with ethyl acetate (3 × 15mL). The organic layers were combined, dried with anhydrous Na2SO4, filtered, and the solvent evaporated under reduced pressure. The residue was purified on silica gel (1 : 1 hexane : ethyl acetate) to yield 6k as a white solid (178 mg, 84% yield). 1H NMR (400 MHz, CD3OD) δ 7.93-7.91 (m, 2H), 7.79 (s, 1H), 7.59-7.48 (m, 4H), 7.31-7.29 (m, 2H), 1.55 (s, 6H); 13C NMR (100 MHz, CD3OD) δ 167.50, 150.36, 138.11, 134.91, 131.42, 128.19, 128.05, 127.18, 120.57, 119.06, 117.45, 71.52, 30.47; HRMS (APCI): calculated for C16H17NO2Na 279.1184 [M+Na]+, found 279.1179 (−1.9 ppm).
3-([1,1'-biphenyl]-3-yl)pentan-3-ol (8g)
A modification of Hartmann’s procedure20 was used: methyl 3-phenylbenzoate (499 mg, 2.35 mmol) was dissolved in 5 mL THF and cooled to −78 °C EtMgCl (2 M in THF, 3.5 mL, 7.5 mmol) was added and stirred for 5 h at this temperature at which point TLC indicated completion. The reaction was quenched by the addition of saturated aq. NH4Cl, and warmed to room temperature. Following aqueous workup and column chromatography (5% EtOAc in hexanes), 8g was obtained as a colorless oil (560 mg, 94%); 1H NMR (500 MHz, CDCl3) δ 7.77 (s, 1H), 7.72–7.74 (m, 2H), 7.40–7.56 (m, 6H), 2.14 (br s, 1H), 1.93–2.03 (m, 4H), 0.92 (t, 3H, J = 7.3 Hz); 13C NMR (125 MHz, CDCl3) δ 146.6, 141.7, 141.0, 128.9, 128.6, 127.5, 127.4, 125.3, 124.7, 124.6, 77.7, 35.2, 8.1; HRMS (APCI) calculated for C17H19 [M-OH]+ 223.1481 found 223.1498 (+7.62 ppm).
3-(tert-Butyl)pyridine (21b) and 3-(prop-1-en-2-yl)pyridine (22b)
The method A procedure for 2c described above was applied to 2-(pyridin-3-yl)propan-2-ol (20b, 137 mg, 1 mmol), using 1,2-dichloroethane in place of dichloromethane as solvent and elevating the reaction temperature to 85 °C. This reaction was quenched with 10% NaHCO3 solution, and the resulting mixture was extracted with dichloromethane. The combined extracts were dried over anhydrous Na2SO4, and concentrated to afford an inseparable 79:21 mixture of the desired compound 21b and the undesired elimination product 22b as a light yellow oil (122 mg, 89% mass recovery).
Analytical data for 3-(tert-Butyl)pyridine (21b)
1H NMR (500 MHz, CDCl3) δ 8.67 (d, J = 2.6 Hz, 1H), 8.43 (br.d, J = 4.8 Hz, 1H), 7.71 (ddd, J = 8.1, 2.6, 1.5 Hz, 1H), 7.25 (dd, J = 5.9, 4.8 Hz, 1H), 1.35 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 146.7, 146.2, 133.5, 123.2, 114.4, 31.0.
Analytical data for 3-(prop-1-en-2-yl)pyridine (22b)
1H NMR (500 MHz, CDCl3) δ 8.73 (br.s, 1H), 8.51 (m, 1H), 7.74 (br.d, J = 8.1 Hz, 1H), 7.26 (dd, J = 8.1, 3.0 Hz, 1H), 5.42 (s, 1H), 5.19 (s, 1H), 2.17 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 148.6, 147.2, 146.8, 140.9, 137.4, 133.6, 123.8, 21.5.
(3-ethyl-3-methylpentyl)benzene (26)
This compound was prepared from 25 (250 mg, 1.26 mmol) using the Method A procedure for 2c, providing 26 (178 mg, 74%) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.34–7.37 (m, 2H), 7.23–7.27 (m, 3H), 2.56–2.60 (m, 2H), 1.53–1.57 (m, 2H), 1.38 (q, 4H, J = 7.9 Hz), 0.90–0.94 (m, 9H); 13C NMR (125 MHz, CDCl3) δ 143.9, 128.4, 125.6, 41.0, 35.3, 31.1, 30.4, 24.1, 8.1.
Elemental analysis: calcd for C14H22 %C 88.35, %H 11.65; found %C 88.50, %H 11.66%
Investigation of SOCl2 activation of 3° benzylic carbinols
1-(2-chloropropan-2-yl)-3,5-dimethoxybenzene (16b).22
This compound was prepared in 98% yield from alcohol 6b and concentrated HCl using the same procedure described for the synthesis of chloride 14h en route to methylated product 4h. Treatment of 6b with SOCl2 for 2 h at 0 °C followed by concentration in vacuo gave the identical compound. 1H NMR (500 MHz, CDCl3) δ 6.77 (d, J = 2.2 Hz, 2H), 6.42 (t, J = 2.2 Hz, 1H), 3.84 (s, 6H), 1.99 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 160.5, 148.7, 104.2, 99.0, 69.5, 55.4, 34.2.
NMR Study of reaction of 8h with SOCl2 in CD2Cl2
Alcohol 8h (30 mg, 0.15 mmol) was dissolved in 750 uL CD2Cl2, transferred to an NMR tube and placed in an ice bath. After 5 min, SOCl2 (60 uL, 98 mg, 0.81 mmol) was added and the tube was agitated to achieve mixing. After 2 h the tube was removed from the ice bath and allowed to warm to room temperature. 1H and 13C NMR spectra were taken after 1, 2, 5, and 25 h at room temperature. Mole ratios of chlorosulfite 19h, chloride 15h, and alkene 13h were determined by integration (see Scheme 4).
3-(3-chlorophenyl)pentan-3-ol (8h)
1H NMR (500 MHz, CD2Cl2) δ 7.32 (t, J = 2.1 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.16 (dt, J = 7.8, 1.6 Hz, 1H), 7.12 (ddd, J = 7.4, 2.2, 1.6 Hz, 1H), 1.65–1.78 (m, 4H), 0.65 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz, CD2Cl2) δ 148.7, 134.3, 129.6, 126.6, 126.3, 124.3, 77.4, 35.5, 7.9.
(E)-1-chloro-3-(pent-2-en-3-yl)benzene (13h)
Resonances deduced from examination of a 73:27 mixture of 15h and 13h. 1H NMR (500 MHz, CD2Cl2) δ 7.22–7.23 (m, 1H), 7.13–7.15 (m, 3H), 5.66 (q, J = 6.9 Hz, 1H), 2.41 (q, J = 7.6 Hz, 2H), 1.71 (d, J = 6.9 Hz, 3H), 0.88 (t, J = 7.6 Hz, 3H); 13C NMR (125 MHz, CD2Cl2) δ 145.6, 141.6, 134.3, 129.9, 126.70, 126.67, 124.8, 123.9, 22.8, 14.2, 13.4.
1-chloro-3-(3-chloropentan-3-yl)benzene (15h, contains 3 mol% elimination product 13h)
1H NMR (500 MHz, CD2Cl2) δ 7.42 (t, J = 2.0 Hz, 1H), 7.28 (ddd, J = 7.8, 1.9, 1.3 Hz, 1H), 7.21 (dt, J = 7.8, 0.5 Hz, 1H), 7.17 (ddd, J = 7.9, 2.0, 1.3 Hz, 1H), 2.06 (q, J = 7.2 Hz, 4H), 0.78 (t, J = 7.2 Hz, 6H); 13C NMR (125 MHz, CD2Cl2) δ 145.6, 134.4, 129.7, 127.49, 127.47, 125.3, 80.3, 37.6, 9.3.
3-(3-chlorophenyl)pentan-3-yl sulfochloridite (chlorosulfite 19h)
1H NMR (500 MHz, CD2Cl2) δ 7.32 (t, J = 2.1 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.16 (dt, J = 7.8, 1.6 Hz, 1H), 7.12 (ddd, J = 7.4, 2.1, 1.6 Hz, 1H), 1.66–1.78 (m, 4H), 0.65 (t, J = 7.4 Hz, 6H); 13C NMR (125 MHz, CD2Cl2) δ 148.7, 134.3, 129. 9, 126.7, 126.3, 124.3, 77.7, 35.5, 8.0. Note: although the pyramidal geometry at sulfur should render the CH2 and CH3 carbons diastereotopic, we could only resolve a single CH2 resonance and a single CH3 resonance in the 13C NMR spectrum at 125 MHz.
Attempted activation/ethylation of diethylarylcarbinol 8c
1-(3-ethylpentan-3-yl)-3-methoxybenzene (23c)
The Method A procedure for 2c described above was applied to diethylarylcarbinol 8c (255 mg, 1.31 mmol), using triethylaluminum (1.0 M in hexanes, 2.6 mL, 2.6 mmol) in place of trimethylaluminum. This procedure afforded an inseparable 77:23 mixture of the desired compound 23c and the undesired hydride addition product 21c as a light yellow oil (245 mg, 94%). Analytical data for 23c: 1H NMR (500 MHz, CDCl3) δ 7.21 (t, 1H, J = 8 Hz), 6.94 (t, 1H, J = 8 Hz), 6.87 (s, 1H), 6.73 (t, 1H, J = 8 Hz), 4.20 (s, 3H), 1.65 (q, 6H, J = 7.0 Hz), 0.65 (t, 9H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 159.4, 149.4, 128.6, 119.6, 113.9, 109.5, 55.1, 43.9, 28.8, 8.1; HRMS (APCI): calculated for C14H23O 207.1743 [M+H]+, found 207.1738 (−2.75 ppm).
1-methoxy-3-(pentan-3-yl)benzene (24c)
The Method A procedure for 2c described above was applied to diethylarylcarbinol 8c (246 mg, 1.26 mmol), using diethylaluminum chloride (1 M in hexanes, 3.16 mL, 3.16 mmol) in place of trimethylaluminum. This procedure afforded a 9:91 ratio of 23c and 24c as a colorless oil (228 mg, 71%). Analytical data for 21c: 1H NMR (500 MHz, CDCl3) δ 7.212 (t, 1H, J = 7.7 Hz), 6.71–6.76 (m, 3H), 3.80 (s, 3H), 2.27–2.31 (m, 1H), 1.66–1.74 (m, 2H), 1.51–1.58 (m, 2H), 0.78 (t, 3H, J = 7.4 Hz); 13C NMR (125 MHz, CDCl3) δ 159.6, 147.7, 129.0, 120.4, 113.8, 110.7, 55.1, 49.8, 29.3, 12.3; HRMS: calculated for C12H19O 179.1430 [M+H]+, found 179.1439 (+4.97 ppm)
Supplementary Material
Acknowledgment
We thank the National Institutes of Health for financial support of this work (1R01AI082581- 01), and Mr. Eugene Camerino for help with analytical characterization.
Footnotes
Supporting Information Available. NMR spectra (1H and 13C) of all new compounds, and 1H NMR spectra of known compounds described in Table 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carlier PR, Anderson TD, Wong DM, Hsu DC, Hartsel J, Ma M, Wong EA, Choudhury R, Lam PC-H, Totrov MM, Bloomquist JR. Chemico-Biol. Interact. 2008;175:368–375. doi: 10.1016/j.cbi.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Faldt A, C. Krebs F, Thorup N. J. Chem. Soc. Perkin Trans. 1997;2:2219–2228. [Google Scholar]
- 3.Carpenter MS, Easter WM, Wood TF. J. Org. Chem. 1951;16:586–617. [Google Scholar]
- 4.Austin M, Egan OJ, Tully R, Pratt AC. Org. Biomol. Chem. 2007;5:3778–3786. doi: 10.1039/b711620a. [DOI] [PubMed] [Google Scholar]
- 5.Reetz MT, Westermann J, Kyung SH. Chem. Ber. 1985;118:1050–1057. [Google Scholar]
- 6.Reetz MT, Westerman J. J. Org. Chem. 1983;48:254–255. [Google Scholar]
- 7.Nikas SP, Grzybowska J, Papahatjis DP, Charalambous A, Banijamali AR, Chari R, Fan PS, Kourouli T, Lin SY, Nitowski AJ, Marciniak G, Guo Y, Li XY, Wang CLJ, Makriyannis A. AAPS J. 2004;6:1–13. doi: 10.1208/aapsj060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reetz MT, Wenderoth B, Peter R, Steinbach R, Westermann J. J. Chem. Soc. Chem. Comm. 1980:1202–1204. [Google Scholar]
- 9.Tanaka H, Shishido Y. Bioorg. Med. Chem. Lett. 2007;17:6079–6085. doi: 10.1016/j.bmcl.2007.09.053. [DOI] [PubMed] [Google Scholar]
- 10. Treatment of 3-hydroxy substrates 6d, 7d and 8d with SOCl2 was performed at −20 °C
- 11.Lewis ES, Boozer CE. J. Am. Chem. Soc. 1952;74:308–311. [Google Scholar]
- 12.Schreiner PR, Schleyer PvR, Hill RK. J. Org. Chem. 1993;58:2822–2829. [Google Scholar]
- 13.Olah GA, Wu AH, Farooq O. J. Org. Chem. 1989;54:1375–1378. [Google Scholar]
- 14.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books: Sausalito, CA; 2006. pp. 421–488. [Google Scholar]
- 15.Blanch JH. J. Chem. Soc. B. 1966 [Google Scholar]
- 16.Miller DB. J. Org. Chem. 1966;31:908–912. [Google Scholar]
- 17.Hatano M, Matsumura T, Ishihara K. Org. Lett. 2005;7:573–576. doi: 10.1021/ol047685i. [DOI] [PubMed] [Google Scholar]
- 18.Hatano M, Ito O, Suzuki S, Ishihara K. J. Org. Chem. 2010;75:5008–5016. doi: 10.1021/jo100563p. [DOI] [PubMed] [Google Scholar]
- 19.Djura P, Sargent MV. J. Chem. Soc. Perkin Trans. I. 1978:395–400. [Google Scholar]
- 20.Hartmann RW, Kranzfelder G, Von Angerer E, Schoenenberger H. J. Med. Chem. 1980;23:841–848. doi: 10.1021/jm00182a006. [DOI] [PubMed] [Google Scholar]
- 21.Jeffery E, Meisters A, Mole T. Aust. J. Chem. 1974;27:2569–2576. [Google Scholar]
- 22.Shih N-Y, Mangiaracina P. Lipoxygenase Inhibitors. Int. Pat. Appl. CP/US90/05824 Oct. 17, 1990. Chem Abstr. 1991;115:114147. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.