

Abstract
Melanoma differentiation-associated gene-5 (MDA5) detects viral double-stranded RNA in the cytoplasm. RNA binding induces MDA5 to activate the signalling adaptor MAVS through interactions between the caspase recruitment domains (CARDs) of the two proteins. The molecular mechanism of MDA5 signalling is not well understood. Here, we show that MDA5 cooperatively binds short RNA ligands as a dimer with a 16–18-basepair footprint. A crystal structure of the MDA5 helicase-insert domain demonstrates an evolutionary relationship with the archaeal Hef helicases. In X-ray solution structures, the CARDs in unliganded MDA5 are flexible, and RNA binds on one side of an asymmetric MDA5 dimer, bridging the two subunits. On longer RNA, full-length and CARD-deleted MDA5 constructs assemble into ATP-sensitive filaments. We propose a signalling model in which the CARDs on MDA5–RNA filaments nucleate the assembly of MAVS filaments with the same polymeric geometry.
Keywords: DExD/H-box helicase, filament, innate immune receptor, RIG-I-like receptor, RNA recognition
Introduction
Innate immune receptors detect broadly conserved microbial structures throughout the cell and induce a rapid inflammatory response. RIG-I (retinoic acid-inducible gene-I) and MDA5 (melanoma differentiation-associated gene-5) recognize cytosolic double-stranded RNA (dsRNA) delivered or generated during infection by certain viruses (Kang et al, 2002; Yoneyama et al, 2004, 2005; Kato et al, 2006). RIG-I and MDA5 have similar sequences and overall architectures, with two N-terminal caspase recruitment domains (CARDs), a DExD/H-box helicase, and a C-terminal domain (CTD; also known as the regulatory or repressor domain). Binding of dsRNA to the helicase and CTD induces changes in the conformation and oligomerization state of RIG-I and MDA5 (Saito et al, 2007; Binder et al, 2011) such that the CARDs recruit and activate the CARD-containing signalling adaptor MAVS (CARDIFF, VISA, IPS-1) (Kawai et al, 2005; Meylan et al, 2005; Seth et al, 2005; Xu et al, 2005). MAVS propagates the signalling cascade, which culminates in activation of NF-κB and IRF-3 followed by type 1 interferon production (Kang et al, 2002; Yoneyama et al, 2004). Cellular exposure to interferon increases expression levels of RIG-I and MDA5, providing a positive feedback mechanism for the rapid amplification of the inflammatory response upon infection (Kang et al, 2004; Yoneyama et al, 2004, 2005). RIG-I and MDA5 signalling is regulated in part by LGP2, a RIG-I-like receptor (RLR) lacking CARDs (Yoneyama et al, 2005; Saito et al, 2007; Satoh et al, 2010). The inflammatory response induced by MDA5 can affect various cellular processes. MDA5 inhibits growth of human melanoma cells by inducing apoptosis (Kang et al, 2002), and rare variants of human MDA5 lower the risk of type 1 diabetes by improving β-cell survival (Nejentsev et al, 2009; Aida et al, 2011).
RIG-I and MDA5 recognize distinct sets of ligands. The 5′-triphosphate cap in the genomes of most plus-strand RNA viruses and a blunt dsRNA end are required but not sufficient for a full RIG-I-dependent signalling response (Hornung et al, 2006; Pichlmair et al, 2006). The phosphorylated blunt end binds to a conserved positively charged surface on the RIG-I and LGP2 CTDs (Takahasi et al, 2008; Li et al, 2009b; Lu et al, 2010, 2011; Wang et al, 2010). In contrast, a different conformation of the RNA recognition loop in the MDA5 CTD relative to the RIG-I and LGP2 CTDs results in a different shape of the putative RNA-binding surface, and MDA5 does not recognize 5′-triphosphate caps (Takahasi et al, 2009; Li et al, 2009a).
Recent crystal structures of RIG-I show that the RIG-I helicase domains also contribute to ligand binding (Kowalinski et al, 2011; Luo et al, 2011; Jiang et al, 2011a), and biochemical data suggest similar functions for the MDA5 helicase domains (Yoneyama et al, 2005). RNA binding does not require ATP hydrolysis, although in the presence of ATP RIG-I can unwind dsRNA substrates (Takahasi et al, 2008) and MDA5 ATPase activity is RNA dependent (Kang et al, 2002). The optimal length for RIG-I ligands is between 30 and 300 basepairs (bp) (Kato et al, 2008; Takahasi et al, 2008; Ranjith-Kumar et al, 2009; Binder et al, 2011), while the highest levels of MDA5 activation require longer dsRNA ligands, up to 2 kb (Kato et al, 2008; Pichlmair et al, 2009). RNA recognition by RLRs is not sequence specific (Kato et al, 2008; Takahasi et al, 2008; Pichlmair et al, 2009; Ranjith-Kumar et al, 2009). Poly(I:C) RNA, which is mostly double-stranded but may contain some single-stranded regions and branched structures, potently activates MDA5. Branched or higher-order structures generated during viral infection appear to be required for MDA5 activation (Pichlmair et al, 2009).
In the absence of structural information for full-length MDA5, it remains unclear how ligand binding affects the structure and oligomeric state of MDA5, and how these conformational changes lead to the recruitment of signalling effectors. We have performed a structural and biophysical analysis of MDA5 and its complexes with different ligands. Based on X-ray crystal and solution structures, we provide the first picture of MDA5 bound to dsRNA. A synthesis of this work with available structural, biochemical and cell signalling data allows us to propose a new model of MDA5 activation.
Results
MDA5 binds short nucleic acids as a dimer with a 16–18-bp footprint
In electrophoretic mobility shift assays (EMSAs), full-length MDA5 bound to both dsDNA and dsRNA, although binding to RNA was tighter (Figure 1A). Quantitation of dsRNA binding (Figure 1B) revealed similar binding affinities of MDA5 for 20 bp AU20 and 24-bp AU24 dsRNA oligonucleotides, with dissociation equilibrium constants (Kd) of 287±13 and 360±22 nM, and Hill coefficients (n) of 1.7 and 1.5, respectively. This binding is 5- to 10-fold tighter than for the MDA5 CTD alone (Li et al, 2009a) and 10-fold weaker than RIG-I binding to similar dsRNA oligonucleotides (McCoy et al, 2007). MDA5 constructs lacking the CARDs or CTD also bound dsRNA (Supplementary Figure S1), although the helicase-only construct bound AU20 RNA with a substantially lower affinity and no cooperativity (Kd=1.45±0.05 μM, n=1; Figure 1B). Mutation of a lysine residue on the dsRNA binding surface of the CTD (K983E) had no effect on the RNA-binding affinity and cooperativity of full-length MDA5 (Kd=277±14.8 nM, n=2.1; Figure 1B). The K983E mutation was shown to abrogate RNA binding by the CTD alone (Li et al, 2009a). MDA5 also binds cooperatively to longer poly(I:C) dsRNA ligands (Kd=652±34 nM, n=2.26±0.25; Supplementary Figure S1). The cooperativity of MDA5 binding to RNA was confirmed by sedimentation experiments (see below).
Figure 1.

MDA5 binds short nucleic acid ligands with a 16–18-bp footprint. (A) Native PAGE showing the mobility shift of 20- and 24-bp RNA and DNA oligonucleotides (stained with SYBR Green) by full-length MDA5. MDA5–oligonucleotide complexes (bracket) migrate more slowly than free oligonucleotides (arrow). (B) Binding curves based on densitometry of bands in EMSA. MDA5 bound AU20 with Kd=287±13 nM and a Hill coefficient, n=1.7 (black squares). MDA5 bound AU24 with Kd=360±22 nM, n=1.5 (open squares). MDA5 K983E bound AU20 with Kd=277±14.8 nM, n=2.1 (red). The MDA5 helicase construct bound AU20 with Kd=1.45±0.05 μM, n=1 (blue). Each curve is the mean±s.e.m. of three to four experiments. (C) Poly(I:C) RNase protection with full-length and CARD-deleted MDA5 on denaturing PAGE. Both constructs protect poly(I:C) from RNases I and V1 in 16–18-bp increments with the smallest band at ∼50 nt. Poly(I:C) (− lane) is completely degraded by RNases (+ lane). Left lane, 22-bp DNA marker. (D) High-resolution gel showing the fine structure of bands in the RNase protection experiment from (C). See also Supplementary Figure S1.
Two species could be distinguished in the EMSAs with AU20. A lower molecular weight MDA5–RNA complex was present at subsaturating concentrations of MDA5, while the complex migrated as a larger species at higher MDA5 concentrations (Figure 1A). This pattern indicates that two or more MDA5 molecules assemble sequentially on one or more oligonucleotide molecules. MDA5 is a monomer in the absence of ligand (see below). Surprisingly, the AU24–MDA5 complex (but not AU20–MDA5 or DNA–MDA5 complexes) formed even larger species at high MDA5 concentrations (Figure 1A). Higher-order oligomers were also observed with CARD-deleted MDA5 constructs (Supplementary Figure S1A).
To determine the footprint of MDA5 on dsRNA, we treated full-length or CARD-deleted MDA5 bound to poly(I:C) RNA with single-stranded RNA and dsRNA endonucleases (Figure 1C). In the presence of MDA5, endonuclease digestion of poly(I:C) produced a ladder of equally spaced bands. We conclude that each band corresponds to an RNA molecule protected by a different number of protein molecules. High-resolution gels were used to accurately measure the size of the footprint at 16–18 bp (Figure 1D), with the smallest protected species at 50 bp. Additionally, bands of lower intensity were visible with a spacing of 8–9 bp from the main bands (Figure 1C). The alternating high- and low-intensity band pattern for MDA5 suggests that MDA5 preferentially binds dsRNA as a dimer with a footprint of 16–18 bp, or 8–9 bp per monomer. Recent crystal structures of RIG-I show that the footprint of a RIG-I monomer is also ∼9 bp (Jiang et al, 2011a; Kowalinski et al, 2011; Luo et al, 2011).
Short dsRNA induces cooperative dimerization of CARD-deleted MDA5
To obtain a more direct and quantitative model of MDA5–RNA complex formation, we measured the sedimentation rate of CARD-deleted MDA5 at different concentrations in the presence of AU20 dsRNA by sedimentation velocity analytical ultracentrifugation (SV-AUC). CARD-deleted MDA5 sedimented as a monomer, with a sedimentation coefficient of 4.2 S and a frictional coefficient of 1.5. Full-length MDA5 was also monomeric, sedimenting at 4.7 S. With increasing concentrations of CARD-deleted MDA5, the sedimentation profiles of AU20 revealed a dynamic equilibrium between free and protein-bound RNA (Figure 2A). At substoichiometric concentrations of MDA5, free RNA sedimented at 2.4 S while a second sedimentation boundary with a concentration-dependent S-value was observed (Figure 2A). This behaviour is a hallmark of reversible associations with an offrate (koff) similar to the timescale of SV-AUC (10−3–10−4 s−1). At high MDA5 concentrations (>3 μM), the sedimentation coefficient of the fast sedimenting boundary approached 7.4 S, consistent with a complex consisting of two MDA5 molecules and either one or two RNA duplexes. To discriminate between these two stoichiometries, we calculated sedimentation profiles of the RNA and protein components based on the refined spectral properties for each component (Figure 2B). From the ratio of the areas under the 7.4 S peaks in these profiles, the MDA5:AU20 molar ratio was 2.6:1. This favours the 2:1 stoichiometry since a 7.4 S species is inconsistent with a complex containing more than two MDA5 subunits.
Figure 2.

Short dsRNA induces rapid and cooperative dimerization of CARD-deleted MDA5. (A) The sedimentation coefficient distributions, c(S), calculated from SV-AUC with 1 μM AU20 RNA monitored at 260 nm. Top and bottom panels, c(S) for MDA5 concentrations below and above 1 μM, respectively. Free AU20 sediments at 2.4 S and free MDA5 at 4.2 S. The limiting peak at 7.4 S (bottom panel) is consistent with a 2:1 MDA5:AU20 complex. (B) Multisignal SV-AUC with 15 μM MDA5 and 1 μM AU20 monitored at 260 nm and with interference optics indicates a 2.6:1 MDA5:AU20 stoichiometry within the complex, based on integration of the areas under the 7.4 S peaks. (C) Global isotherm analysis using the parameters from the curves in (A). The weight-averaged sedimentation coefficient (top), reaction boundary sedimentation coefficient (middle) and population signals of the undisturbed and reaction boundaries (bottom) yielded dissociation constants of 187 and 124 nM for association to AU20 of the first and second MDA5 subunits, respectively. Models enforcing completely independent binding sites (Kd2/Kd1=4) had significantly worse fits (Supplementary Figure S2).
The concentration dependence of the populations and weight-averaged sedimentation coefficients in reversible interactions can be used to test association models and determine binding constants by isotherm analysis (Dam and Schuck, 2005). Figure 2C shows the global fit of the model R+P → RP+P → RP2 (R, AU20; P, MDA5) to the total weight-averaged sedimentation coefficient (Sw), the sedimentation coefficient of the reaction boundary (Sfast) and the signal corresponding to the undisturbed and reaction boundaries. A cooperative model yielded a Kd1 value of 187 nM (with a 1-σ confidence interval of 92–368 nM) for the first binding step and Kd2=124 nM for the second binding step (with a 1-σ confidence interval of 83–173 nM). Since independent, non-cooperative sites would predict Kd2/Kd1=4 (see Supplementary data) our data indicate that MDA5 binds to RNA with positive cooperativity. A fit to a non-cooperative model was significantly worse (Supplementary Figure S2). The refined S-values for the 1:1 and 2:1 complexes were 5.6 and 7.6 S, respectively.
Crystal structure of the MDA5 helicase-insert domain
The lack of detailed structural information for RLR helicase domains limits our understanding of RNA recognition by MDA5. The most divergent sequence in MDA5 is an insertion within Hel2 (Hel2i; residues 547–693). Sequence analysis of RLRs and an electron microscopy (EM) structure of LGP2 suggest that the RLR Hel2i domain may be similar to an insertion of archaeal DNA repair helicase Hef (Murali et al, 2008). The sequence identity between the MDA5 and the Pyrococcus furiosus Hef Hel2i regions is, however, insignificant. Hef does have sequence similarity to Fanconi anaemia-associated helicase FANCM (Meetei et al, 2005) and the two DNA helicases have similar in vitro activities (Whitby, 2010). FANCM is important in eukaryotes for resolving stalled replication forks by binding and remodelling branched DNA junctions (Whitby, 2010).
To gain structural insight on MDA5, we determined the crystal structure of MDA5 Hel2i (Figure 3). The overall fold is a five-helix bundle with similar shape, dimensions and topology as Hef Hel2i and RIG-I Hel2i (Figure 3C and D). The N- and C-termini are positioned within only 8.5 Å of one another, consistent with Hel2i being inserted within a loop in Hel2. Helices 2–4 form a surface with relatively high-sequence conservation across vertebrate MDA5 sequences (Figure 3B). In the Hef crystal structure and in one of the RIG-I structures, this surface faces the RNA-binding cleft and forms contacts with Hel2 (Murali et al, 2008; Jiang et al, 2011a). Helices 2 and 3 are shorter in MDA5 Hel2i than in RIG-I (Figure 3D), which is notable because this region interacts with CARD2 in the full-length duck RIG-I structure. It has been suggested that this interaction keeps RIG-I in an inactive state in the absence of RNA (Kowalinski et al, 2011). Moreover, a phenylalanine residue that is critical for this interaction in duck RIG-I (F540) is not conserved in MDA5 (Figure 3D). Together, these differences between the RIG-I and MDA5 Hel2i structures suggest that MDA5 Hel2i interacts with its CARDs differently than in RIG-I, or not at all. The adjacent helix 4 is connected to helix 5 by a largely disordered 30-residue loop in MDA5, forming a slight concavity at the distal end of the domain. This loop contains between 7 and 15 acidic residues in MDA5 sequences, but none in RIG-I or LGP2, which have shorter loops. There is no complementary basic region predicted on the surface the MDA5 CARDs.
Figure 3.

Crystal structure of the MDA5 helicase-insert (Hel2i) domain. (A) Cartoon representation of MDA5 Hel2i (blue to red, N- to C-terminus). The disordered loop containing the ΔL2 loop deletion is shown as a dashed line. (B) Surface representation coloured according to sequence homology across vertebrate MDA5 sequences (green is conserved, yellow is variable). The view is the same as in (A). (C) MDA5 Hel2i (magenta) has the same overall topology as P. furiousus Hef Hel2i (yellow), with an RMSD of Cα atom positions of 3.8 Å. (D) Comparison of MDA5 Hel2i and duck RIG-I Hel2i (blue). Two residues important for binding of Hel2i to the CARDs in RIG-I (F540 and F571) are shown in stick representation with the corresponding residues for MDA5 (R596 and F630). The extended α3 helix in RIG-I also contributes to the CARD interface. See also Supplementary Table SI.
Solution structure of MDA5 domains by small-angle X-ray scattering
To gain insight on the overall MDA5 domain architecture, we collected small-angle X-ray scattering (SAXS) data for four MDA5 constructs: the helicase domains (Hel1, Hel2 and Hel2i), CARD-deleted MDA5, the CARDs and full-length MDA5. Each construct had a loop deletion within Hel2i to enhance stability (Supplementary Figure S3; Supplementary Table SII). Dummy atom (DA) ab initio models were calculated using the SAXS data for each construct. For the helicase domains, the average DA model had a two-lobed shape similar to that of Hef (Figure 4A and B). The presence of AMPPNP or ATPγS had no significant effect on the overall scattering and shape parameters of the helicase domain (not shown). To complement the DA modelling, we generated a homology model of MDA5 helicase based on the structures of Hef (PDB 1WP9), MDA5 Hel2i, and the partial MDA5 Hel1 domain (PDB 3B6E). The relative positions of the helicase subdomains were then refined against the SAXS curve with movements only allowed along normal modes calculated from the initial model. This refinement increased the angle of the cleft formed by the Hel1 and Hel2i domains, resulting in a more open conformation than in the Hef or RIG-I structures (Figure 4B). To complete our model, residues 866–888 between Hel2 and the CTD, predicted by primary sequence analysis to form an amphipathic helix (Hel-link), were manually docked onto the helicase. The best fit of Hel-link was with its hydrophobic face against a hydrophobic surface formed mainly by the last helix and β-strand of Hel1, modelled using Hef as a template. Consistent with a Hel-link/Hel1 interaction, helicase constructs lacking either Hel-link or Hel1 were unstable and tended to aggregate during purification, while a construct spanning Hel2 and Hel2i (but lacking Hel-link) was stable. The homology model refined against the SAXS data (χ=2.8) was highly consistent with one enantiomer of the averaged DA model (Figure 4B). The homologous so-called pincer or bridging helices in RIG-I also pack against Hel1, although they are positioned differently relative to Hel1.
Figure 4.

SAXS structures of unliganded MDA5 fragments. (A) DA model of the MDA5 helicase domains. (B) Homology model of the Hel1, Hel2i and Hel2 helicase domains (light to dark grey, respectively) refined against the SAXS curve. The helix between Hel2 and the CTD (Hel-link; yellow) was docked manually prior to refinement. The envelope of the DA model is shown superimposed. (C) DA model of CARD-deleted MDA5 with the additional lobe corresponding to the CTD in cyan. (D) Superposition of the six best rigid-body models of CARD-deleted MDA5 consistent with the SAXS data. Coloured regions were constrained by flexible loops; the helicase domains (grey) were fixed. The positions of the CTDs are indicated by ovals and a representative cartoon (cyan). (E) DA model of the CARDs. All DA models represent an average of 15 models. (F) Calculated one-dimensional X-ray scattering of MDA5 CARD homology models in prototypical death domain family interactions (red and blue) fitted against the observed scattering (black). The ‘b’ indicates a swap of the two CARD domains across the interface. The curves were vertically offset for clarity and the best fit is indicated in blue. (G) Cartoon representation of the CARD homology model with the type II interface (blue to red, N- to C-terminus). See also Supplementary Figure S3 and Supplementary Table SII.
For CARD-deleted MDA5, the average DA model resembled that of the helicase but contained a third lobe corresponding to the CTD (Figure 4C). To generate an atomic model of CARD-deleted MDA5, we performed simultaneous rigid-body fitting of the helicase model developed above and the MDA5 CTD crystal structure (PDB 3GA3) with dummy residue modelling of flexible loops (Petoukhov and Svergun, 2005). In the resulting models, the CTD occupied various positions adjacent to Hel1 (Figure 4D). The CTD is also flexibly linked to the helicase domains in RIG-I, as judged by the absence of electron density for the CTD in the crystal structure of unliganded full-length RIG-I (Kowalinski et al, 2011).
The pair distance distribution function and SAXS DA models of the MDA5 CARDs (residues 1–207) reveal a compact structure, indicating that the CARDs associate with one another (Figure 4E). Death domains form three prototypical interfaces (types I–III) (Park et al, 2007), and the structure of the Caenorhabditis elegans CED-4 apoptosome suggests a fourth (type IV) (Qi et al, 2010). The RIG-I CARDs associate through a type II interface (Kowalinski et al, 2011). The MDA5 CARDs were modelled in each configuration and their theoretical solution scattering was compared with the experimental scattering (Figure 4F). The CARD dimer modelled in the type II interface using duck RIG-I as a template was most consistent with the SAXS data (χ=2.9; Figure 4G).
MDA5 CARDs do not interact with the other MDA5 domains
The RIG-I CARDs interact with the Hel2i domain (Kowalinski et al, 2011) and repress ATPase activity until RNA binding releases the CARDs for downstream interactions (Cui et al, 2008). However, an interaction between the CARDs and helicase or CTD has not been shown for MDA5. Our crystal structure of MDA5 Hel2i also shows significant differences in the region of Hel2i that is responsible for binding CARDs in RIG-I. We found that the MDA5 CARDs and CARD-deleted MDA5 did not interact with each other in solution, even at high protein concentrations (>0.1 mM; Figure 5A), whereas RIG-I CARDs and CARD-deleted RIG-I did interact in a more stringent pulldown experiment (Kowalinski et al, 2011). Similarly, wild-type MDA5 Hel2i did not interact with the MDA5 CARDs in solution. We could not rule out, however, that the missing MDA5-specific, 95-residue linker between the CARD2 and Hel1 domains was required for the CARDs to interact with other MDA5 domains. This linker has no predicted secondary structure and its sequence is not conserved across species. To determine whether the CARDs interact with the other MDA5 domains in the presence of the CARD2–Hel1 linker, we collected SAXS data for full-length MDA5. The molecular mass calculated from Io (116 kDa) indicated that the SAXS data were not contaminated by dimeric species. The Kratky plot indicated a high degree of flexibility and the presence of unstructured regions (Figure 5B). Assuming the increased flexibility relative to CARD-deleted MDA5 was due to the CARD2–Hel1 linker, we performed ensemble optimization of random linker structures to match the solution scattering. The optimized ensemble had bimodal Rg and Dmax distributions, with a more abundant compact set of structures and a less abundant extended set (Figure 5C). The CARDs adopted many different positions in the selected models, and the dimensions of the structures in the extended set are incompatible with a CARD–Hel2i interaction, leading to the conclusion that the CARD2–Hel1 linker is highly flexible. However, we cannot rule out that in the compact set of the MDA5 SAXS ensemble, the CARDs and Hel2i form a weak or transient interaction, which only occurs in the context of the full-length protein.
Figure 5.

The MDA5 helicase and CTD domains do not associate with the CARDs and bind RNA as a dimer. (A) The MDA5 CARDs and CARD-deleted MDA5 elute separately from a size-exclusion column (blue). Elution curves of the two components loaded separately are shown in grey. (B) Plateau in the Kratky plot of full-length MDA5 indicates substantial flexibility within the molecule. (C) Histograms of the radius of gyration (Rg) and maximum dimension (Dmax) of full-length MDA5 models (orange) selected to fit the SAXS data with a five-curve minimal ensemble from a pool of 105 random domain arrangements (black). (D) Single-phase DA model of CARD-deleted MDA5 in complex with AU20 using data to 0.168 Å−1 (average of 15 models). (E) Three-phase DA model (average of seven) of the same complex as in (D) with two protein phases (cyan and green) and one RNA phase (magenta). (F) Rigid-body model using knowledge-based restraints of the same complex as in (D, E). See Supplementary Figures S4 and S5.
SAXS of CARD-deleted MDA5 bound to dsRNA
To gain insight into the ligand-induced dimerization of MDA5, we collected SAXS data on CARD-deleted MDA5 in complex with AU20 dsRNA. Based on I0, the molecular weight of the complex was 192 kDa, which is consistent with the expected molecular weight of 173 kDa, since I0 was calculated using a protein standard even though the complex contains RNA, which scatters more strongly than protein. DA modelling assuming constant electron density within the complex and using data to 0.168 Å−1 produced a model with approximate two-fold symmetry (Figure 5D). However, enforcing P2 symmetry decreased the stability of the solutions significantly. Using data to 0.3 Å−1 and three separate phases, one for each MDA5 subunit and one for the RNA, yielded DA models with the RNA phase on one side of an MDA5 dimer, across the dimer interface (Figure 5E). The overall shape of the average three-phase structure was similar to the shape of the single-phase structure. While the shapes of the protein phases were consistent, their relative positions around the RNA varied in individual models (Supplementary Figure S4). As a result, one protein phase in the averaged three-phase model had relatively poor shape definition in the average three-phase DA model (Figure 5E), making it impossible to dock a homology model or interpret these models beyond the conclusion that two MDA5 molecules surround a central RNA duplex.
To gain more detailed insight into the mode of MDA5 binding to RNA, we sought a complementary and independent modelling approach that explicitly exploited knowledge of helicase structure. Superfamily 2 helicases bind RNA and nucleotide through eight conserved motifs, with the RecA-like Hel1 and Hel2 domains contacting the RNA 3′ and 5′ ends, respectively (Pyle, 2008). Threonines in motifs Ib and V contact phosphates at the i and i+3 positions of the tracking RNA backbone. Using these threonine–RNA interactions as constraints, we placed helicase domain models in every possible position along a 20-bp poly(A:U) model dsRNA. The domains were modelled in a conformation based on known nucleic acid-bound helicase structures. Simulated annealing using the SAXS data was performed to refine the helicase domain positions, place the CTDs and score the models (Supplementary Figure S5A). The best resulting models were consistent with the overall shapes of the DA models, with the RNA bound on one side of the MDA5 dimer. In these models, the helicase domains were located at either end of opposite RNA tracking strands such that the RNA did not extend over the bridging (or pincer) domains. The Hel2i and CTD domains formed dimer contacts in the rigid-body models with the apposing molecule (Figure 5F). Interestingly, the CTDs in the models did not interact with the RNA, and constraints to force an interaction significantly worsened the fit. We note, however, that the SAXS models do not contain enough predictive power to firmly exclude the possibility that either the CTD or Hel2i domain may still bind RNA directly.
In contrast to our SAXS model of MDA5 bound to RNA, in the structures of RIG-I the CTD clamps the 5′ end of the RNA helix against the three helicase domains (Jiang et al, 2011a; Kowalinski et al, 2011; Luo et al, 2011). Several lines of evidence support the differences between the RNA-bound structures of MDA5 reported here and the RIG-I structures: (1) the binding affinity of the MDA5 CTD for RNA is 3 μM, which is 20- to 30-fold lower than for RIG-I and LGP2 (Li et al, 2009a). (2) The MDA5 CTD does not inhibit signalling and CTD-deleted MDA5 is not constitutively active, in contrast to RIG-I (Saito et al, 2007). (3) The cooperativity of MDA5 ligand binding that we observed is consistent with protein–protein contacts between RNA-bound MDA5 molecules, and deletion of the CTD significantly affects cooperativity (Figure 1B). No binding cooperativity has been reported for RIG-I. (4) Homology models of an MDA5 dimer based on recent crystal structures of RIG-I either have serious steric clashes (model based on Luo et al, 2011), or do not fit the SAXS data (χ=22.3; Supplementary Figure S5B–D; models based on Jiang et al, 2011a and Kowalinski et al, 2011). We also note that a 22-bp RNA can accommodate two bound RIG-I molecules, while an 18-bp ligand induces only partial RIG-I dimerization (Luo et al, 2011). Moreover, the threshold length for 5′-triphosphate, blunt ended dsRNA to induce significant RIG-I-dependent signalling is 19–21 bp (Takahasi et al, 2008; Schlee et al, 2009). Since RIG-I dimerization does not enhance ATPase activity, binding of RIG-I to such short ligands is thought to differ from binding to internal sites on long RNAs (Luo et al, 2011). Indeed, the crystal structures of RIG-I involve multiple contacts with the 5′ end of the RNA that are incompatible with binding to an internal site, and the RIG-I helicase is not fully engaged with the ligand in at least one of the structures (Luo et al, 2011).
MDA5 forms ATP-sensitive filaments on long dsRNA
Natural MDA5 ligands are as long as 2 kb and may contain branched or higher-order structures (Kato et al, 2008; Pichlmair et al, 2009). To relate our solution binding data to such ligands, we examined MDA5 in complex with genomic dsRNA from the Pseudomonas bacteriophage ϕ6 by EM. Full-length or CARD-deleted MDA5 formed long filamentous structures along the RNA (Figure 6A). The genome of ϕ6 is composed of three segments of 2.9, 4.1 and 6.4 kb, which if fully extended would be 0.8, 1.1 and 1.8 μm long, respectively. Many of the observed filaments had lengths near these expected sizes, although some were shorter. Most filaments were continuous with few breaks along their length. The filaments were 8–10 nm thick, consistent with a protein coat surrounding the dsRNA core (2.6 nm). Filaments with CARD-deleted MDA5 were slightly thinner (7–8 nm) and in some cases aggregated into bundles (Supplementary Figure S6). The MDA5 helicase construct did not form filaments with ϕ6 RNA. Assembly of MDA5 into filaments along dsRNA is consistent with the RNase protection pattern of poly(I:C) by MDA5 (Figure 1C and D). Since CARD-deleted MDA5 forms filaments, MDA5 polymerization does not require the CARDs. Significantly, MAVS was recently shown to aggregate through its CARD into prion-like fibrils similar in appearance to the MDA5 filaments (Hou et al, 2011).
Figure 6.

MDA5 assembles into regular filaments on long dsRNA ligands. (A) Negative-stain electron micrograph (EM) of MDA5 mixed with bacteriophage ϕ6 genomic dsRNA shows filaments 8–10 nm in diameter. Filaments have few breaks along their length and can be up to 1.8 μm long. Inset: close-up of the boxed region. (B) 1 mM ATP induces breaks in the filaments. At 5 mM ATP, no filaments were observed. Inset: close-up of the boxed region. (C) Nuclease protection assay shows ϕ6 RNA is not protected by MDA5 when ATP is present (left). The three ϕ6 genome segments (2.9, 4.1 and 6.4 kb) are degraded by dsRNA endonuclease V1 (V1), but not ssRNA endonuclease 1 (R1). MDA5 reduces the mobility of ϕ6 and poly(I:C) RNA on agarose gel electrophoresis and protects them from RNases. Increasing ATP levels decrease RNase protection by MDA5 of ϕ6 RNA (left) but not of poly(I:C) RNA (right). At >2.5 mM ATP, free poly(I:C) is also protected. (D, E) EM of poly(I:C) and MDA5 without ATP (D), and with 2.5 mM ATP (E). Filaments incubated with ATP tend to aggregate, most likely from enhanced filament crosslinking. This effect is not observed with AMPPNP. See Supplementary Figure S6 for protein-only and RNA-only controls.
Surprisingly, no filaments were observed with full-length or CARD-deleted MDA5 and ϕ6 RNA after addition of 5 mM ATP. With 1 mM ATP, filaments were visible but appeared fragmented (Figure 6B). ADP or AMPPNP had no effect on filament formation. This suggests that ATP hydrolysis causes filament disassembly, perhaps by inducing translocation along the RNA or by reducing binding affinity. However, we found no significant difference in the binding affinity of MDA5 for AU20 or AU24 dsRNA in the presence of AMPPNP or ATP in EMSAs. To confirm the ATP sensitivity of filaments in solution, we treated MDA5–RNA filaments with double-stranded endonuclease RNase V1. As expected, protected supershifted RNA was detected by native agarose gel electrophoresis in the absence of ATP, but the RNA was increasingly susceptible to hydrolysis with increasing ATP concentrations and was completely digested in 5 mM ATP (Figure 6C). We note that a recent independent study also reported that MDA5 assembles into ATP-sensitive, segmented filaments (Peisley et al, 2011).
In contrast, ATP did not increase the RNase sensitivity of poly(I:C) RNA bound to MDA5. The amount of protected supershifted species remained constant with increasing ATP concentrations. Indeed at >2.5 mM ATP, free poly(I:C) was also protected (Figure 6C), probably due to exclusion of RNases by extensive crosslinking of the RNA. Similar protection patterns were observed for CARD-deleted MDA5. However, helicase-only constructs did not induce protection of poly(I:C), nor were filaments observed by EM. EM of poly(I:C) with MDA5 in the absence of ATP revealed short filaments consistent with the average length of the poly(I:C) RNA (300–500 bp; Figure 6D). With increasing ATP (but not AMPPNP) concentrations, filaments became shorter and lost definition (Figure 6E), with large aggregated material appearing. With 5 mM ATP, only large unevenly stained aggregates were visible on EM grids (not shown), suggesting that the filaments were heavily crosslinked to each other.
Discussion
A synthesis of our data with previous signalling and ligand-binding studies on RLRs allows us to propose a new model of MDA5 activation (Figure 7). In the absence of ligand, MDA5 is monomeric and adopts an open conformation with flexible linkers between the CARD2 and Hel1 domains, and between the Hel2 and CTD domains. Unlike models of RIG-I activation (Cui et al, 2008), in our model the CARDs do not specifically associate with the helicase but MDA5 is in a conformational equilibrium between compact and extended forms, with compact forms favoured in the unliganded state. In the crystal structure of full-length duck RIG-I, the CARDs interact with Hel2i in the unliganded state and this interaction is strong enough that the domains are able to bind to one another despite stringent washing when not connected by a linker (Kowalinski et al, 2011). However, Phe540, which is important in the RIG-I CARD2–Hel2i interface, is an arginine residue in all MDA5 sequences. Mutation of Phe540 in duck RIG-I abrogates CARD–helicase interactions and increases basal signalling in overexpressing cells (Kowalinski et al, 2011). Together, these data lead us to propose that in contrast to RIG-I, the MDA5 CARDs do not form a stable interaction with the helicase domains in the absence of RNA, and that the signalling activity of MDA5 is regulated differently than that of RIG-I.
Figure 7.

Proposed model for MDA5 ligand binding and activation. MDA5 binds dsRNA cooperatively as a dimer with a 16–18-bp footprint. The dimer contacts are formed by the helicase domains (grey) and the CTD (cyan). The antiparallel arrangement of the subunits within the dimers is based on the homology model of a CARD-deleted MDA5 dimer bound to RNA refined against SAXS data. The binding mode of full-length MDA5 may differ from that shown. Full-length (or CARD-deleted) MDA5 assembles into helical or linear polymeric filaments on long dsRNA ligands. The CARDs (orange) on the outside of filaments recruit the CARD of MAVS (red) on the mitochondrial membrane. Through this CARD–CARD interaction, the regular helical or linear arrangement of MDA5 CARDs is imposed on MAVS, thereby inducing the formation of MAVS fibrils, which propagate the signalling response (Hou et al, 2011).
CARDs belong to the death domain superfamily, some of which form large helical or circular assemblies during signalling, including MAVS CARD (Park et al, 2007; Lin et al, 2010; Qi et al, 2010; Hou et al, 2011; Yuan et al, 2011). MDA5 may follow this paradigm since MDA5 signalling requires oligomerization (Childs et al, 2009). Our finding that the MDA5 CARDs are preassembled in a prototypical CARD interaction suggests that MDA5 may also form large oligomeric signalling assemblies. The discovery that MDA5 polymerizes into filaments along dsRNA supports a signal transduction model in which MDA5 filaments establish a helical or linear array of CARDs capable of activating downstream signalling. This model is especially compelling in the light of a recent study showing that MAVS aggregates through its CARD (independently of ATP) into prion-like fibrils with potent signalling activity (Hou et al, 2011). We propose that the regular array of MDA5 CARDs on the outside of MDA5–RNA filaments recruit the MAVS CARD. Through this CARD–CARD interaction, the regular helical or linear arrangement of MDA5 CARDs is imposed on MAVS, thereby inducing the formation of MAVS polymers or fibrils, which propagate the signalling response (Figure 7). In this model, the MDA5 CARD does not act as a repressor of the helicase, but rather as a polymeric scaffold for MAVS recruitment. Our SAXS structure of the MDA5–RNA complex is consistent with this model since both ends of the dsRNA are exposed, implying that multiple copies of MDA5 could bind in the same manner to a long dsRNA ligand. In the structure of duck RIG-I in complex with a 19-bp dsRNA, the RNA formed a nearly continuous, filament-like structure through crystal packing. One helicase was bound to the end of each RNA molecule with no contacts between protein molecules (Kowalinski et al, 2011). Moreover, a 60-bp dsRNA with a 5′-triphosphate cap only bound two RIG-I molecules, consistent with a lack of ligand-induced multimerization of RIG-I (Kowalinski et al, 2011). We suggest that the propensity for MDA5 to form filaments on long dsRNA and the cooperativity of MDA5 binding to RNA are important for the ability of MDA5 to selectively recognize much longer RNA ligands than RIG-I. Moreover, we conclude that MDA5 does not recognize the 5′ or 3′ ends of RNA, in contrast to RIG-I, which depends on at least one 5′ end for optimal ligand recognition.
The crystal structure of the MDA5 Hel2i domain demonstrates an evolutionary relationship between MDA5 and archael Hef DNA repair helicases. Since Hef and FANCM have similar sequences and activities, including branch point migration, fork regression and D-loop dissociation (Whitby, 2010), MDA5 may catalyse similar processes on RNA. Our results seem to support this since in the presence of ATP, MDA5 crosslinks poly(I:C) RNA, which is a heterogeneous enzymatic preparation and contains short single-stranded regions, overhangs and branched junctions. Branched or higher-order structures generated during viral infection are required to activate MDA5 signalling (Pichlmair et al, 2009).
The role of ATPase activity in MDA5 signalling remains controversial. Ectopically expressed ATPase-deficient MDA5 mutants have been reported either to have no effect on intrinsic signalling (Yoneyama et al, 2005) or to produce constitutively active signalling (Bamming and Horvath, 2009). In contrast, ATPase mutations in RIG-I produce a dominant negative protein (Yoneyama et al, 2004; Bamming and Horvath, 2009). We have presented evidence that ATP causes MDA5 filaments to disassemble from exclusively double-stranded ligands (ϕ6 RNA) and to crosslink more heterogeneous ligands (poly(I:C) RNA). The ability of poly(I:C) to activate MDA5 is well established, and ϕ6 genomic RNA segments were recently shown to induce MDA5 signalling in human dendritic cells (Jiang et al, 2011b). MDA5 recognition of dsRNA and branched structures are not mutually exclusive. We speculate that both types of ligands may contribute to microbial detection depending on the ATP concentration in the cell. Indeed, we found that the ATP sensitivity for filament formation occurred within the physiological concentration range of ATP (1–5 mM) and some viruses are known to manipulate host metabolism (Vastag et al, 2011). Future studies on the intersection of metabolism and innate immunity, and on MDA5 recognition of branched versus linear RNA ligands, may reveal new strategies for modulating host responses to infection.
Materials and methods
Nucleic acids
AU20 and AU24 have the palindromic sequences 5′-GCAAAUAAGCGCUUAUUUGC and 5′-GCAUAAAUAAGCGCUUAUUUAUGC, respectively. dAU20 and dAU24 are the corresponding DNA oligonucleotides. Oligonucleotides were self-annealed by slow cooling from 90 to 30°C. Poly(I:C) RNA was purchased from Midland Certified Reagents. ϕ6 RNA, a gift from Paul Turner (Yale University), was prepared with a QIAamp viral RNA kit (Qiagen) from phage grown on Pseudomonas syringae.
Expression and purification of MDA5 constructs
The residue ranges of the mouse MDA5 constructs were as follows: full-length 1–1026; CARDs 1–207; CARD-deleted 304–1026; helicase 304–898; and Hel2i 545–697. In all constructs, residues 639–669 were replaced with GSGSG (ΔL4 helicase loop deletion), except in Hel2i and CARD-deleted MDA5 in which residues 646–663 were deleted (ΔL2 loop deletion). Each construct was expressed from pET28a in Escherichia coli Rosetta (DE3) cells (Novagen). Selenomethionine-substituted Hel2i was expressed by metabolic inhibition of E. coli methionine biosynthesis. MDA5 proteins were purified by nickel-affinity, cation exchange and size-exclusion chromatography. For proteins containing the CTD, an additional anion exchange chromatography step was necessary to remove contaminating E. coli RNA.
Electrophoretic mobility shift assays
AU20 and AU24 were incubated with MDA5 in SPG buffer pH 7.4 (49 mM NaH2PO4, 49 mM glycine, 14 mM succinic acid), 0.1 M KCl, 2 mM MgCl2 for 1 h at 20°C and analysed by PAGE. For Kd determination, protein was titrated from 50 nM to 5 mM into a solution of 0.1 μM RNA. The fraction of bound nucleic acid was determined by integrating the signal intensity in each lane. At low protein concentrations, ligand depletion was corrected for by subtracting the concentration of bound RNA from the total protein concentration. Data for three titrations of MDA5 with AU20 and AU24 were averaged and the fraction of RNA bound, Y, was fitted to the following equation:
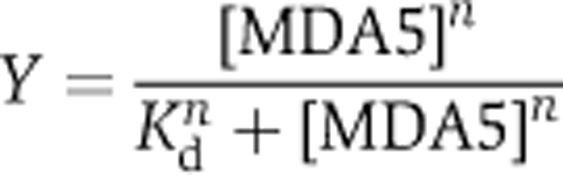 |
where n is the Hill coefficient.
RNase protection assay
In each reaction, 1 μg ϕ6 RNA or 5 μg of Poly(I:C) were mixed with 3 μg of full-length or CARD-deleted MDA5, 0.2 U of RNase V1 and 0.1 U of RNase 1 (Ambion). Reactions were performed in SPG pH 7, 0.1 M KCl, 10 mM MgCl2 and 0–5 mM ATP. After incubation at 20°C for 2 h, reactions were run on a 0.8% agarose gel, or phenol:chloroform-extracted and ethanol precipitated and run on a 20% polyacrylamide gel containing 8 M urea.
Analytical ultracentrifugation
CARD-deleted MDA5 and 1 μM AU20 were centrifuged at 20°C and 45–50 k.r.p.m. in an An-60 Ti rotor (Beckman-Coulter). Absorption at 260 nm and interference fringe displacement were measured for 6–10 h. Data were fitted to a c(S) sedimentation coefficient distribution calculated with SEDFIT (Brown et al, 2009) and regularized using the maximum entropy method with a value of 0.7. The protein vbar, buffer density and viscosity were calculated with SEDNTERP (http://rasmb.bbri.org). In the multisignal sedimentation velocity (MSSV) analysis (Figure 2B), SV-AUC data of the individual components and the mixture were collected with absorption and interference optics. The spectral properties of the individual components were refined by global sedimentation profile fitting of the SV-AUC data with SEDPHAT (Dam and Schuck, 2005). A sedimentation profile consistent with each component's spectral properties was then calculated for the mixture using global fitting. The stoichiometry of the complex was calculated from the ratio of the areas under the peaks in the profile.
The weight-averaged sedimentation coefficient, reaction boundary sedimentation coefficient and population isotherms were calculated from the c(S) distributions from 260 nm SV-AUC data for each mixture with SEDFIT. For the 1.5 μM CARD-deleted MDA 1 μM AU20 mixture, a Bayesian prior probability was applied to select c(S) profiles consistent with a free protein peak at 4.2 S to resolve the undisturbed and reaction boundaries. Global fitting of isotherms was performed with SEDPHAT using the extinction coefficients determined in the MSSV experiment. See Supplementary data for further details.
Crystallization and structure determination of the MDA5 helicase-insert domain (Hel2i)
Crystals of selenomethionine-substituted MDA5 Hel2i grew by hanging drop vapour diffusion at 16–20°C. Hel2i at 10–20 g l–1 in 20 mM HEPES 7.5, 0.2 M KCl, 5 mM β-mercaptoethanol was mixed with an equal volume of SPG buffer pH 8.5–9.5 and 1.9–2.2 M ammonium sulphate. Crystals belonged to space group P212121 with one molecule per asymmetric unit. Data were collected at 100 K and phases were determined by multiple-wavelength anomalous diffraction. Selenium sites were located and experimental phases were calculated with HKL2MAP (Pape and Schneider, 2004). Buccaneer (Cowtan, 2006) was used for automated model building. The model was improved with rounds of model building, refinement and water placement with Coot (Baum et al, 2008) and REFMAC5 (Murshudov et al, 1997). After final refinement, Rwork and Rfree were 19 and 22%, respectively. In all, 98.4% of residues were in the most favoured regions of the Ramachandran plot. See Supplementary Table SI for data collection and refinement statistics. The atomic coordinates and structure factors have been deposited in the Protein Data Bank under PDB ID 3TS9.
Structure determination of MDA5 constructs by SAXS
Data were collected at beamline X9A of the National Synchrotron Light Source. For each data set, a dilution series was performed (Supplementary Table SII) and triplicate measurements were collected. Bovine serum albumin was used as a molecular weight standard. For data sets for which Io/c and Rg increased with concentration, linear extrapolation was used to calculate zero-concentration values for these parameters.
DA modelling, rigid-body modelling and ensemble optimization were performed with ATSAS (Volkov and Svergun, 2003; Petoukhov and Svergun, 2005; Franke and Svergun, 2009). See Supplementary data for homology modelling details. DA models were calculated using data to a q-value of 8/Rg. Models with NSD greater than twice the variation were discarded during averaging. For the three-phase modelling, the volume of the protein phases was set based on SAXS data for CARD-deleted MDA5 alone and the volume of the RNA phase was calculated based on a 20-bp poly(A:U) model RNA. A starting DA ellipsoid was then refined against SAXS data for the protein:RNA complex, protein alone and simulated RNA alone.
For ensemble optimization of the full-length MDA5 data set, 105 structures were generated with flexible CARD–Hel1 and Hel2–CTD linkers. A genetic selection algorithm in ATSAS was used to select an ensemble of curves to fit the SAXS data. The optimal ensemble size was 5–10 structures.
A rigid-body model of the MDA5:AU20 complex was generated by superimposing the modelled MDA5 helicase domains on known nucleic acid-bound helicase structures (PDB IDs 2P6R, 3O8C, 3RC8) such that the conserved threonines in RNA-binding motifs Ib and V overlapped optimally. All possible configurations with contacts between these threonines and phosphates at the i and i+3 positions of the RNA were generated by applying screw translations to the protein along the RNA backbone. In all, 16 non-clashing model combinations were refined with various constraints (see Supplementary Figure S5A).
EM of MDA5–RNA complexes
In all, 50 ng of ϕ6 RNA, 20 ng MDA5 and 0–5 mM ATP in SPG buffer containing 10 mM MgCl2 were mixed, incubated on ice for 20 min and applied to freshly glow-discharged carbon-coated grids (Electron Microscopy Sciences) for 3 min. Grids were washed for 5 s in water and stained with 3% uranyl acetate for 3 min before blotting dry. Grids were examined on a Zeiss EM-900 microscope at 80 kV and images were recorded with an Olympus Megaview G2 camera. The same procedure was applied for poly(I:C).
Supplementary Material
Acknowledgments
We thank Marc Allaire and Lin Yang of the NSLS X9 beamline for assistance in SAXS data collection. Joy Zhao (NIH) provided advice on SV-AUC data analysis. This work was supported by Grant P01 GM022778 from NIH/NIGMS, and a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease award to YM.
Author contributions: ICB performed all the experiments. YM participated in the crystallographic data collection and structure determination of MDA5 Hel2i and devised the study. YM and ICB wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aida K, Nishida Y, Tanaka S, Maruyama T, Shimada A, Awata T, Suzuki M, Shimura H, Takizawa S, Ichijo M, Akiyama D, Furuya F, Kawaguchi A, Kaneshige M, Itakura J, Fujii H, Endo T, Kobayashi T (2011) RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes 60: 884–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamming D, Horvath CM (2009) Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J Biol Chem 284: 9700–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum J, Tonkin CJ, Paul AS, Rug M, Smith BJ, Gould SB, Richard D, Pollard TD, Cowman AF (2008) A malaria parasite formin regulates actin polymerization and localizes to the parasite-erythrocyte moving junction during invasion. Cell Host Microbe 3: 188–198 [DOI] [PubMed] [Google Scholar]
- Binder M, Eberle F, Seitz S, Mucke N, Huber CM, Kiani N, Kaderali L, Lohmann V, Dalpke A, Bartenschlager R (2011) Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid inducible gene-I (RIG-I). J Biol Chem 286: 27278–27287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PH, Balbo A, Schuck P (2009) On the analysis of sedimentation velocity in the study of protein complexes. Eur Biophys J 38: 1079–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs KS, Andrejeva J, Randall RE, Goodbourn S (2009) Mechanism of mda-5 Inhibition by paramyxovirus V proteins. J Virol 83: 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 62: 1002–1011 [DOI] [PubMed] [Google Scholar]
- Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP (2008) The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell 29: 169–179 [DOI] [PubMed] [Google Scholar]
- Dam J, Schuck P (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: sedimentation coefficient distributions c(s) and asymptotic boundary profiles from Gilbert-Jenkins theory. Biophys J 89: 651–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 42: 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G (2006) 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 314: 994–997 [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional Prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146: 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Patel SS, Marcotrigiano J (2011a) Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Osterlund P, Sarin LP, Poranen MM, Bamford DH, Guo D, Julkunen I (2011b) Innate immune responses in human monocyte-derived dendritic cells are highly dependent on the size and the 5′ phosphorylation of RNA molecules. J Immunol 187: 1713–1721 [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, Fisher PB (2004) Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene 23: 1789–1800 [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB (2002) mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA 99: 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S (2008) Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205: 1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441: 101–105 [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147: 423–435 [DOI] [PubMed] [Google Scholar]
- Li X, Lu C, Stewart M, Xu H, Strong RK, Igumenova T, Li P (2009a) Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch Biochem Biophys 488: 23–33 [DOI] [PubMed] [Google Scholar]
- Li X, Ranjith-Kumar CT, Brooks MT, Dharmaiah S, Herr AB, Kao C, Li P (2009b) The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J Biol Chem 284: 13881–13891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lo YC, Wu H (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465: 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ranjith-Kumar CT, Hao L, Kao CC, Li P (2011) Crystal structure of RIG-I C-terminal domain bound to blunt-ended double-strand RNA without 5′ triphosphate. Nucleic Acids Res 39: 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P (2010) The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18: 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM (2011) Structural insights into RNA recognition by RIG-I. Cell 147: 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37: 958–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Murali A, Li X, Ranjith-Kumar CT, Bhardwaj K, Holzenburg A, Li P, Kao CC (2008) Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J Biol Chem 283: 15825–15833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA (2009) Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324: 387–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Schneider TR (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J Appl Crystallogr 37: 843–844 [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128: 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S (2011) Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA 108: 21010–21015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 89: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis ESC (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′ phosphates. Science 314: 997–1001 [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C (2009) Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83: 10761–10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37: 317–336 [DOI] [PubMed] [Google Scholar]
- Qi S, Pang Y, Hu Q, Liu Q, Li H, Zhou Y, He T, Liang Q, Liu Y, Yuan X, Luo G, Wang J, Yan N, Shi Y (2010) Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell 141: 446–457 [DOI] [PubMed] [Google Scholar]
- Ranjith-Kumar CT, Murali A, Dong W, Srisathiyanarayanan D, Vaughan R, Ortiz-Alacantara J, Bhardwaj K, Li X, Li P, Kao CC (2009) Agonist and antagonist recognition by RIG-I, a cytoplasmic innate immunity receptor. J Biol Chem 284: 1155–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M Jr (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA 104: 582–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA 107: 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J et al. (2009) Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31: 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Takahasi K, Kumeta H, Tsuduki N, Narita R, Shigemoto T, Hirai R, Yoneyama M, Horiuchi M, Ogura K, Fujita T, Inagaki F (2009) Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J Biol Chem 284: 17465–17474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M Jr, Inagaki F, Fujita T (2008) Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell 29: 428–440 [DOI] [PubMed] [Google Scholar]
- Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD (2011) Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog 7: e1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, Hartmann G, Patel DJ (2010) Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat Struct Mol Biol 17: 781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby MC (2010) The FANCM family of DNA helicases/translocases. DNA Repair (Amst) 9: 224–236 [DOI] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19: 727–740 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175: 2851–2858 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
- Yuan S, Yu X, Topf M, Dorstyn L, Kumar S, Ludtke SJ, Akey CW (2011) Structure of the Drosophila apoptosome at 6.9 a resolution. Structure 19: 128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.