

Abstract
In addition to genetic predisposition, environmental and lifestyle factors contribute to the pathogenesis of type 2 diabetes (T2D). Epigenetic changes may provide the link for translating environmental exposures into pathological mechanisms. In this study, we performed the first comprehensive DNA methylation profiling in pancreatic islets from T2D and non-diabetic donors. We uncovered 276 CpG loci affiliated to promoters of 254 genes displaying significant differential DNA methylation in diabetic islets. These methylation changes were not present in blood cells from T2D individuals nor were they experimentally induced in non-diabetic islets by exposure to high glucose. For a subgroup of the differentially methylated genes, concordant transcriptional changes were present. Functional annotation of the aberrantly methylated genes and RNAi experiments highlighted pathways implicated in β-cell survival and function; some are implicated in cellular dysfunction while others facilitate adaptation to stressors. Together, our findings offer new insights into the intricate mechanisms of T2D pathogenesis, underscore the important involvement of epigenetic dysregulation in diabetic islets and may advance our understanding of T2D aetiology.
Keywords: DNA methylation, pancreatic islets, type 2 diabetes
Introduction
Type 2 diabetes (T2D) has developed into a major public health concern. While previously considered as a problem primarily for western populations, the disease is rapidly gaining global importance, as today around 285 million people are affected worldwide (IDF, 2009). Lifestyle and behavioural factors play an important role in determining T2D risk. For example, experimentally induced intrauterine growth retardation as well as nutrient restriction during pregnancy in rats have been shown to result in development of T2D in offspring (Inoue et al, 2009) while chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring (Ng et al, 2010). In humans, a reduced birth weight together with an accelerated growth in infancy has been associated with impaired glucose tolerance (IGT) in adulthood (Bhargava et al, 2004). The pancreatic islets of Langerhans are of central importance in the development of T2D. Under normal conditions, increasing blood glucose levels after a meal trigger insulin secretion from the pancreatic islet β-cells to regulate glucose homeostasis. β-Cell failure marks the irreversible deterioration of glucose tolerance (Cnop et al, 2007b; Tabak et al, 2009) and results in T2D (UKPDSG, 1995). The unbiased genome-wide search for T2D risk genes (Saxena et al, 2007; Scott et al, 2007; Sladek et al, 2007; Zeggini et al, 2007, 2008) has placed the insulin-producing β-cells at centre stage. These approaches have also inadvertently highlighted the complexity of the biological mechanisms critical to T2D development. Most T2D risk genes identified in these genome-wide association studies (GWAS) affect β-cell mass and/or function (Florez, 2008). While the majority of studies in the field have characterised diabetes aetiology on the basis of genetics, new findings suggest the potential involvement of epigenetic mechanisms in T2D as a crucial interface between the effects of genetic predisposition and environmental influences (Villeneuve and Natarajan, 2010). Epigenetic changes are heritable yet reversible modifications that occur without alterations in the primary DNA sequence. DNA methylation and histone modifications are the main molecular events that initiate and sustain epigenetic modifications. These modifications may therefore provide a link between the environment, that is, nutrition and lifestyle, and T2D but only few studies so far have documented aberrant DNA methylation events in T2D (Ling et al, 2008; Park et al, 2008).
DNA methylation occurs as 5-methyl cytosine mostly in the context of CpG dinucleotides, so-called CpG sites. It is the best-studied epigenetic modification and governs transcriptional regulation and silencing (for review, see Suzuki and Bird, 2008). Unlike the relatively study genome, the methylome changes in a dynamic way during development, tissue differentiation and aging. Pathologically altered DNA methylation is well described in various cancers (reviewed in Jones and Baylin, 2007) and its role is starting to be revealed in several other diseases such as multiple sclerosis (Casaccia-Bonnefil et al, 2008), Alzheimer's disease (Mastroeni et al, 2009) and systemic lupus erythematosus (Javierre et al, 2010). About 75% of human gene promoters are associated with CpG islands (CGIs) (Jones and Baylin, 2007; Suzuki and Bird, 2008), which are clusters of 500 bp to 2 kb length with a comparatively high frequency of CpG dinucleotides. They usually harbour low levels of DNA methylation but can become hypermethylated; this CGI hypermethylation was demonstrated to abrogate transcription of tumour suppressor genes during tumourigenesis (Jones and Baylin, 2007). Lately, DNA methylation changes in CpG sites adjoining yet outside of CGIs, so-called CGI shores (Irizarry et al, 2009), are gaining increased attention. Intriguingly, CpG sites in these shore sequences, in addition to those within CGIs, are proposed to display differential DNA methylation between cancer and normal cells as well as between cells of different tissues (Irizarry et al, 2009).
The goal of the present work was to clarify the hitherto poorly understood connection between DNA methylation and T2D pathogenesis and to determine whether identified epigenetic changes translate into functional effects that impinge on pancreatic β-cell function. For this, we have explored DNA methylation landscapes in islets isolated from T2D patients and non-diabetic individuals.
Results
Identification of the T2D-related differential DNA methylation profile
We performed DNA methylation profiling to analyse the methylomes of freshly isolated islets from 16 human cadaveric donors of similar age, BMI and ethnicity (5 diabetic and 11 non-diabetic Caucasian donors; Table I). Using electron microscopy (EM), we examined the purity of the islet preparations (see Supplementary data). In diabetic islets, decreases in the percentage of β-cells have been reported (Sakuraba et al, 2002; Rahier et al, 2008). As shifts in the composition of islet cell types (especially β-cells that constitute about two thirds of the islet cell mass) with different epigenomes might overlay T2D-related changes in the cells’ DNA methylation patterns, we used EM to estimate the percentage of β-cells. The amount of β-cells in three randomly chosen control islet preparations was 66.3±0.9%. Diabetic islets (n=3, randomly chosen) contained only marginally less β-cells, accounting for 59.7±1.7% of total islet cell number (cf. Supplementary Table S1, Materials and methods, Supplementary data).
Table 1. Pancreatic islet donor characteristics.
| CTL | T2D | |
|---|---|---|
| Number of samples | 11 | 5 |
| Age (years) | 57.2±15.2 | 64.4±5.3 (P=0.33) |
| BMI (kg/m2) | 27.0±1.1 | 26.5±2.2 (P=0.8) |
| Glucose-induced insulin secretion (ratio of secretion at 16.7 mM/3.3 mM glucose) | ||
| 3.59±1.05 | 1.24±0.3 (P<0.001) | |
To perform DNA methylation profiling, we used the recently developed Infinium Methylation Assay (Illumina® Infinium® HumanMethylation27 BeadChip; Supplementary Figure S1; Bibikova et al, 2009). This assay interrogates the methylation status of 27 578 CpG sites corresponding to 14 475 consensus coding sequences and well-known cancer genes (Bibikova et al, 2009). Although the Infinium methylation assay is not a genome-wide DNA methylation technology, it is a useful screening tool that is sensitive, specific and highly reproducible (Bibikova et al, 2009) allowing for analysis of a defined set of CpG sites in a large number of samples. The analysed CpGs are primarily located in proximal promoter regions (and preferentially inside CGIs) and the array encompasses probes with an average coverage of 1.9 CpG sites per promoter.
As an initial step, we evaluated whether DNA methylation changes in T2D were sufficient to distinguish the diabetic from the control samples when comparing complete methylation profiles. For this, an unsupervised hierarchical clustering was performed which placed the diabetic islet samples as one self-contained group distinct from the control samples in the resulting dendrogram (Supplementary Figure S2). This outcome highlights two facts: first, diabetic DNA methylation profiles are more similar to each other than to the methylation profile of any control sample, indicating the possibility of a T2D-specific DNA methylation profile; second, the existence of a single branch containing the five diabetic samples shows that the DNA methylation changes are sufficiently pronounced (even in the unfiltered data sets) to distinguish diabetic from control samples. To further substantiate the correlation of variations of DNA methylation with T2D, we performed a principal component analysis on the data set (Materials and methods; Supplementary Figure S3) and compared the resulting principal components with the T2D state in a point-biserial correlation (Lowry, 1998–2011). We found that PC #1 and PC #3, together accounting for 37% of the variance in the data set, correlate well with T2D state (Supplementary Figure S3). This is indicative of distinct, T2D-specific changes in the epigenome of pancreatic islets.
Following this initial data analysis, we identified T2D-related methylation changes by filtering the data sets for CpG sites showing significant differences in DNA methylation levels between control and T2D groups (cf. Materials and methods and Supplementary Table S2). The results of the filtering are shown as a heatmap (Figure 1A). The depicted methylation profiles discriminate between control samples (left side, indicated by yellow bar above heatmap) and T2D samples (right side, indicated by blue bar). It is already apparent from the above data that there are marked DNA methylation changes in T2D islets. The number of differentially methylated CpG loci in T2D islets is in the same range as reported for other non-malignant conditions analysed with the same technology platform (19 in T1D-related nephropathy; Bell et al, 2010); 84 and 360, respectively, in analyses of ageing in cells and tissue specimens (Bork et al, 2009; Rakyan et al, 2010).
Figure 1.
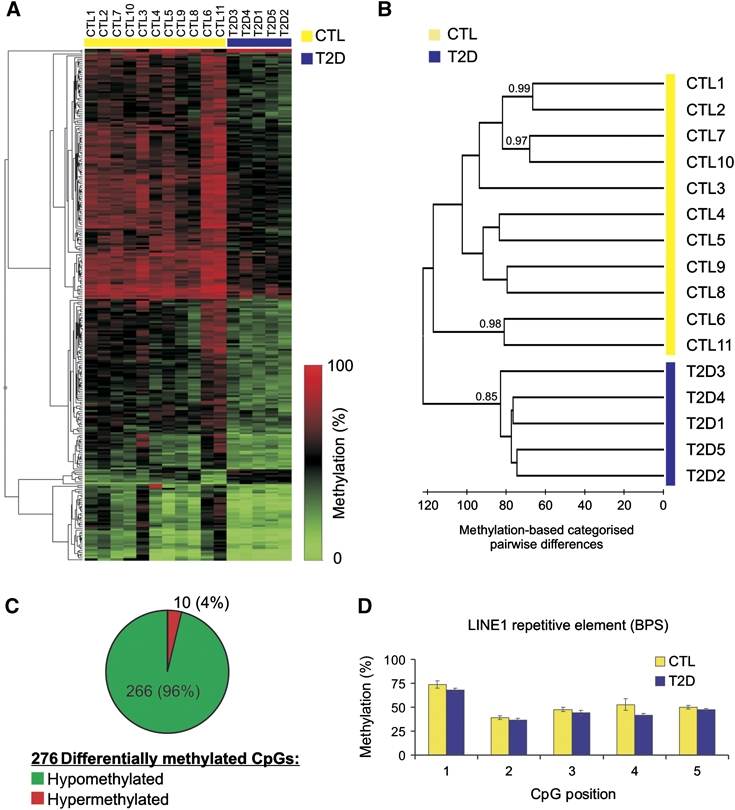
Hierarchical profile-based clustering and evaluation of T2D islet DNA methylation. (A) A heatmap of the differentially methylated CpG loci shows distinct patterns separating data sets obtained from CTL (yellow line above heatmap) and T2D samples (blue line). (B) Supervised clustering based on the differentially methylated CpG loci separates T2D from control samples. As an indication of statistical significance, bootstrap values (>0.7) are shown next to the branches. Note that the order of samples in the heatmap in Figure 1A follows the one established by supervised clustering. (C) Pie chart depicting the 276 CpG sites showing differential methylation between T2D and control samples (see also Supplementary Table S2). Note the high proportion of hypomethylation events as compared with hypermethylation events. (D) LINE-1 repetitive element DNA methylation for CTL and T2D samples by BPS (n=11 for CTL; n=5 for T2D).
We then set out to evaluate the descriptive power of the CpG sites in the filtered data set to differentiate diabetic from non-diabetic specimens in sample-wise comparisons. We therefore extracted the methylation values for each sample and performed a supervised clustering (Figure 1B, cf. Materials and methods). As expected, the resulting dendrogram shows that samples group together in two clusters containing exclusively control (CTL, yellow bar) or diabetic (T2D, blue bar) samples, indicating that class identity (CTL, T2D) is the most important separation criterion (Figure 1B, left-most branch). To assess clustering confidence in an unbiased way and to overcome inherently subjective visual interpretation of the results depicted in the heatmap (Figure 1A), a bootstrapping analysis was carried out after dendrogram computation (cf. Materials and methods). The obtained bootstrap of 0.85 indicates significant statistical support for the bipartite distribution between diabetic and non-diabetic samples based on the analysis of the CpGs contained in the filtered data set. The occasional high bootstrap values adjoined to sample pairs illustrate similarities in the DNA methylation profiles of these samples. These data demonstrate that human pancreatic islets undergo DNA methylation alterations in T2D that are discernible by means of DNA methylation profiles.
T2D-related aberrations encompass mostly promoter-specific DNA hypomethylation
The above experiments enabled us to collect the first comprehensive DNA methylation data set for T2D human islets. We identified 276 CpG sites, affiliated to 254 gene promoters, showing differential methylation between normal and diseased samples (Figure 1C; Supplementary Table S2). Strikingly, 266 of these 276 CpGs (96%) showed decreased methylation levels, while only 10 were hypermethylated (Figure 1C). This unexpected finding contrasts with the well-known DNA methylation changes observed in cancers, where gene-specific hypomethylation and hypermethylation are more or less evenly distributed (Jones and Baylin, 2007). With respect to global DNA methylation, cancers generally display hypomethylation (Jones and Baylin, 2007; Tost, 2010), primarily in repetitive DNA.
To test whether the observed T2D-related changes are gene specific or whether they reflect global hypomethylation in the genome of islet cells, we measured DNA methylation levels of the repetitive LINE-1 element in control and diabetic samples with bisulphite pyrosequencing (BPS). Analysing DNA methylation of LINE-1, which makes up ∼20% of human genome, provides an accurate estimate of global DNA methylation changes (Yang et al, 2004). Figure 1D shows that repetitive elements are not differentially methylated in T2D, as substantiated by the strong overlap between CTL and T2D samples, indicating the absence of global hypomethylation in T2D islets.
As an additional quality control, we examined the set of 276 differentially methylated CpG sites for overlap with known single-nucleotide polymorphism (SNP) positions to be excluded from further data analysis. We found no overlap with the reported 180 potentially problematic CpG sites contained in the Humanmethylation27 array (Bell et al, 2011) and therefore continued our analyses with the full set of 276 CpGs.
BS validation of T2D-related differential DNA methylation
To corroborate the observed Infinium measurements (cf. Figure 1 and Supplementary Table S2), we applied BPS and in some cases conventional BS to randomly selected, differentially methylated CpG sites. In all 19 cases tested, differential DNA methylation at the respective CpG sites was confirmed by BPS (Figure 2; Supplementary Figure S4). Where implemented, BS also confirmed the DNA methylation profiling data (Figure 2A; Supplementary Figure S4A). Figure 2A depicts an exemplary analysed gene, ALDH3B1, for which the Infinium data were confirmed by BS and BPS. Additional validated genes are shown in Figure 2B (CASP10) and Figure 2C (PPP2R4 alias PP2A). We discovered two differentially methylated CpG sites inside a CGI in the IGF2/IGF2AS locus. The differential methylation of one of the CpGs in this region was tested and confirmed by BPS (Figure 2D). Further examples are shown in Supplementary Figure S4. A direct comparison of methylation percentages obtained by the Infinium Methylation assay and BPS (Figure 2E) yielded a highly positive correlation (Pearson's correlation R=0.927) confirming the validity of the data. BPS analysis of three negative controls constituting high (>90%), intermediate (∼40%) and low (<10%) levels of DNA methylation showed expectedly no differential DNA methylation between sample groups (Supplementary Figure S4M–O). Of note, CpG sites neighbouring the Infinium-assayed site are often encompassed in the BPS experiments (cf. Figure 2 and Supplementary Figure S4); these adjacent CpG sites also displayed similar DNA methylation levels. Hence, our validation experiments indicate that individual CpGs from the Infinium Methylation array can be used as informative markers for the methylation status of the respective surrounding regions. Taken together, BPS and BS results confirm the methylation values obtained from the Infinium-based assay, thus validating the T2D-specific DNA methylation profiles.
Figure 2.

Validation of aberrantly methylated loci in T2D islets. (A) An exemplary locus, ALDH3B1, is shown. Methylation data for the indicated CpGs obtained with the Infinium assay as well as by conventional bisulphite sequencing (BS), and by bisulphite pyrosequencing (BPS), are compared. Detected DNA methylation levels at other loci (B–D) are also in good agreement between the Infinium methylation assay (bar charts above the gene scale) and BPS used for validation (bar charts below gene scale) (see Supplementary Figure S4 for further examples). CpG islands are indicated by a green line below gene scale. (E) Validation of the Infinium DNA methylation data by BPS. Methylation values obtained by Infinium assay and BPS show high correlation (Pearson's correlation coefficient R=0.927); (n=3–8 for CTL; n=3–5 for T2D).
Genomic features associated with differential DNA methylation in T2D
Having shown that the observed differential DNA methylation in T2D is gene-specific rather than global (cf. Figure 1D), we set out to determine whether this aberrant DNA methylation is (a) located within or outside CGIs, (b) prevalent in distinct promoter classes (that are based on CpG density) or (c) correlated with specific regulatory elements.
Previous work that investigated the localisation of differential DNA methylation in cancer and different tissues has suggested that substantial DNA methylation differences occur in CGI shores (Doi et al, 2009; Irizarry et al, 2009). We used bioinformatic tools to determine the distance of differentially methylated CpGs to the nearest CGI. Utilising CGI prediction (Bock et al, 2007), we found that ∼50% of the differentially methylated CpG sites are located >2 kb from the nearest CGI (‘other CpGs’ in Figure 3A and B and Supplementary Table S3). Additionally, about one quarter of CpGs resides in CGI shores (1–2000 bp from a CGI border) while another quarter of CpG sites was located inside CGIs (Figure 3A; Supplementary Tables S2 and S3). A more detailed representation of the CpG distribution reveals the sites of differential methylation inside the CGI shores to be located preferentially close to the CGIs (bar chart in Figure 3B). This distribution of differentially methylated sites inside CGI shores is similar to regions showing differential methylation in cancer (c-DMRs; Irizarry et al, 2009), between tissues (t-DMRs; Irizarry et al, 2009) and during differentiation/cell reprogramming (r-DMRs; Doi et al, 2009). However, an overall comparison of differential methylation locations between these DMRs (Doi et al, 2009; Irizarry et al, 2009) and our data (Figure 3A and B) shows significant disparity, with a predominance of DNA methylation changes >2 kb away from CGIs (‘other CpGs’ in Figure 3A and B and Supplementary Table S3), thus distinguishing the localisation of the DNA methylation profile in T2D islets from those found in tumours, between tissues and in stem cells.
Figure 3.

Classification of differentially methylated CpG sites and regulatory element analysis of affected genes. (A) Classification of the CpG sites according to their location relative to CpG islands. Most of the differentially methylated CpG sites are affiliated to genes not possessing a CpG island or are >2 kb away from the nearest CpG island (termed ‘other CpGs’); only about one quarter of the affected CpGs is located inside a CGI (‘CGI’) and about one quarter is located in CGI shores (‘CGI shore’), that is, distance to the CGI is between 1 and 2000 bp. The observed distribution of differentially methylated CpGs is significantly different from that of the Infinium array (χ2 goodness-of-fit test P<2.2 × 10−16). (B) Representation as bar plot with spatial resolution of the CGI shore shows that differential methylation in CGI shores occurs predominantly close to CGI borders (0–0.5 and 0.5–1 kb). (C) Classification of the promoters affiliated to the CpG sites based on the promoter CpG content. The observed distribution of promoter classes among genes associated with differentially methylated CpGs is significantly distinct from that of the entirety of genes represented by CpG sites on the Infinium array (χ2 goodness-of-fit test P=7.1 × 10−14) (for numerical representation of classifications in A and C: cf. Supplementary Table S3). (D) Direct plotting of CpG ratio (cf. Materials and methods) shows T2D-associated differential methylation (blue bars) predominantly in low CpG density promoters. (E) Prediction of putative TF binding sites using the set of low CpG differentially methylated gene promoters (CpG ratio <0.5) with the Pscan transcription factor motif analysis software. Statistically significant overrepresented binding sites have been found for members of the GATA transcription factor family.
We next analysed the occurrence of these differentially methylated CpG sites in relation to CpG density of the affiliated gene promoters. Saxonov et al (2006) discovered a bipartite distribution of gene promoters with minor overlap between both classes when categorising promoter sequences by means of their CpG content. They discovered that promoters are either relatively depleted of CpG sites (low CpG promoters, LCPs) or enriched in CpG sites (high CpG promoters, HCPs), preferentially around the transcription start site (TSS). Weber et al (2007) introduced a third class of promoters called intermediate CpG promoters (ICPs), to account for the overlap between the classes mentioned above. They also developed precise classification criteria for the three classes, which we utilised in an adapted form: in this study, we considered positions −700 to +500 relative to the TSS for the promoter classification (cf. Materials and methods), since both CpG density and differential DNA methylation are distributed symmetrically around the TSS (Saxonov et al, 2006). Figure 3C shows that most of the differentially methylated CpG sites from T2D islets are located in LCP and ICP class promoters. ICP class promoters have been described as regions of dynamic DNA methylation changes (Weber et al, 2007), while LCP class promoters have seldomly been investigated. Their role as sites of hypomethylation in T2D therefore remains to be explored.
Looking directly at the CpG ratio of the promoter sequences (cf. Materials and methods), the Infinium array resembles the distribution found in a comprehensive set of human promoters (cf. Weber et al, 2007 and Figure 3D, red bars). The CpG ratio of the promoters displaying differential methylation in T2D islets (Figure 3D, blue bars) is clearly distinct from the probes represented on the Infinium array and also contrasts with the CpG distribution found in promoters throughout the genome. Following the distribution of the blue bars in Figure 3D, it becomes apparent that most T2D-linked differentially methylated promoters are relatively depleted in CpG sites (CpG ratio <0.5) whereas only few CpG-rich promoters gain or lose methylation in T2D islets.
In terms of genomic features, we third evaluated whether the observed aberrant DNA methylation in T2D islets correlated with specific regulatory elements. The use of computational approaches to extract functional meaning from the annotated genome has gained importance in recent years. Roider et al (2009) underscored the need to first separate gene promoters on the basis of their CpG content before analysing the presence of enriched transcription factor-binding motifs. These authors found that promoters depleted of CpG sites often contain tissue-specific transcription factor-binding sites. Therefore, we first extracted CpG-depleted promoters by selecting all differentially methylated promoters with a CpG ratio lower than 0.5 (Saxonov et al, 2006). We then used the extracted set of 172 CpG-poor promoters as a starting point to detect putative transcriptional regulatory signals using the Pscan (Zambelli et al, 2009) software and the TRANSFAC transcription factor motifs database (Matys et al, 2006). As shown in Figure 3E and Supplementary Figure S5A, we identified significant enrichment of GATA transcription factor family binding sites, namely GATA1 and GATA2 binding sites, as well as a common binding site assigned to all GATA proteins (P-values for enrichment ranged from 2.38 × 10−15 to 3.28 × 10−13; Supplementary Figure S5). We then repeated this analysis using all differentially methylated LCP and ICP promoters (cf. Supplementary Tables S2 and S3) to enlarge the above mentioned set of 172 ‘CpG ratio <0.5’ promoters, yielding similar results. To increase precision and to assess validity of our predictions, we performed a negative control by conducting the detection routine described above with 100 sets of 172 promoters randomly sampled from CpG-depleted genes (CpG ratio <0.5) that are not differentially methylated but present on the Infinium array. Importantly, the highest significance of enrichment (best P-value) encountered in the searches with these random gene promoter sets never exceeded the best P-value obtained with the set of differentially methylated gene promoters (Supplementary Figure S5B). Combined, these data reveal a statistically significant enrichment of GATA transcription factor-binding motifs in our set of promoters (CpG ratio <0.5) differentially methylated in T2D islets.
Differential DNA methylation observed in T2D islets is not inducible by high glucose
The differential DNA methylation observed in T2D islets might be either causative or instead secondary to the hyperglycaemia inherent to diabetes. Recent reports have shown that transient exposure of aortic endothelial cells to high glucose induced persistent epigenetic changes (DNA methylation, H3K4 mono-methylation, H3K9/H3K16 acetylation; El-Osta et al, 2008; Pirola et al, 2011). To determine whether high-glucose stress modifies DNA methylation in islets, we exposed non-diabetic human islets to 28 mM glucose for 72 h and subsequently analysed DNA methylation. Similar culture conditions (30 mM glucose, 48 h) have been demonstrated to result in determinate DNA methylation changes in vascular cells (Pirola et al, 2011). We chose CpG sites that displayed alterations in DNA methylation in T2D islets and which we previously confirmed by BPS (cf. Figure 2 and Supplementary Figure S3). Islets from the same donor incubated for the same duration at 6 mM glucose served as control. As shown in Figure 4A, the high-glucose treatment did not significantly affect DNA methylation of 16 CpGs in 13 gene promoters tested, particularly in relation to the magnitude of DNA methylation change observed in T2D islets (Figure 4A, right). Figure 4B illustrates representative genes, namely SIRT6, GRB10 and CHAC1, for which methylation changes measured between islets exposed to high glucose and control islets were minor compared with the absolute levels of methylation at these positions. To assess global DNA methylation after high-glucose stress, we analysed the repetitive LINE-1 element by BPS. As shown above for the gene-specific methylation changes, we did not find significant alterations in LINE-1 methylation between islets incubated under normal glucose conditions and those exposed to high-glucose concentrations (Figure 4C). Together, these findings suggest that the T2D-related differential DNA methylation is probably not secondary to hyperglycaemia.
Figure 4.

Differential DNA methylation after glucose stress. Differential DNA methylation was measured by BPS after culture in high glucose-containing medium (28 mM glucose for 72 h). (A) Differences of DNA methylation in high glucose treated islets versus islets from the same donor incubated under normal glucose conditions. The CpG sites analysed in this experiment correspond to the genomic positions interrogated by the Infinium assay that were demonstrated to display differential DNA methylation in T2D islets (cf. Figure 2 and Supplementary Figure S4). Gene symbols listed twice represent genes with two Infinium probes both of which were tested for glucose-induced differential DNA methylation. Light and dark grey bars indicate two independent experiments. For comparison, the differences in DNA methylation in T2D versus non-diabetic islets as determined by BPS are given (cf. Figure 2). (B) Three examples depicting BPS results in the two independent experiments. (C) Global DNA methylation, as assessed by repetitive element LINE-1, shows no significant changes after exposure of islets to high glucose levels (norm. Glc: 6 mM glucose in medium; high Glc: 28 mM glucose in medium).
The T2D-related differential methylation pattern is a feature of diseased islets
An important question regarding the differential DNA methylation detected in T2D islets is whether these epigenetic changes are unique for pancreatic islets or whether they are part of a general pattern occurring in different tissues.
To examine this, we analysed peripheral blood leukocytes from 12 T2D patients and compared their DNA methylation profiles with 12 non-diabetic controls matched for age and BMI (Supplementary Table S4). The two groups had significantly different glycaemia and HbA1c (Supplementary Table S4). We quantified DNA methylation levels of 11 CpG loci in 9 gene promoters that exhibited differential methylation in T2D islets and were validated by BPS (cf. Figure 2 and Supplementary Figure S4). We detected no significant differences in DNA methylation levels between control and T2D blood samples in any of the 11 CpG loci analysed (Supplementary Figure S6). This impelled us to examine DNA methylation broadly, using again the Infinium Humanmethylation27 array already employed for the islets. Differently from the islets, however, both groups displayed very similar DNA methylation profiles (linear regression R=0.9989; Supplementary Figure S7A). As a matter of fact, we detected almost no T2D-related differential DNA methylation in blood surpassing the cutoff (±15%, P<0.01). Only one CpG locus in the promoter of the CIDEB gene showed significant hypermethylation (+15.9%, P=0.00066). CIDEB influences obesity and liver steatosis and is a negative regulator of insulin sensitivity (Li et al, 2007). Regarding the 276 CpG loci differentially methylated in T2D islets, these showed very limited DNA methylation changes between non-diabetic and T2D blood cells (Supplementary Figure S7B).
In conclusion, the T2D-related DNA methylation changes detected in pancreatic islets are essentially absent from whole blood DNA. Indeed, we detected no T2D-related differential methylation satisfying our significance criterion except for a single CpG in the promoter of CIDEB that, in turn, displays no significant differential DNA methylation in T2D pancreatic islets. These data suggest that (1) the methylation pattern observed in islets is apparently not a general phenomenon; (2) blood is not a suitable surrogate tissue for studying T2D-related epigenetic changes in pancreatic islets.
Differential DNA methylation can be correlated with changes in gene expression in a subset of genes
Only few studies to date have reported gene expression profiling in human pancreatic islets. An example of such a study is the work by Bhandare et al (2010) that described gene expression in islets from 31 non-diabetic donors. It was of interest to examine whether the differential DNA methylation observed in our study occurred in promoters of expressed genes identified by Bhandare et al (2010) or whether it was associated with inactive genes that may become transcriptionally activated when hypomethylated. By comparison of Entrez gene IDs, 196 genes (266 probes) from the reported expression array could be matched to our set of differentially methylated genes (for expression data cf. Supplementary Table S2 columns AP ff.). Expression of all matched genes was above background levels, asserting that differential methylation occurs at promoters of genes that are active in islets. As expected, their absolute expression levels covered several orders of magnitude with no significant correlation between expression and promoter methylation level (cf. Supplementary Table S2), that is, highly active genes in islets are not necessarily devoid of DNA methylation in their promoters. Our comparison of methylation and expression data therefore strongly suggests that the observed changes in promoter DNA methylation levels are not restricted to silent or lowly expressed genes but are also occurring in promoters of expressed genes. In T2D islets, we observed hypomethylation in the promoters of these active genes.
A recent study assessed gene expression in different islet cell types including the insulin-producing β-cells (Dorrell et al, 2011). A comparison showed that 240 of our 254 genes are covered by the microarray used by these authors. In all, 170 of these genes have a positive presence call in β-cells. This indicates that the majority of the genes we detected as differentially methylated in T2D islets are expressed in non-diabetic β-cells to a sufficient amount to be reliably detected by microarrays, that is, these are genes actively transcribed in β-cells.
We next examined whether the altered promoter DNA methylation observed in T2D as compared with control islets could be correlated with changes in gene expression. For this purpose, we analysed gene expression in six islet preparations from male islet donors (three non-diabetic and three T2D) using an Affymetrix HG-U133A microarray (cf. Materials and methods). Of the 254 genes showing differential DNA methylation in T2D (cf. Figure 2, Supplementary Figure S4 and Supplementary Table S2), 181 had associated probes on the microarray, with a total of 320 matches between Infinium and expression assays (due to multiple methylation or expression probes affiliated to the same genes; Supplementary Table S4). Forty-one of these genes showed significant differential gene expression between CTL and T2D islets (BH-adjusted P<0.05), of which about 80% (34 genes) displayed an anticorrelation between changes in DNA methylation and expression, for example, a decrease of DNA methylation coinciding with an increase of expression (Figure 5A and B; Supplementary Table S6). This suggests that, at least for a subset of T2D-modified genes, changes in the promoter DNA methylation correlate inversely with the transcriptional activity of the gene. Regarding genomic features, the distribution of the differentially methylated CpGs in the promoters of these genes relative to the TSS is similar to the distribution of all 276 differentially methylated CpGs in our study (Supplementary Figure S8A). Also, the distribution of promoter classes is comparable between the 254 differentially methylated genes and the subgroup displaying an inverse correlation of methylation and expression (Supplementary Figure S8B; cf. also Figure 3B and Supplementary Table S3).
Figure 5.

Correlation between DNA methylation and gene expression changes in T2D islets. (A) Out of 41 genes that are differentially methylated and differentially expressed (adjusted P<0.05; cf. Supplementary Table S4), 34 genes, ≈83%, show anticorrelation between methylation and expression (33 are hypomethylated and overexpressed, PER2 is hypermethylated and its expression is decreased in T2D islets; Supplementary Table S5) while 7 (≈17%) show a positive correlation. (B) Boxplots of DNA methylation levels and gene expression in 41 genes showing differential expression in T2D islets. Prevalent promoter hypomethylation is accompanied by overexpression of the associated genes. (C) Three exemplative genes with indicated promoters, methylation probes and expression probes. ALDH3B1 methylation is measured at promoter #2 and is correlated with expression data of a common exon that is present in all but one of the ALDH3B1 mRNAs. In case of hypomethylation uniquely occurring at promoter #2 (promoters #1 & #3 are not interrogated by the Infinium assay), the effect of this promoter hypomethylation on gene expression would be abated by the unchanged expression of isoforms transcribed from the other two promoters. In the case of PER2, methylation at promoter #1 and expression of the long isoform is assessed. *There are several Affymetrix probes for CASP10 that demonstrate differential expression but fail to reach the significance criterion of P<0.05 (BH-corrected P of Student's t-test; cf. Supplementary Table S4).
The observation that only a subset of genes displays such inverse correlation between DNA methylation and expression changes could be partially explained by the fact that comparisons between promoter DNA methylation and expression of the affiliated genes are often hampered by positioning of array probes. For instance, the Infinium array achieves an average coverage of 1.9 CpG sites per promoter. However, due to the occasionally high number of alternative promoters in one gene, in most cases the Humanmethylation27 array does not interrogate all promoters of a given gene (see Figure 5C for examples). On the other hand, microarray probes are mainly positioned in common exons that are present in most or all alternative transcripts of a gene, thereby potentially masking expression changes in one or few isoforms (Dai et al, 2005).
Consequently, our data are consistent with other studies that were either unable to establish a connection between differential DNA methylation and gene expression (Illingworth et al, 2008) or found a small but significant set of genes with anticorrelation between changes in promoter methylation and gene expression (Rakyan et al, 2008). Our work is thus in agreement with the emerging notion that, for most genes, the methylation–expression relationship cannot be broken down to the simple formula that decreased methylation equals elevated expression (Eckhardt et al, 2006; Illingworth et al, 2008; Suzuki and Bird, 2008).
Biological pathways associated with differential DNA methylation in diabetic islets: at least some of the uncovered differentially methylated genes possess pathophysiological significance for β-cell function
To determine the biological relevance of the differential DNA methylation in pancreatic islets, we followed a two-winged strategy consisting of a combined computational and literature approach on one hand, and on the other hand in-vitro experiments to uncover the functions of genes that were previously unknown in the context of pancreatic islets and/or β-cells.
For the first approach, we assessed whether the differentially methylated genes have any overlap or other association with known T2D risk genes. Then, we carried out an Ingenuity Pathway Analysis (IPA; Figure 6A) to identify pathways that are epigenetically affected in T2D islets according to our methylation profiling data. This was augmented by a manual search for the differentially methylated genes in scientific literature reporting on the general biology as well as T2D-related functions of these genes or the pathways they are part of (Figures 6 and 7). For the second approach, we knocked down expression of several genes by RNA interference and tested the functional consequence of their depletion in β-cells (Figure 8). For two selected genes, we explored their functional role more extensively in isolated β-cells and human islets (Figure 9).
Figure 6.

Biological pathways associated with differentially methylated loci. (A) Ingenuity pathway analysis reveals canonical pathways significantly enriched in T2D pancreatic islets. Measure of significance is indicated by Benjamini-Hochberg-corrected P-value (abbreviated as ‘BH P-value’ in the x axis label of the depicted chart). (B) Manual curation of the biological functions associated with the differentially methylated genes unravelled three broad categories of cellular responses that might be affected in T2D islets. Some of these genes are part of processes leading to β-cell dysfunction and cell death while others seem to facilitate β-cell survival and adaptation to the T2D environment.
Figure 7.

Biological functions of differentially methylated genes in T2D islets from the existing literature. Biological functions are highlighted for a selection of the differentially methylated loci that we hypothesise to play important roles in diabetic islets. Note that several genes involved in different processes can negatively affect insulin secretion. The referenced figures at the end of the descriptions of several genes depict the methylation analyses of these genes' promoters.
Figure 8.

Screening of biological function of eight differentially methylated genes in β-cells. Cell death was evaluated in INS-1E cells transfected with different specific siRNAs and treated 2 days later with 0.5 mM palmitate or 25 μM CPA for 16 h (n=3–7). siRNAs were used targeting Mknk2 (MAP kinase-interacting serine/threonine kinase 2), Guca2b (guanylate cyclase activator 2B), Per2 (period homologue 2), Sfrs2ip (splicing factor, arginine/serine-rich 2, interacting protein), Chac1 (ChaC, cation transport regulator-like 1), Nr4a1 (nuclear receptor subfamily 4, group A, member 1), Bcl2 (B-cell lymphoma 2) and Niban. Cell survival was measured by neutral red staining and data were expressed as cell death relative to cells transfected with negative siRNA (siCTL) and cultured under control condition (CTL). *P<0.05 against cells under the same treatment transfected with siCTL, #P<0.05 as indicated.
Figure 9.

Role of differentially methylated NIBAN and CHAC1 in β-cell apoptosis. (A) NIBAN mRNA expression in human islets treated for 24 h with oleate or palmitate (left panel) and with synthetic ER stressors thapsigargin (THA), tunicamycin (TUN) or brefeldin (BRE). CTL denotes control. Results represent mean±s.e. of 3–5 independent experiments. (B) Niban mRNA expression in rat INS-1E cells transfected with negative siRNA (siCTL) or Niban siRNA (siNiban) and treated for 16 h with palmitate or cyclopiazonic acid (CPA). Results represent mean±s.e. of 3–4 independent experiments. (C) Percentage of apoptosis in siRNA-transfected INS-1E cells after 16 h of palmitate or CPA treatment. Lower panel: western blot quantification of cleaved caspase 3 as an additional apoptosis marker. The blot is representative of four independent experiments. (D) CHAC1 mRNA expression in human islets treated for 24 h with oleate or palmitate (left panel) and with synthetic ER stressors. Results represent mean±s.e. of 3–5 independent experiments. (E) Chac1 mRNA expression in rat INS-1E cells transfected with negative siRNA (siCTRL) or Chac1 siRNA (siChac). (F) Apoptosis in siRNA-transfected INS-1E cells after 16 h of palmitate or CPA treatment. Results represent mean±s.e. of 4–10 independent experiments. Lower panel: western blot quantification of cleaved caspase 3 as an additional apoptosis marker. The blot is representative of four independent experiments. *P<0.05 against untreated (CTL) cells, #P<0.05 by paired ratio t-test.
At the outset, we analysed the differentially methylated genes with regard to their reported functions and the biological pathways they are part of. To date, large-scale GWAS have uncovered a number of T2D susceptibility loci (Saxena et al, 2007; Scott et al, 2007; Sladek et al, 2007; Zeggini et al, 2007, 2008). Several of these genes have a role in β-cells. Therefore, we compared our data with the known T2D risk genes and asked whether these loci are also targets of epigenetic dysregulation. None of the established T2D susceptibility loci (Voight et al, 2010) revealed significant differential DNA methylation in our analyses. The only gene previously detected in GWAS as a potential T2D-associated locus is GRB10 (Di Paola et al, 2006; Rampersaud et al, 2007), an imprinted gene (cf. Supplementary data) that displays hypomethylation in diabetic islets (Supplementary Figure S4L). We did find aberrantly methylated genes with similar biological functions to the known T2D susceptibility genes (Supplementary Table S2). For example, KCNQ1 and KCNJ11 (SNP variants of which are associated with higher T2D incidence; Scott et al, 2007) were not significantly altered in their methylation levels but three other potassium channel genes, KCNE2, KCNJ1 and KCNK16, had changed promoter methylation states (Supplementary Table S2). Other examples of T2D susceptibility loci for which we identified genes with related functions are SLC30A8 and CDKAL1. In our data sets, SLC30A8 was not differentially methylated but two other zinc transporter genes, SLC39A5 and ZIM2, were hypomethylated. For CDKAL1 we found its methylation state unchanged in T2D islets, while its target gene CDK5R1 exhibited pronounced hypomethylation (Supplementary Figure S4C). In summary, although the promoter methylation of established T2D susceptibility loci was unchanged in our profiling approach, other genes with similar biological function (e.g., potassium and zinc transporters) or part of the same regulatory networks (e.g., CDK5R1 in the CDK5 pathway and GRB10 in the insulin signalling pathway) displayed aberrant DNA methylation.
The analyses described above found only few common T2D candidate genes among the differentially methylated genes uncovered in this study. This could imply that T2D pathogenesis in islets is partially mediated by previously unappreciated genes. To decipher their roles in the context of T2D islets, as a first step we performed an IPA to determine which canonical pathways were overrepresented in our set of genes (Figure 6A). Inflammation-related processes were highly enriched, in particular the acute phase response and IL-8 signalling. Other enriched pathways, such as apoptosis and death receptor signalling, emphasise the role of β-cell loss in T2D. Enrichment for pathways involved in metabolism and internal and external cell structure (e.g., actin cytoskeleton and integrin signalling) may be indicative of altered islet function and architecture.
Second, we performed an extensive manual curation according to a previously described β-cell-targeted annotation (Kutlu et al, 2003; Ortis et al, 2010). In partial agreement with the IPA, we found these genes to fall into three broad categories: (1) genes related to β-cell dysfunction and death, (2) genes potentially facilitating the adaptation of the pancreatic islets to the altered metabolic situation in T2D and (3) genes whose role in disease pathogenesis remains to be unearthed (Figure 6B). The adaptation-related gene category contains few metabolism-associated genes (e.g., HK1, FBP2; Figure 6B, right part, Figure 7) and many more genes involved in signal transduction or encoding hormones, growth factors (e.g., EGF, FGF1, IGF2/IGF2AS; Figure 7), or transcription factors involved in important regulatory networks (for instance, FOXA2/HNF3B, PAX4 and SOX6) (Figure 6B, right part, Figure 7). In the β-cell dysfunction and death category, there were hypomethylated genes related to DNA damage and oxidative stress (e.g., GSTP1, ALDH3B1; Figure 7), the endoplasmic reticulum (ER) stress response (NIBAN, PPP2R4, CHAC1), and apoptosis (CASP10, NR4A1, MADD; Figure 6B, left part, Figure 7).
Some genes of interest from the highlighted categories are depicted in Figure 7. Their annotated functions provide possible explanations of how the epigenetic dysregulation of these genes in diabetic islets is connected to T2D pathogenesis. Numerous genes that were identified by our methylation profiling approach have been functionally implicated in insulin secretion. Examination of the available literature on the function of these genes revealed three aspects of insulin secretion with which they interfere: some of these genes influence the expression of the insulin gene, like MAPK1 and SOX6, or its post-translational maturation, like PPP2R4 (cf. Figure 7 and references therein). Others can deregulate the process of insulin secretion itself (SLC25A5, Ahuja et al, 2007; RALGDS, Ljubicic et al, 2009) or influence synthesis as well as secretion (vitronectin, Kaido et al, 2006). A third group of differentially methylated genes affects (i) signalling processes in the β-cell leading to insulin secretion or (ii) glucose homeostasis in β-cells, thereby modulating insulin response upon stimulation. GRB10 (Yamamoto et al, 2008), FBP2 and HK1 (Figure 7) are examples for these genes. Additional genes found in our study have been implicated in the β-cells' capability to secrete insulin, though the mechanisms have not yet been fully established. The putative functions of these genes indicate a potential epigenetic impact on insulin secretion at multiple levels, namely signalling, expression/synthesis and secretion.
Functional analysis of differentially methylated genes reveals their involvement in specific biological processes in islets and β-cells
We next sought to provide experimental proof that altering the activity of some of the differentially methylated genes translates into functional effects that impinge on β-cell viability. We first performed a screening for cellular outcome after knockdown of eight selected differentially methylated genes. β-Cell survival was examined under basal conditions and after exposure to the saturated fatty acid palmitate or the ER stress-inducer cyclopiazonic acid (CPA, a specific inhibitor of the sarcoendoplasmic reticulum Ca2+ ATPase pump (SERCA; Pahl et al, 1996)) following silencing of MKNK2 (MAP kinase-interacting serine/threonine kinase 2 isoform 2), GUCA2B (guanylate cyclase activator 2B), PER2 (period isoform 2), SFRS2IP (splicing factor arginine/serine-rich 2, interacting protein), NR4A1, BCL2, CHAC1 and NIBAN by RNA interference (Figure 8). Knockdown of BCL2, an antiapoptotic protein (Gurzov and Eizirik, 2011), NR4A1, an immediate-early response gene, and MKNK2, a kinase that functions downstream of p38 and ERK, controlling cell survival and negatively regulating global protein synthesis, resulted in increased basal cell death in the rat β-cell line INS-1E (Figure 8). NR4A1, BCL2 and NIBAN also were antiapoptotic under lipotoxic conditions, as their knockdown sensitised β-cells to palmitate. Four of the selected genes exerted proapoptotic functions in β-cells, illustrated by the improved survival following their knockdown and palmitate or CPA exposure. These were the clock gene PER2, GUCA2B, a ligand for guanylate cyclase receptor C that transduces signals for insulin secretion and growth and differentiation, SFRS2IP, a protein required for pre-mRNA splicing, and CHAC1.
The biological role of two of these differentially methylated genes with previously unknown function in β-cells, namely NIBAN and CHAC1, was further studied. NIBAN and CHAC1 have been suggested to be part of the ER stress response in other cell types (Sun et al, 2007; Mungrue et al, 2009). Such ER stress response can initiate apoptosis and has been implicated in β-cell demise in diabetes (Eizirik et al, 2008).
To explore the role of NIBAN and CHAC1 in ER stress in greater depth, human islets were treated with the physiological ER stressors oleate and palmitate (Cunha et al, 2008) or the synthetic ER stressors thapsigargin (THA), tunicamycin (TUN) or brefeldin (BRE). Expression of NIBAN was induced about two-fold by the saturated fatty acid palmitate (Figure 9A, column 3) but not with oleate (Figure 9A, column 2), which is a less potent inducer of ER stress (Cunha et al, 2008). A similar induction of expression was observed for CHAC1 when islets were treated with palmitate (Figure 9D, column 3) but not with oleate (Figure 9D, column 2). The synthetic ER stressors THA, TUN and BRE also increased NIBAN and CHAC1 mRNA expression in human islets (Figure 9A and D, right panels). THA, which causes ER calcium depletion by blocking the SERCA pump (Pahl et al, 1996), TUN, which blocks glycosylation of nascent proteins (Hickman et al, 1977), and especially BRE, which inhibits ER-to-Golgi transport (Misumi et al, 1986), induced NIBAN and CHAC1 gene expression. The magnitude of ER stress induced by these three chemicals and palmitate, as measured by ATF3, CHOP and BiP mRNA expression (Supplementary Figure S9), was closely correlated with the NIBAN and CHAC1 induction.
We next determined the functional role of NIBAN and CHAC1 expression on the outcome of ER stress in β-cells. For this purpose, we exposed INS-1E cells to palmitate or CPA. ER stress induced by these agents increased Niban mRNA expression (columns 1, 3, 5 in Figure 9B). As shown in Figure 9B, expression of Niban is efficiently diminished by a specific small interfering RNA (siRNA) (siNiban; compare columns 1 and 2, 3 and 4, 5 and 6). RNAi-mediated knockdown of Niban increased apoptosis induced by palmitate (Figure 9C; columns 3 and 4) as well as by CPA (columns 5 and 6 in Figure 9C). This is mirrored by an elevated caspase 3 activation (Figure 9C, lower panel). Taken together, the increase of Niban expression during ER stress and augmented apoptosis after its knockdown indicates an antiapoptotic role for Niban during the β-cell ER stress response.
Chac1 mRNA expression was also induced by palmitate and CPA in INS-1E cells (Figure 9E). Chac1 knockdown by siRNA (siChac) significantly reduced its expression upon incubation with palmitate (Figure 9E, columns 3 and 4) or with CPA (columns 5 and 6 in Figure 9E). We next analysed whether the knockdown of Chac1 affected ER stress-induced apoptosis stimulated by palmitate and CPA (Figure 9F). We found that Chac1 knockdown protected β-cells from apoptosis (cf. columns 3, 4 and 5, 6 in Figure 9F), which was confirmed by a lessened activation of caspase 3 (Figure 9F, lower panel). The induction of Chac1 expression by ER stressors and the fact that its knockdown protected β-cells from ER stress-induced apoptosis suggest that Chac1 is involved in apoptosis triggered after the different branches of ER stress response fail to re-establish ER homeostasis in β-cells.
These experiments investigated two novel genes with opposite effects on ER stress-induced apoptosis in human islets, CHAC1 and NIBAN. Expression of both genes is strongly induced by ER stress, elicited by different agents. While NIBAN protects β-cells against apoptosis, the activation of CHAC1 indicates failure of the processes restoring β-cell functionality and heralds ER stress-induced apoptosis (Eizirik et al, 2008; Eizirik and Cnop, 2010).
In conclusion, by looking at genes known to be involved in T2D/islet pathology as well as by loss of function experiments, our analyses indicate that we did not pick up some irrelevant variations in DNA methylation levels but that the uncovered epigenetic changes indeed affect genes vital for β-cell function and survival. These genes might therefore be important, and provide previously unappreciated players in T2D pathogenesis.
Discussion
In this study, we performed the first comprehensive DNA methylation profiling of human T2D pancreatic islets identifying 276 differentially methylated CpG sites that are affiliated to 254 genes. These generated data illustrate, for the first time, the epigenetic dimension of T2D in islets and how this is associated with transcriptional alterations. The pathophysiological significance of eight genes found to be differentially methylated in their promoters was validated by RNAi experiments.
It has been suggested that progressively occurring DNA methylation errors lead to diminished gene responsiveness to external stimuli and might thus contribute to the development of T2D (Gallou-Kabani and Junien, 2005). Our findings of prevalent promoter hypomethylation in T2D islets are indicative of active biological processes involved in adaptation to the diabetic environment as well as biological pathways associated with β-cell dysfunction and apoptosis (Figures 6B and 7). The functional relevance of some of the differentially methylated genes in β-cells was documented by screening for β-cell survival/death following RNAi and subsequent exposure to stresses relevant to T2D (Figure 8). Given the increased evidence that ER stress-induced apoptosis is one of the mechanisms of β-cell loss in T2D (Eizirik et al, 2008), it was of interest to further assess the biological functions of two putative ER stress-related genes that we found to be hypomethylated in T2D islets, namely NIBAN and CHAC1. We observed that these two genes are upregulated by synthetic ER stressors and by the more physiologically relevant saturated fatty acid palmitate in human islets, while knockdown of their expression by specific RNAi demonstrated their modulatory role in apoptosis (cf. Figure 9). While NIBAN protects against ER stress-induced apoptosis, CHAC1 seems to contribute to cell death. The hypomethylation observed at both genes could be explained by competing proapoptotic and antiapoptotic processes during ER stress response in diabetic islets. NIBAN is a negative regulator of translation initiation factor eIF2α (Sun et al, 2007). Therefore, its hypomethylation may indicate an attempt to re-establish ER homeostasis by reduction of protein synthesis (Eizirik et al, 2008). Pending the outcome of these attempts, ER stress-induced apoptosis may be triggered by CHAC1 and other proapoptotic genes.
An important question with regard to epigenetic changes is: are the observed DNA methylation changes reflected in gene activity? By comparing the obtained DNA methylation profiles with microarray gene expression data, we were able to determine that a high proportion of genes in whose promoter T2D-related differential DNA methylation occurs are actively transcribed in pancreatic islets. A comparison with expression data of islet cell types (Dorrell et al, 2011) showed that most of the differentially methylated genes are expressed in β-cells. This allowed us to conclude that T2D-related aberrant DNA methylation partially happens in the promoters of active genes. One has to keep in mind though that the expression studies in islets as well as in the β-cells analysed non-diabetic material. We observed mostly DNA hypomethylation in diabetic islets, not infrequently accompanied by elevated gene expression. Therefore, it can be assumed that the T2D-related hypomethylation leads, in part, to the induction of formerly silent genes.
Regarding differential gene expression in T2D islets, we observed an inverse correlation between differential promoter methylation and differential gene expression for a subset of genes. It is worth mentioning that for a significant proportion of differentially methylated genes, no statistically significant differential expression was observed (compare Supplementary Tables S4 and S5). This may partially be due to the incompatibilities between methylation assays and the design of expression microarrays mentioned above that hamper in-depth comparisons between methylation and expression. However, for many genes the link between differential methylation and gene activity may be quite complex. Methylation of the cytosine base changes the topology of the major DNA groove, which may affect the binding of many transcription factors and DNA-binding proteins. Supporting this possibility, binding of two transcription factors whose target genes are differentially methylated in T2D islets has been described as methylation sensitive, namely CTCF (which binds to IGF2/IGF2AS, Bell and Felsenfeld, 2000; Filippova et al, 2005) and YY1 (which binds to ZIM2, Kim et al, 2003). Additionally, we found several genes encoding chromatin-associated proteins and transcription factor genes differentially methylated in T2D islets (cf. Figure 6B and Supplementary Table S2). Should the expression of these genes and/or their binding motifs be influenced by differential DNA methylation in T2D islets, it might further add to the complexity of the methylation–expression relationship. This could potentially explain the observations made in this study as well as others (Eckhardt et al, 2006; Illingworth et al, 2008; Suzuki and Bird, 2008) that the relationship between DNA methylation and gene expression is rather complex.
In terms of genomic features, we detected T2D-associated differential DNA methylation mainly in LCP and ICP, while HCPs are underrepresented. Analysing LCP and a subset of ICP genes (CpG ratio <0.5), we discovered GATA family transcription factors that are predicted to regulate a significant subset of these genes. Interestingly, the GATA transcription factor family members are critical regulators in endocrine development, function and pathologies (Viger et al, 2008). The physiological roles of many differentially methylated loci in T2D can be described as genes responding to (external) stimuli and to stress. Of note, Saxonov et al (2006) found that a disproportionately high percentage of genes affiliated to these biological functions possess promoters with a low CpG density. This might indicate a general principle with regard to the promoter class of the differentially methylated gene loci: while in chronic diseases such as T2D and lupus (Javierre et al, 2010) LCP genes are overrepresented, in diseases associated with cellular overgrowth (such as cancer) there is increased prevalence of HCP and relatively few LCP genes (Richter et al, 2009; Martin-Subero et al, 2009a, 2009b). Further studies are required to test this intriguing possibility.
A key issue is whether the methylation changes we report play a causal role in T2D or are secondary to the diabetic condition. Indeed, the hypomethylation observed in oxidative stress, ER stress and apoptotic pathways may result from chronic exposure to the stressful metabolic environment of T2D, for example, high-glucose concentrations (Cnop et al, 2005). An interesting example in this respect is CASP10: we found significant hypomethylation in its promoter (Figure 2B) and since caspase 10 is inducible by advanced glycation end products (Lecomte et al, 2004; Obrenovich and Monnier, 2005), this hypomethylation could be indicative of gene activation caused by chronically elevated blood glucose levels and consequently heightened non-enzymatic glycosylation events. Interestingly, experimental exposure of islets from non-diabetic donors to high-glucose concentrations (28 mM) for 72 h did not induce differential DNA methylation in any of the genes that display methylation changes in T2D islets. Even though these findings do not exclude an impact of chronic exposure to stressors like hyperglycaemia on the islet epigenome, they do make it unlikely that the observed alterations in DNA methylation are merely a consequence of relatively short metabolic insults. By inferring from the functions of the differentially methylated genes, it is possible that some of the identified epigenetic changes play a role in the progressive islet dysfunction in T2D, that is, they have potentially been acquired at different time points during pathological decline. Thus, the hypomethylation observed at some genes, like CASP10, may be a consequence of T2D and severe and long-lasting hyperglycaemia. On the other hand, some genes, for example, those related to insulin secretion, may have obtained alterations in promoter methylation much earlier. For instance, defects in acute insulin response to glucose (AIRg) are among the earliest impairments and even precede the onset of pre-diabetic IGT (Bogardus and Tataranni, 2002; Fukushima et al, 2004; Leahy, 2005; Bunt et al, 2007). If aberrant AIRg arises from epigenetic aberrations at genes involved in insulin secretion (which is an established function for many genes in our study), these defects should manifest ahead of clinical T2D development. Whether such changes can be designated ultimately causal for the decline into T2D will remain to be proven. Overall, from the data at hand the changed methylation in the promoters of some genes identified in our study might thus be consequential and represent reactions to the diabetic environment. At other genes, the methylation aberrations could be interpreted to play a causal role, driving the islet dysfunction and T2D pathogenesis. Future, large-scale studies involving multiple stages of T2D development will be needed to elucidate the role of the epigenetic changes in the various stages of T2D pathogenesis. Due to medical ethics, it is impossible to obtain repeated pancreatic biopsies. Therefore, these studies will need to rely on surrogate tissues that remain to be validated. The availability of the presently described human islet methylation profiling will allow future search and validation of surrogate tissues.
However, identification and validation of tissues whose T2D-related DNA methylation profiles can serve as a proxy for pancreatic islets might prove difficult. The apparent absence of significant T2D-related differential DNA methylation in blood raises the possibility that T2D-related epigenetic aberrations are tissue-specific although more tissues will have to be screened to substantiate this. The finding of almost no differential DNA methylation in blood cells of T2D patients versus the significant changes in pancreatic islets implies the question whether the observed blood–islet difference is attributable to the different lifespan of the cells, for blood cells being days to months while β-cells have a lifespan of many decades (Cnop et al, 2010). The validity of blood for epigenetic analysis has, however, been established by previous studies that uncovered differential methylation in DNA isolated from whole blood of individuals that were prenatally exposed to famine (Heijmans et al, 2008; Tobi et al, 2009). Further investigations into T2D-related epigenetic changes in surrogate tissues for pancreatic islets might elucidate their causative role or expose them as consequences of the disease.
A possible confounding factor for the identification of T2D-related epigenetic profiles is the medication that T2D patients receive and that may influence gene regulation. Histone deacetylase inhibitors (HDACi), for example, have been demonstrated to increase insulin sensitivity in muscle and liver and partially thwart diabetic nephropathy and retinopathy (Christensen et al, 2011). It is possible that diabetes drugs like rosiglitazone, a PPAR-γ agonist, or metformin will alter gene activity patterns and confound profiling approaches. Adequately powered epigenetic profiling studies of surrogate tissues that consider the patients' medication may yield new insight of relevance for drug-based T2D therapy.
As acknowledged by McCarthy and Zeggini (2009), the >40 gene variants of T2D susceptibility genes known to date cannot fully explain T2D predisposition. Our study points to the involvement of epigenetic alterations in T2D thus underscoring the previously established contribution of lifestyle habits to its development. Combining the advantages of genome-scanning techniques and epigenome analyses might pave the way to better comprehend the pathogenesis of T2D. It will be of great interest to examine SNPs in the differentially methylated genes in T2D described in this study since the interplay between SNPs and differential (allele-specific) DNA methylation has recently been described (Shoemaker et al, 2010). Linked to the topic of allele-specific DNA methylation, it is noteworthy that a number of the genes found to display differential methylation are also reported to be imprinted (Supplementary data). It could hence be speculated that at least a partial loss of imprinting occurs in T2D islets.
In conclusion, we report the first comprehensive and detailed analysis of epigenetic changes in T2D, specifically an altered DNA methylation profile in the pancreatic islets of T2D patients with a major preponderance of hypomethylation in sequences outside CGIs. These aberrant methylation events affect over 250 genes, a subset of which is also differentially expressed. The dysregulation of these genes in T2D may notably be linked to β-cell functionality, cell death and adaptation to metabolic stress. Examination of two genes identified by methylation profiling, NIBAN and CHAC1, revealed their biological functions in distinct processes of the ER stress response. Furthermore, our data highlight genes belonging to biological processes whose involvement in T2D is not yet fully understood, such as inflammation and ion transporters/channels/sensors. Importantly, it can be envisaged that the uncovered DNA methylation changes might be, on one part, indicative of reactions of the islet cells to the diabetic condition and on another part, might be causal of T2D. A challenge in the future is to provide further evidence for the primary effects of methylation changes in the diabetic condition. Taken together, our DNA methylation study on human islets thus lays the ground to further unravel the biological complexity of T2D and outlines an unexpected level of epigenetic regulation in islets, which must be taken into account in future studies aiming to understand the pathogenesis of T2D.
Materials and methods
Isolation of pancreatic islets
From September 2004 to November 2009, pancreatic islets of Langerhans were isolated from pancreata of 5 T2D and 11 non-diabetic male cadaveric donors in Pisa, Italy, with the approval of the local Ethical Committee, and as described previously (Del Guerra et al, 2005). Glucose-induced insulin secretion was measured as described. The diagnosis of T2D was based on the previously described clinical criteria (ADA, 1997; Genuth et al, 2003).
Islet purity and β-cell content
Reliable purity assessment for diabetic islets is challenging. In T2D, the degranulated β-cells contain less insulin and zinc (Ostenson et al, 2006) and the qualitative dithizone assessment (which targets zinc) therefore underestimates T2D islet purity. Hence, we used EM to analyse islet purity in some of the samples used in the methylation profiling after >2 days in culture (Supplementary data; n=3 for T2D and non-diabetic samples, respectively) as described (Welsh et al, 2005).
Blood sampling
After obtaining written informed consent, blood was sampled from 12 male T2D patients and 12 male age- and BMI-matched controls in K2EDTA tubes.
Methylation profiling using the Infinium assay
Genomic DNAs were isolated from pancreatic islets using the Wizard® SV Genomic DNA kit (Promega Corp.) and from 200 μl of blood using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany). In all, 1 μg of genomic DNA was treated with sodium bisulphite using the EZ DNA Methylation™ kit (Zymo Research) according to manufacturer's procedure, respecting the recommended alterations in protocol for consecutive Infinium methylation analysis. Methylation status of 27 578 distinct CpG sites was analysed using the Infinium HumanMethylation27 BeadChip array (Illumina, Inc., San Diego) according to manufacturer's protocol. Data acquisition was done with the Illumina BeadArrayReader and quality control was performed using the Methylation module (version 1.0.5) of the GenomeStudio™ software (version 1.0.2.20706). Data normalisation was omitted due to resulting in essentially unchanged data sets (background normalisation) or due to inapplicability (Loess, quantile, Bayes) to methylation datasets because of their heteroscedasticity (Cancer Genome Atlas Research Network, 2011). For quality control, a standard quality analysis was performed for each array assessing Bisulphite conversion efficiency, hybridisation efficiency and specificity, single base extension rate, target removal as well as staining for negative and non-polymorphic probes (GenomeStudio Controls Dashboard). Data handling, comparisons and so on were performed with the Methylation module of the GenomeStudio software package (Illumina), MS Excel, R 2.8.0-2.11.1, Openstat and MicroarrayAnalyse v1.0 (Graessler, 2008).
CGI and promoter class annotation
Annotations for the Infinium HumanMethylation27 provided by Illumina were augmented with respect to (i) the position of the analysed CpG relative to the nearest CGI (inside a CGI, in CGI shore or >2 kb away from an island) and (ii) the promoter class of the gene affiliated to the evaluated CpG (high/intermediate/low CpG content promoter). For all annotations, the human genome build 36.1 (hg18, March 2006) provided the basis.
For classification of the CpG position relative to CGIs, the CGI map provided by Bock et al (2007) (combined epigenetic score >0.5; genome assembly hg18/NCBI36) was used as reference; the CpGs were classified into three categories according to Irizarry et al (2009). Designation of the CpGs is as follows: ‘inside CGI’ if the CpG was inside a CGI, ‘CGI shore’ if the CpG was located within a 2-kb region around a CGI and ‘other CpG’ otherwise (distance to closest CGI >2 kb).
Promoters of the gene loci affiliated to the analysed CpG sites were classified according to their CpG content. First, we extracted sequences ranging from positions −700 to +500 relative to the TSS from UCSC genome browser database, then calculated the CpG ratio and the GC content of these sequences in sliding windows of 500 with 5 bp offsets. For classification criteria, we followed the definition by Weber et al (2007). In short, promoters were defined as HCPs if at least one 500 bp window contained a CpG ratio >0.75 together with a GC content >0.55 whereas in LCPs no 500 bp window reached a CpG ratio of at least 0.48. All promoters not fitting in either of the above promoter classes were termed ICPs. Five differentially methylated gene promoters (and a total of 54 gene promoters on the Infinium array) could not be classified due to great distance to TSS or lack of annotation.
Hierarchical clustering
For unsupervised hierarchical clustering, the data sets were filtered for probes/CpG sites with a detection P-value of <0.05. CpGs not fitting into this criterion in at least one sample were excluded from further analysis. The clustering was performed with the average β-values (equals methylation percentage/100) of 24 422 CpGs for each sample using the cluster analysis function in GenomeStudio applying Euclidean distance metrics.
For UPGMA (Unweighted Pair Group Method using Arithmetic averages) clustering, the data sets for computation were assembled as follows: a group-wise ∣Δβ∣>0.15 and P<0.01 (Mann–Whitney) were set as filtering criteria. In all, 276 probes fitted into these criteria. The methylation percentage of the CpG site corresponding to each probe was extracted for each sample. Then, the methylation values were categorised into 10 equal classes and imported into MEGA4 (Tamura et al, 2007) in which the phylogenetic analysis was conducted. The dendrogram was computed using the UPGMA method applying the ‘number of differences’ model (Sneath and Sokal, 1973). To determine the validity of the sample clustering based on the methylation data, a bootstrap test (10 000 sampling steps) was used to calculate the percentage of replicate trees in which the associated samples clustered together (Felsenstein, 1985). Bootstrap values of 0.7 or higher were considered significant and are shown next to the branches in Figure 1B.
Conventional BS and BPS
In all, 750 ng genomic DNA was subjected to bisulphite conversion using the Epitect® Bisulfite Kit (Qiagen) or the EZ DNA Methylation kit (Zymo Research) according to manufacturer's protocol. Elution of the converted DNA was generally performed with 26 μl elution buffer and 8 μl of the eluted DNA were used as template in subsequent PCRs. To ensure sufficient amount of product, amplifications were generally performed as nested PCRs.
PCR and sequencing primers for BPS were deduced using the PyroMark® Assay Design 2.0 software (Qiagen). Primers for pre-amplification and conventional BS were designed manually or with the help of BiSearch primer design tool (http://bisearch.enzim.hu) and evaluated using the GeneRunner software (v3.05 Hastings Software, Inc.). Primers were obtained from Eurogentec S.A. or Sigma-Aldrich Corp. Biotinylated primers were ordered HPLC purified, all other primers desalted. PCR and sequencing primers are listed in Supplementary Table S7.
The pre-amplification PCR was conducted with primers (see EF, ER primers in Supplementary Table S7) amplifying 400–720 bp spanning the CpG of interest and additionally as many as possible neighbouring CpG sites. CpG sites in the annealing positions of the PCR primers were avoided where possible; otherwise primers were ordered with ambiguities at the respective positions. PCR was conducted with 3 mM MgCl2, 1 mM of each dNTP, 12% (v/v) DMSO, 500 nM of each primer and optionally 500 mM Betaine in heated-lid thermocyclers under the following conditions: 95°C 3:00; 25 × (94°C 0:30; 51°C 0:40; 72°C 1:30); 72°C 5:00 and cooled afterwards to 10°C.
For conventional BS, nested PCRs were conducted as described above using 35 PCR cycles and a decreased elongation time of 1 min (for PCR primers cf. Supplementary Table S7). PCR products were separated on a 1% agarose gel and single bands were cut and eluted from the gel. Cloning of the nested PCR products was performed with TOPO TA Cloning® kit (Invitrogen Corp.) and the plasmids were sequenced by Genoscreen.
For pyrosequencing, a nested PCR was performed with primers designed by the PyroMark Assay Design software (Qiagen) using the HotStarTaq PCR kit (Qiagen) according to manufacturer's recommendations. Reactions were performed in heated-lid thermocyclers under the following conditions: 95°C 15:00; 45 × (94°C 0:30; 55°C 0:30; 72°C 0:30); 72°C 10:00 and finally cooled to 8°C. Sample preparation and pyrosequencing reactions were performed with the Pyromark Q24 system (Qiagen). For validation of Infinium assay-derived DNA methylation by BPS, usually three to five randomly chosen samples from each group (CTL and T2D) were analysed and DNA methylation degrees were averaged.
Microarray gene expression studies
In all, 100 ng total RNA was prepared and hybridised onto Affymetrix Human HG-U133A chips, according to the protocols described in the Affymetrix GeneChip® Expression Analysis Manual (Affymetrix, Santa Clara, CA). Chips were scanned in an Affymetrix GeneChip Scanner 3000 and their quality verified with the Microarray Analysis Suite 5.0 (Affymetrix) software and functions from the R/Bioconductor affy and affyPLM packages (Gautier et al, 2004; Bolstad et al, 2005) (R version 2.8.0; Bioconductor version 2.3). Raw gene expression data were normalised using the Robust Multiarray Average (RMA) method described by Irizarry et al (2003) implemented in the affy package. Gene expression ratios were calculated using the limma package by fitting a linear model on each gene (Smyth, 2005).
For comparison of differentially methylated loci with expression profiles of non-diabetic islets (Bhandare et al, 2010), we utilised the extracted expression data (downloaded from ArrayExpress; accession number E-MTAB-191; http://www.ebi.ac.uk/microarray-as/ae/).
INS-1E cell and human islet culture
The rat insulin-producing INS-1E cell line (a kind gift from Professor C Wollheim, Centre Medical Universitaire, Geneva, Switzerland) was cultured in RPMI-1640 (with 2 mM GlutaMAX-I) containing 5% FBS (Asfari et al, 1992; Cnop et al, 2007a) and used at passages 59–73. Human islets were isolated from 11 organ donors (age 69±6 years; body mass index 26±1 kg/m2) in Pisa, Italy, as described above. The islets were cultured in Ham's F-10 medium containing 6.1 or 28 mM glucose as previously described (Cunha et al, 2008; Igoillo-Esteve et al, 2010; Ladriere et al, 2010). The percentage of β-cells, assessed in dispersed islet preparations following staining with mouse monoclonal anti-insulin antibody (1:1000, Sigma) and donkey anti-mouse IgG Rhodamine (1:200, Jackson Immuno Research Europe, Soham, Cambridgeshire, UK), was 53±3%. Palmitate and oleate (Sigma-Aldrich, Schnelldorf, Germany) were dissolved in 90% ethanol, and used at a final concentration of 0.5 mM in the presence of 1% BSA (Cunha et al, 2008). The chemical ER stressors THA (diluted in DMSO and used at a final concentration of 1 μM), CPA (diluted in DMSO and used at final concentration of 25 μM), TUN (diluted in PBS and used at a final concentration of 5 μg/ml) and BRE (diluted in ethanol and used at a final concentration of 0.1 μg/ml) were obtained from Sigma-Aldrich. The control condition contained similar dilutions of vehicle.
Assessment of β-cell death
Cell death was measured using the neutral red kit (Sigma, TOX4) following manufacturer's instructions. Briefly, cells were incubated with 17 mM neutral red for 3 h at 37°C, washed and the dye extracted for absorbance measurement in a spectrophotometer. Quantitative evaluation of INS-1E cell apoptosis was done by fluorescence microscopy following staining with the DNA-binding dyes propidium iodide (5 μg/ml) and Hoechst 33342 (5 μg/ml) (Cnop et al, 2007a). Caspase 3 activation was assessed by western blot, as previously described (Gurzov et al, 2009), using anti-cleaved caspase 3 antibody (1:1000; from Cell Signaling, Beverly, MA, USA).
RNA interference
Genes were knocked down using siRNA. The Niban siRNA was SMARTpool (L-080179-01 from Dharmacon, Chicago, IL, USA). Stealth RNAi (Invitrogen, Carlsbad, CA) was used for CHAC1 (RSS324745), MKNK2 (RSS312292), PER2 (RSS329646), BCL2 (RSS340652) and NR4A1 (RSS330510). The siRNAs for SFRS2IP (s161636) and GUCA2B (s133784) were Silencer Select from Ambion (Austin, TX). A negative control (siCTL) of 21 nucleotide duplex RNA with no known sequence homology was obtained from Qiagen. Lipid–RNA complexes were formed in Optimem1 with 1.5 μl Lipofectamine 2000 (Invitrogen) to 150 nM siRNA and added at a final concentration of 30 nM siRNA for transfection as described (Cunha et al, 2008). Transfected cells were cultured for 2 days and subsequently treated. The achieved knockdown was 74.8±10.4% for MKNK2 (P=0.03), 86.4±3.5% for GUCA2B (P=0.003), 34.2±12.9% for PER2 (P=0.098), 58.3±5.9% for SFRS2IP (P=0.04), 72.4±3.2% for CHAC1 (P=0.02), 72.5±3.6% for NR4A1 (P=0.001), 47.2±3.8% for Bcl-2 (P=0.01) and 50.7±4.8% for NIBAN (P=0.02), as measured at the mRNA level, except for Bcl-2 that was measured by western blotting.
Real-time PCR
Poly(A)+ RNA was isolated and reverse transcribed as previously described (Chen et al, 2001). The PCR was done in 3 mM MgCl2, 0.5 μM forward and reverse primers, 2 μl SYBR Green PCR master mix (Qiagen) and 2 μl cDNA. Standards for each gene were prepared using appropriate primers in a conventional PCR. The samples were assayed on a LightCycler instrument (Roche Diagnostics, Mannheim, Germany) and their concentration was calculated as copies per μl using the standard curve (Overbergh et al, 1999). The expression level of the gene of interest was corrected for the expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh, for INS-1E cells) or β-actin (for human islets). PCR primers are listed in Supplementary Table SXXX. The different treatments utilised in the study did not change expression of the housekeeping gene (Igoillo-Esteve et al, 2010; data not shown).
Statistics
Significance of group-wise differences in DNA methylation profiles was measured by Mann–Whitney rank-sum test, P<0.01 was considered significant. Taking into account inter-individual differences in methylation levels and following Illumina Inc. recommendations, a 15% group-wise difference of methylation levels was set as a cutoff additional to PMann–Whitney<0.01. Correlation between methylation values by Infinium and BPS was computed using Pearson's correlation test. Differences between distributions (CpG localisation, promoter class) were calculated with χ2 goodness-of-fit test (R 2.11.1); P-values were estimated from the resulting χ2 value. Significance of gene expression differences was tested by Bayes moderated t-test and P-values were FDR adjusted using Benjamini-Hochberg method (R package limma; Smyth, 2005); adj. P<0.05 was considered significant. Differences in glucose-stimulated insulin secretion, methylation as analysed by BPS and gene expression as analysed by RT–qPCR were assessed by Student's t-test, P<0.05 was considered significant. Data are represented as mean±s.d. unless indicated otherwise.
Accession codes
DNA methylation data sets for pancreatic islets and whole blood have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession numbers GSE21232 and GSE34008, respectively.
Supplementary Material
Acknowledgments
We thank Drs Francoise Fery and Bernard Corvilain for their precious help in the recruitment of T2D patients and healthy controls. This work was supported by grants from the Action de Recherche Concertée (ARC04/09-309 and ARC 2010-2015–Eizirik), the Interuniversity Attraction Poles (IUAP P6/28 and P6/40), the F.N.R.S. (1.1.371.10.F, 1.2.136.06.F, 3.4559.05, 3.4.542.09.F, 7.4559.05 and 3.4583.12), the Belgian Télévie, the European Union (FP6 and FP7) projects Eurodia (LSMH-CT-2006-518153), CEED3 (FP7-HEALTH-F2-2008-223211) and Naimit (HS-F2-2009-241447) and the EFSD/Novo Nordisk Programme 2011.
Author contributions: MV designed experiments, performed research and interpreted data. Infinium methylation assays were done by EC and SD; MV and MD interpreted the Infinium methylation assay data. Bisulphite genomic sequencing and bisulphite pyrosequencing were done by MV, RD and EC; MV, UV, MD and NN conducted bioinformatic and statistical analyses. MB and PM performed the array analysis. DAC conducted gene functional analyses. For clinical samples: selection was done by SDG, PM and MC; preparation was done by SDG, MB, PM; characterisation was done by DC, MIE, SDG, MM, PM and MC. MV, MC and DLE designed experiments and interpreted data. MC, DLE and FF directed the study. MV, MNN, MC, DLE and FF wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- ADA (1997) Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 20: 1183–1197 [DOI] [PubMed] [Google Scholar]
- Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E (2007) Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem 282: 33583–33592 [DOI] [PubMed] [Google Scholar]
- Anuradha CV, Balakrishnan SD (1999) Taurine attenuates hypertension and improves insulin sensitivity in the fructose-fed rat, an animal model of insulin resistance. Can J Physiol Pharmacol 77: 749–754 [PubMed] [Google Scholar]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB (1992) Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130: 167–178 [DOI] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405: 482–485 [DOI] [PubMed] [Google Scholar]
- Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA (2010) Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK (2011) DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 12: R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandare R, Schug J, Le Lay J, Fox A, Smirnova O, Liu C, Naji A, Kaestner KH (2010) Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res 20: 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava SK, Sachdev HS, Fall CH, Osmond C, Lakshmy R, Barker DJ, Biswas SK, Ramji S, Prabhakaran D, Reddy KS (2004) Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N Engl J Med 350: 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL (2009) Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 1: 177–200 [DOI] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, Lengauer T (2007) CpG island mapping by epigenome prediction. PLoS Comput Biol 3: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogardus C, Tataranni PA (2002) Reduced early insulin secretion in the etiology of type 2 diabetes mellitus in Pima Indians. Diabetes 51(Suppl 1): S262–S264 [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Collin F, Brettschneider J, Simpson K, Cope L, Irizarry RA, Speed TP (2005) Quality assessment of Affymetrix GeneChip data. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S (eds). Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer: New York. pp 33–47 [Google Scholar]
- Bork S, Pfister S, Witt H, Horn P, Korn B, Ho AD, Wagner W (2009) DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 9: 54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun T, Franklin I, St-Onge L, Biason-Lauber A, Schoenle EJ, Wollheim CB, Gauthier BR (2004) The diabetes-linked transcription factor PAX4 promotes b-cell proliferation and survival in rat and human islets. J Cell Biol 167: 1123–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunt JC, Krakoff J, Ortega E, Knowler WC, Bogardus C (2007) Acute insulin response is an independent predictor of type 2 diabetes mellitus in individuals with both normal fasting and 2-h plasma glucose concentrations. Diabetes Metab Res Rev 23: 304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Pandozy G, Mastronardi F (2008) Evaluating epigenetic landmarks in the brain of multiple sclerosis patients: a contribution to the current debate on disease pathogenesis. Prog Neurobiol 86: 368–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL (2001) Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1β-exposed human and rat islet cells. Diabetologia 44: 325–332 [DOI] [PubMed] [Google Scholar]
- Christensen DP, Dahllöf M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, Grunnet LG, Mandrup-Poulsen T (2011) Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med 17: 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, Gunter JH, de Koning EJ, Walls GV, Gray DW, Johnson PR, Hansen BC, Morris JF, Pipeleers-Marichal M, Cnop I, Clark A (2010) The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 53: 321–330 [DOI] [PubMed] [Google Scholar]
- Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, Eizirik DL (2007a) Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem 282: 3989–3997 [DOI] [PubMed] [Google Scholar]
- Cnop M, Vidal J, Hull RL, Utzschneider KM, Carr DB, Schraw T, Scherer PE, Boyko EJ, Fujimoto WY, Kahn SE (2007b) Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care 30: 677–682 [DOI] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2): S97–S107 [DOI] [PubMed] [Google Scholar]
- Contreras JL, Smyth CA, Bilbao G, Eckstein C, Young CJ, Thompson JA, Curiel DT, Eckhoff DE (2003) Coupling endoplasmic reticulum stress to cell death program in isolated human pancreatic islets: effects of gene transfer of Bcl-2. Transpl Int 16: 537–542 [DOI] [PubMed] [Google Scholar]
- Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, Moore F, Rasschaert J, Cardozo AK, Bellomo E, Overbergh L, Mathieu C, Lupi R, Hai T, Herchuelz A, Marchetti P, Rutter GA, Eizirik DL, Cnop M (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 121: 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33: e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S, Marchetti P (2005) Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54: 727–735 [DOI] [PubMed] [Google Scholar]
- Di Paola R, Ciociola E, Boonyasrisawat W, Nolan D, Duffy J, Miscio G, Cisternino C, Fini G, Tassi V, Doria A, Trischitta V (2006) Association of hGrb10 genetic variations with type 2 diabetes in Caucasian subjects. Diabetes Care 29: 1181–1183 [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP (2009) Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 41: 1350–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Lin CF, Canaday PS, Fox AJ, Smirnova O, Bonnah R, Streeter PR, Stoeckert CJ Jr, Kaestner KH, Grompe M (2011) Transcriptomes of the major human pancreatic cell types. Diabetologia 54: 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Magi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R et al. (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38: 1378–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29: 42–61 [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Cnop M (2010) ER stress in pancreatic beta cells: the thin red line between adaptation and failure. Sci Signal 3: pe7. [DOI] [PubMed] [Google Scholar]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205: 2409–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J (1985) Confidence limits in phylogenies: an approach using the bootstrap. Evolution 39: 783–791 [DOI] [PubMed] [Google Scholar]
- Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD, Disteche CM (2005) Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell 8: 31–42 [DOI] [PubMed] [Google Scholar]
- Florez JC (2008) Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia 51: 1100–1110 [DOI] [PubMed] [Google Scholar]
- Fukushima M, Suzuki H, Seino Y (2004) Insulin secretion capacity in the development from normal glucose tolerance to type 2 diabetes. Diabetes Res Clin Pract 66(Suppl 1): S37–S43 [DOI] [PubMed] [Google Scholar]
- Gallou-Kabani C, Junien C (2005) Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 54: 1899–1906 [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315 [DOI] [PubMed] [Google Scholar]
- Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, Nathan D, Palmer J, Rizza R, Saudek C, Shaw J, Steffes M, Stern M, Tuomilehto J, Zimmet P (2003) Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 26: 3160–3167 [DOI] [PubMed] [Google Scholar]
- Graessler J (2008) Microarray Analyse v1.0
- Guo J, Qian Y, Xi X, Hu X, Zhu J, Han X (2010) Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol Cell Biochem 338: 283–290 [DOI] [PubMed] [Google Scholar]
- Gurzov EN, Eizirik DL (2011) Bcl-2 proteins in diabetes: mitochondrial pathways of beta-cell death and dysfunction. Trends Cell Biol 21: 424–431 [DOI] [PubMed] [Google Scholar]
- Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, Eizirik DL (2009) Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ 16: 1539–1550 [DOI] [PubMed] [Google Scholar]
- Hart AW, Baeza N, Apelqvist A, Edlund H (2000) Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 408: 864–868 [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 105: 17046–17049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Kulczycki A Jr, Lynch RG, Kornfeld S (1977) Studies of the mechanism of tunicamycin in hibition of IgA and IgE secretion by plasma cells. J Biol Chem 252: 4402–4408 [PubMed] [Google Scholar]
- IDF (2009) IDF Diabetes Atlas, 4th edn IDF Executive Office [Google Scholar]
- Igoillo-Esteve M, Marselli L, Cunha DA, Ladriere L, Ortis F, Grieco FA, Dotta F, Weir GC, Marchetti P, Eizirik DL, Cnop M (2010) Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53: 1395–1405 [DOI] [PubMed] [Google Scholar]
- Iguchi H, Ikeda Y, Okamura M, Tanaka T, Urashima Y, Ohguchi H, Takayasu S, Kojima N, Iwasaki S, Ohashi R, Jiang S, Hasegawa G, Ioka RX, Magoori K, Sumi K, Maejima T, Uchida A, Naito M, Osborne TF, Yanagisawa M et al. (2005) SOX6 attenuates glucose-stimulated insulin secretion by repressing PDX1 transcriptional activity and is down-regulated in hyperinsulinemic obese mice. J Biol Chem 280: 37669–37680 [DOI] [PubMed] [Google Scholar]
- Iguchi H, Urashima Y, Inagaki Y, Ikeda Y, Okamura M, Tanaka T, Uchida A, Yamamoto TT, Kodama T, Sakai J (2007) SOX6 suppresses cyclin D1 promoter activity by interacting with b-catenin and histone deacetylase 1, and its down-regulation induces pancreatic b-cell proliferation. J Biol Chem 282: 19052–19061 [DOI] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, Humphray S, Cox T, Langford C, Bird A (2008) A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 6: e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Kido Y, Asahara S, Matsuda T, Shibutani Y, Koyanagi M, Kasuga M (2009) Effect of intrauterine undernutrition during late gestation on pancreatic beta cell mass. Biomed Res 30: 325–330 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Sohn D, Totzke G, Schulze-Osthoff K (2006) Caspase-10 in mouse or not? Science 312: 1874. [DOI] [PubMed] [Google Scholar]
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O'Hanlon TP, Rider LG, Jacinto FV, Lopez-Longo FJ, Dopazo J, Forn M, Peinado MA, Carreno L, Sawalha AH, Harley JB, Siebert R, Esteller M et al. (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 20: 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128: 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaido T, Yebra M, Cirulli V, Rhodes C, Diaferia G, Montgomery AM (2006) Impact of defined matrix interactions on insulin production by cultured human beta-cells: effect on insulin content, secretion, and gene transcription. Diabetes 55: 2723–2729 [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Kimura Y, Kimura H, Niki I (2006) L-cysteine inhibits insulin release from the pancreatic beta-cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes 55: 1391–1397 [DOI] [PubMed] [Google Scholar]
- Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine JC, de Cabo R, Cohen HY (2008) Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett 582: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136: 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebede M, Favaloro J, Gunton JE, Laybutt DR, Shaw M, Wong N, Fam BC, Aston-Mourney K, Rantzau C, Zulli A, Proietto J, Andrikopoulos S (2008) Fructose-1,6-bisphosphatase overexpression in pancreatic b-cells results in reduced insulin secretion: a new mechanism for fat-induced impairment of b-cell function. Diabetes 57: 1887–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, Steinberg HA, Chaibub Neto E, Kleinhanz R, Turner S, Hellerstein MK, Schadt EE, Yandell BS, Kendziorski C, Attie AD (2008) A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 18: 706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kollhoff A, Bergmann A, Stubbs L (2003) Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum Mol Genet 12: 233–245 [DOI] [PubMed] [Google Scholar]
- Kurada BR, Li LC, Mulherkar N, Subramanian M, Prasad KV, Prabhakar BS (2009) MADD, a splice variant of IG20, is indispensable for MAPK activation and protection against apoptosis upon tumor necrosis factor-alpha treatment. J Biol Chem 284: 13533–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu B, Cardozo AK, Darville MI, Kruhoffer M, Magnusson N, Orntoft T, Eizirik DL (2003) Discovery of gene networks regulating cytokine-induced dysfunction and apoptosis in insulin-producing INS-1 cells. Diabetes 52: 2701–2719 [DOI] [PubMed] [Google Scholar]
- Ladriere L, Igoillo-Esteve M, Cunha DA, Brion JP, Bugliani M, Marchetti P, Eizirik DL, Cnop M (2010) Enhanced signaling downstream of ribonucleic acid-activated protein kinase-like endoplasmic reticulum kinase potentiates lipotoxic endoplasmic reticulum stress in human islets. J Clin Endocrinol Metab 95: 1442–1449 [DOI] [PubMed] [Google Scholar]
- Lawrence M, Shao C, Duan L, McGlynn K, Cobb MH (2008) The protein kinases ERK1/2 and their roles in pancreatic beta cells. Acta Physiol (Oxf) 192: 11–17 [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McGlynn K, Naziruddin B, Levy MF, Cobb MH (2007) Differential regulation of CHOP-10/GADD153 gene expression by MAPK signaling in pancreatic beta-cells. Proc Natl Acad Sci USA 104: 11518–11525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy JL (2005) Pathogenesis of type 2 diabetes mellitus. Arch Med Res 36: 197–209 [DOI] [PubMed] [Google Scholar]
- Lecomte M, Denis U, Ruggiero D, Lagarde M, Wiernsperger N (2004) Involvement of caspase-10 in advanced glycation end-product-induced apoptosis of bovine retinal pericytes in culture. Biochim Biophys Acta 1689: 202–211 [DOI] [PubMed] [Google Scholar]
- Lee CS, Sund NJ, Vatamaniuk MZ, Matschinsky FM, Stoffers DA, Kaestner KH (2002) Foxa2 controls Pdx1 gene expression in pancreatic beta-cells in vivo. Diabetes 51: 2546–2551 [DOI] [PubMed] [Google Scholar]
- Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z, Li Q, Wen Z, Peng Li P (2007) Cideb regulates diet-induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56: 2523–2532 [DOI] [PubMed] [Google Scholar]
- Liang B, Song X, Liu G, Li R, Xie J, Xiao L, Du M, Zhang Q, Xu X, Gan X, Huang D (2007) Involvement of TR3/Nur77 translocation to the endoplasmic reticulum in ER stress-induced apoptosis. Exp Cell Res 313: 2833–2844 [DOI] [PubMed] [Google Scholar]
- Ling C, Del Guerra S, Lupi R, Ronn T, Granhall C, Luthman H, Masiello P, Marchetti P, Groop L, Del Prato S (2008) Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51: 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic S, Bezzi P, Vitale N, Regazzi R (2009) The GTPase RalA regulates different steps of the secretory process in pancreatic beta-cells. PLoS One 4: e7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry R (1998–2011) Point Biserial Correlation Coefficient. ( http://faculty.vassar.edu/lowry/pbcorr.html )
- Maeda H, Rajesh KG, Suzuki R, Sasaguri S (2004) Epidermal growth factor and insulin inhibit cell death in pancreatic beta cells by activation of PI3-kinase/AKT signaling pathway under oxidative stress. Transplant Proc 36: 1163–1165 [DOI] [PubMed] [Google Scholar]
- Malmgren S, Nicholls DG, Taneera J, Bacos K, Koeck T, Tamaddon A, Wibom R, Groop L, Ling C, Mulder H, Sharoyko VV (2009) Tight coupling between glucose and mitochondrial metabolism in clonal b-cells is required for robust insulin secretion. J Biol Chem 284: 32395–32404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchitti SA, Orlicky DJ, Vasiliou V (2007) Expression and initial characterization of human ALDH3B1. Biochem Biophys Res Commun 356: 792–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, Bruggemann M, Bug S, Calasanz MJ, Deckert M, Dreyling M, Du MQ, Durig J, Dyer MJ, Fan JB, Gesk S, Hansmann ML, Harder L, Hartmann S, Klapper W et al. (2009a) A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One 4: e6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, Rosolowski M, Richter J, Lopez-Serra L, Ballestar E, Berger H, Agirre X, Bernd HW, Calvanese V, Cogliatti SB, Drexler HG, Fan JB, Fraga MF, Hansmann ML, Hummel M et al. (2009b) New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood 113: 2488–2497 [DOI] [PubMed] [Google Scholar]
- Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD (2009) Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease. PLoS One 4: e6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Zeggini E (2009) Genome-wide association studies in type 2 diabetes. Curr Diab Rep 9: 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, Bohr VA, Chua KF (2009) SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany, NY) 1: 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8: 2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y (1986) Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem 261: 11398–11403 [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D et al. (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124: 315–329 [DOI] [PubMed] [Google Scholar]
- Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ (2009) CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol 182: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutskov V, Felsenfeld G (2009) The human insulin gene is part of a large open chromatin domain specific for human islets. Proc Natl Acad Sci USA 106: 17419–17424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ (2010) Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 467: 963–966 [DOI] [PubMed] [Google Scholar]
- Oberg-Welsh C, Welsh M (1996) Effects of certain growth factors on in vitro maturation of rat fetal islet-like structures. Pancreas 12: 334–339 [DOI] [PubMed] [Google Scholar]
- Obrenovich ME, Monnier VM (2005) Apoptotic killing of fibroblasts by matrix-bound advanced glycation endproducts. Sci Aging Knowledge Environ 2005: pe3. [DOI] [PubMed] [Google Scholar]
- Ortis F, Naamane N, Flamez D, Ladriere L, Moore F, Cunha DA, Colli ML, Thykjaer T, Thorsen K, Orntoft TF, Eizirik DL (2010) Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes 59: 358–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T (2006) Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 55: 435–440 [DOI] [PubMed] [Google Scholar]
- Overbergh L, Valckx D, Waer M, Mathieu C (1999) Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine 11: 305–312 [DOI] [PubMed] [Google Scholar]
- Pahl HL, Sester M, Burgert HG, Baeuerle PA (1996) Activation of transcription factor NF-kappaB by the adenovirus E3/19K protein requires its ER retention. J Cell Biol 132: 511–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Stoffers DA, Nicholls RD, Simmons RA (2008) Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 118: 2316–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, Beresford-Smith B, Macintyre G, Kelong M, Hongyu Z, Zhu J, El-Osta A (2011) Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 21: 1601–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10(Suppl 4): 32–42 [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD (2010) Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 20: 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, Tomazou EM, Backdahl L, Johnson N, Herberth M, Howe KL, Jackson DK, Miretti MM, Fiegler H, Marioni JC, Birney E, Hubbard TJ, Carter NP, Tavare S, Beck S (2008) An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 18: 1518–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampersaud E, Damcott CM, Fu M, Shen H, McArdle P, Shi X, Shelton J, Yin J, Chang YP, Ott SH, Zhang L, Zhao Y, Mitchell BD, O'Connell J, Shuldiner AR (2007) Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the Old Order Amish: evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes 56: 3053–3062 [DOI] [PubMed] [Google Scholar]
- Reed JC, Doctor K, Rojas A, Zapata JM, Stehlik C, Fiorentino L, Damiano J, Roth W, Matsuzawa S, Newman R, Takayama S, Marusawa H, Xu F, Salvesen G, Godzik A (2003) Comparative analysis of apoptosis and inflammation genes of mice and humans. Genome Res 13: 1376–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Ammerpohl O, Martin-Subero JI, Montesinos-Rongen M, Bibikova M, Wickham-Garcia E, Wiestler OD, Deckert M, Siebert R (2009) Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer 9: 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roider HG, Lenhard B, Kanhere A, Haas SA, Vingron M (2009) CpG-depleted promoters harbor tissue-specific transcription factor binding signals—implications for motif overrepresentation analyses. Nucleic Acids Res 37: 6305–6315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S (2002) Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 45: 85–96 [DOI] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G et al. (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316: 1331–1336 [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL (2006) A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA 103: 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL et al. (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316: 1341–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U, Ukil S, Dimitrova N, Agrawal S (2009) Expression-based network biology identifies alteration in key regulatory pathways of type 2 diabetes and associated risk/complications. PLoS One 4: e8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R, Deng J, Wang W, Zhang K (2010) Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 20: 883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885 [DOI] [PubMed] [Google Scholar]
- Smyth GK (2005) Limma: linear models for microarray data. In: Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S (eds). Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer: New York. pp 397–420 [Google Scholar]
- Sneath PHA, Sokal RR (1973) Numerical Taxonomy: The Principles and Practice of Numerical Classification. San Francisco: W. H. Freeman [Google Scholar]
- Stipanuk MH, Dominy JE Jr, Lee JI, Coloso RM (2006) Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 136: 1652S–1659S [DOI] [PubMed] [Google Scholar]
- Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A (2005a) Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet {beta}-cells from pancreatic duct cells and an increase in functional {beta}-cell mass. J Clin Endocrinol Metab 90: 3401–3409 [DOI] [PubMed] [Google Scholar]
- Suarez-Pinzon WL, Yan Y, Power R, Brand SJ, Rabinovitch A (2005b) Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes 54: 2596–2601 [DOI] [PubMed] [Google Scholar]
- Sun GD, Kobayashi T, Abe M, Tada N, Adachi H, Shiota A, Totsuka Y, Hino O (2007) The endoplasmic reticulum stress-inducible protein Niban regulates eIF2alpha and S6K1/4E-BP1 phosphorylation. Biochem Biophys Res Commun 360: 181–187 [DOI] [PubMed] [Google Scholar]
- Sund NJ, Vatamaniuk MZ, Casey M, Ang SL, Magnuson MA, Stoffers DA, Matschinsky FM, Kaestner KH (2001) Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev 15: 1706–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476 [DOI] [PubMed] [Google Scholar]
- Tabak AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimaki M, Witte DR (2009) Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373: 2215–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599 [DOI] [PubMed] [Google Scholar]
- Tiwari AK, Prasad P, B KT, Kumar KM, Ammini AC, Gupta A, Gupta R (2009) Oxidative stress pathway genes and chronic renal insufficiency in Asian Indians with Type 2 diabetes. J Diabetes Complications 23: 102–111 [DOI] [PubMed] [Google Scholar]
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT (2009) DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet 18: 4046–4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tost J (2010) DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol Biotechnol 44: 71–81 [DOI] [PubMed] [Google Scholar]
- Ubeda M, Kemp DM, Habener JF (2004) Glucose-induced expression of the cyclin-dependent protein kinase 5 activator p35 involved in Alzheimer's disease regulates insulin gene transcription in pancreatic beta-cells. Endocrinology 145: 3023–3031 [DOI] [PubMed] [Google Scholar]
- Ubeda M, Rukstalis JM, Habener JF (2006) Inhibition of cyclin-dependent kinase 5 activity protects pancreatic beta cells from glucotoxicity. J Biol Chem 281: 28858–28864 [DOI] [PubMed] [Google Scholar]
- UKPDSG (1995) U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 44: 1249–1258 [PubMed] [Google Scholar]
- Viger RS, Guittot SM, Anttonen M, Wilson DB, Heikinheimo M (2008) Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol 22: 781–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve LM, Natarajan R (2010) The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol 299: F14–F25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Guanming Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM et al. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42: 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang P, Sun X, Luo Y, Wang X, Ma D, Wu J (2007) Cloning and characterization of a novel caspase-10 isoform that activates NF-kappaB activity. Biochim Biophys Acta 1770: 1528–1537 [DOI] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466 [DOI] [PubMed] [Google Scholar]
- Wei FY, Nagashima K, Ohshima T, Saheki Y, Lu YF, Matsushita M, Yamada Y, Mikoshiba K, Seino Y, Matsui H, Tomizawa K (2005) Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat Med 11: 1104–1108 [DOI] [PubMed] [Google Scholar]
- Welsh N, Cnop M, Kharroubi I, Bugliani M, Lupi R, Marchetti P, Eizirik DL (2005) Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes 54: 3238–3244 [DOI] [PubMed] [Google Scholar]
- Williamson JM, Boettcher B, Meister A (1982) Intracellular cysteine delivery system that protects against toxicity by promoting glutathione synthesis. Proc Natl Acad Sci USA 79: 6246–6249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Ishino F, Kaneko-Ishino T, Shiura H, Uchio-Yamada K, Matsuda J, Suzuki O, Sato K (2008) Type 2 diabetes mellitus in a non-obese mouse model induced by Meg1/Grb10 overexpression. Exp Anim 57: 385–395 [DOI] [PubMed] [Google Scholar]
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP (2004) A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 32: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, Pavesi G (2009) Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 37: W247–W252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P et al. (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40: 638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR et al. (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316: 1336–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.