

Abstract
Despite decades of basic and clinical research, our understanding of how benzodiazepines tend to lose their efficacy over time (tolerance) is at least incomplete. In appears that tolerance develops relatively quickly for the sedative and anticonvulsant actions of benzodiazepines, whereas tolerance to anxiolytic and amnesic effects probably does not develop at all. In light of this evidence, we review the current evidence for the neuroadaptive mechanisms underlying benzodiazepine tolerance, including changes of (i) the GABAA receptor (subunit expression and receptor coupling), (ii) intracellular changes stemming from transcriptional and neurotrophic factors, (iii) ionotropic glutamate receptors, (iv) other neurotransmitters (serotonin, dopamine, and acetylcholine systems), and (v) the neurosteroid system. From the large variance in the studies, it appears that either different (simultaneous) tolerance mechanisms occur depending on the benzodiazepine effect, or that the tolerance-inducing mechanism depends on the activated GABAA receptor subtypes. Importantly, there is no convincing evidence that tolerance occurs with α subunit subtype-selective compounds acting at the benzodiazepine site.
1. Introduction
Shortly after their development in the 1960s, benzodiazepines became very popular as they exerted many desirable effects such as reduction of anxiety, anticonvulsant properties, and myorelaxation combined with a rather low toxicity [1]. However, their use is associated with many side effects precluding their long-term use, including sedation, amnesia, cognitive impairment, and ataxia. Even though guidelines generally recommend limiting benzodiazepines to short-term use, long-term use still often occurs. Chronic benzodiazepine treatment can result in the development of benzodiazepine dependence [2]. DSM-IV criteria for benzodiazepine dependence consist of various psychological (behavioral) and physical symptoms, including tolerance, withdrawal symptoms when drug intake is stopped and dose escalation [3]. Indeed, chronically treated patients become less sensitive to some effects of benzodiazepines (tolerance) which may include anticonvulsant, sedative, hypnotic, and myorelaxant effects of benzodiazepines. Also, benzodiazepine discontinuation may result in the appearance of a characteristic withdrawal syndrome with heightened anxiety, insomnia, and sensory disturbances [4]. In fact, tolerance and withdrawal could be two manifestations of the same compensatory mechanism, with withdrawal occurring when the counterbalancing benzodiazepine effect is absent [5]. This is supported by the fact that acutely induced benzodiazepine effects are opposite to the withdrawal symptoms, and that changes in glucose use in the Papez circuit (including the cingulate cortex and mammillary body) were also observed on withdrawal, implying a common circuitry in the withdrawal process [6]. However, physical dependence (usually defined by withdrawal symptoms) does not require the presence of tolerance, and tolerance may develop without any signs of physical dependence [7].
Presently, despite decades of basic and clinical research, our understanding how benzodiazepines tend to lose their efficacy over time (i.e., tolerance) is at least incomplete. Here we review the current knowledge on the neuroadaptive mechanisms underlying benzodiazepine tolerance. This paper does not specifically address the addictive properties of benzodiazepines and their effects on the dopamine system or their abuse liability potential (including their nonmedical use in popular culture), which are described in detail elsewhere [8–10].
Benzodiazepine tolerance is considered to constitute an adaptive mechanism following chronic treatment, and it may thus be regarded as an example of neuronal plasticity. Efforts have been made to explain tolerance at the molecular or functional level of the GABAA receptor because classical (nonselective) benzodiazepines modulate inhibitory GABAA receptors possessing α 1, α 2, α 3, or α 5 subunits. On the other hand, the excitatory glutamate system has also been implicated to play a role in the development of benzodiazepine tolerance [5]. Enhanced understanding of the dynamic process leading to reduced benzodiazepine efficacy following chronic treatment could accelerate the development of compounds that would maintain efficacy during chronic treatment [11]. Indeed, increasing knowledge on the specific functions of different GABAA receptor subunits has led to a breakthrough of novel and more selective drugs acting at the benzodiazepine site of the GABAA receptor. It is interesting but beyond the scope of the review to draw a comparison between benzodiazepine tolerance and alcohol tolerance as alcohol (albeit with low potency) acts at the GABAA receptor [12].
Firstly, we will discuss the molecular basis of the GABAA receptor system before taking a closer look at the clinical aspects of the development of benzodiazepine tolerance. Then, the putative molecular mechanisms underlying benzodiazepine tolerance will be extensively discussed, followed by a section specifically addressing the issue of tolerance development with novel and more selective benzodiazepines in the light of the putative tolerance mechanisms associated with classical benzodiazepines. From a clinical perspective, the understanding of tolerance is important because long-term benzodiazepine treatment with continuing efficacy—using either existing or novel and more selective drugs—could offer potential benefits to several groups of patients.
2. Benzodiazepines and the GABAA System
2.1. GABAA Receptors
GABAA receptors constitute the major fast inhibitory neurotransmitter system in the brain. They are composed of five transmembrane-spanning subunits that assemble to form a ligand-gated chloride channel with various possible subunits (α 1–6 , β 1–3 , γ 1–3 , δ, ε, θ, and π) resulting in GABAA receptor heterogeneity [13]. Binding of GABA to the GABAA receptor increases the influx of negatively charged chloride ions, resulting in an inhibitory postsynaptic signal (IPSP). Although in theory a vast number of subunits combinations could be expected, GABAA receptors are found in typical subunit compositions with the most common receptor subtype being composed of two α, two β, and one γ subunit [14] (Figure 1). In situ hybridization and immunohistochemical studies have shown that GABAA receptor subunits display a distinct CNS distribution with a differential cellular localization pattern, suggesting that GABAA receptor subunits have a specialized function (Table 1) [14]. Overall, a high expression of GABAergic subunits is present in the cortex, hippocampus, and basal ganglia [15]. Of the GABAergic subunits, α 1, β 1, β 2, β 3, and γ 2 subunits are found throughout the brain. In contrast, the α 2, α 3, α 4, α 5, α 6, γ 1, and δ subunits have a specific regional expression pattern. The α 1 subunit is highly coassembled with β 2 and γ 2 subunits and is synaptically located on neuronal cell bodies. GABAA receptors that contain an α 2 or α 3 subunit are less abundant and are codistributed with the β 3 and γ 2 subunits. The α 2 subunit is present in the cortex, hippocampus, amygdale, and hypothalamus, and often its expression is negatively correlated with the expression of α 1 subunits. The expression of the α 3 subunit is highest in the cortex, hippocampus, amygdala, thalamus, and brainstem, although it is also expressed in monoaminergic neurons (e.g., the raphe nuclei and the locus coeruleus in the brainstem) and cholinergic neurons in the forebrain. α 5 subunits are predominantly expressed in the hippocampus where they comprise 15–20% of the diazepam-sensitive GABAA receptors [16]. Regarding cellular localization, cortical and hippocampal pyramidal cells receive input from morphologically distinct GABAergic interneurons that innervate different pyramidal cell parts depending on the type of interneuron (e.g., chandelier and basket cells) with a specialized postsynaptic expression of α subunits [17, 18].
Figure 1.
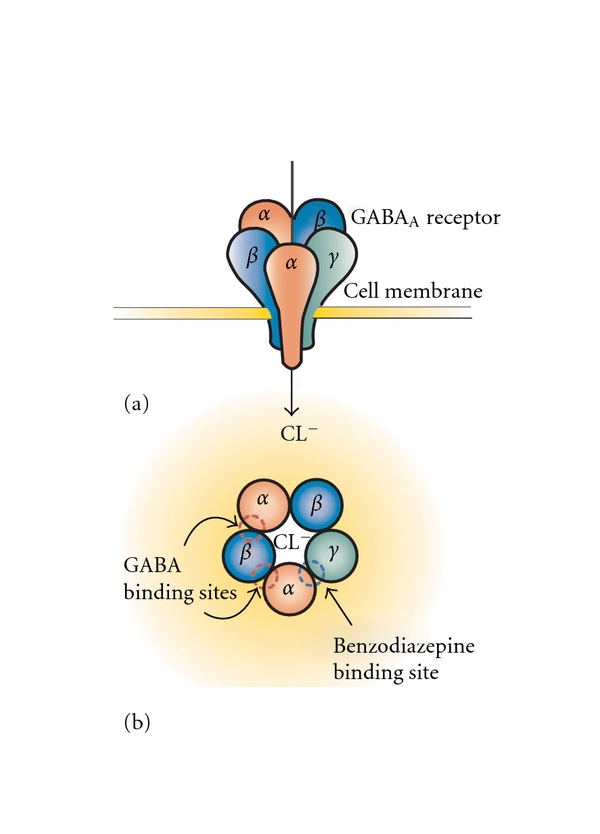
Representation of the GABAA receptor structure. The inhibitory GABAA receptor consists of five subunits that together form a ligand-gated chloride (Cl−) channel (a). When GABA binds (between the α and the β subunit of the GABAA receptor), chloride ions flow into the neuron, resulting in a hyperpolarization of the cell membrane (a). Classical nonselective benzodiazepines allosterically enhance the inhibitory actions of GABA by binding between the α 1, α 2, α 3, or α 5 subunit and the γ subunit (b). Although the GABAA receptor displays a large molecular heterogeneity depending on the subunit composition, the most common subtype is a pentamer with 2α, 2β, and 1γ subunit.
Table 1.
Localization of common GABAA receptor subtypes in the brain (adapted from [19]).
| Subtype | Frequency | Localization |
|---|---|---|
| α 1 β 2 γ 2 | Major (60%) synaptic | Cerebral cortex (layer I–VI), hippocampus, striatum, cerebellum, amygdala, brainstem. |
| α 2 β n γ 2 | Minor (15–20%) synaptic | Cerebral cortex (layers I–IV), hippocampus, striatum, hypothalamus, amygdala. |
| α 3 β n γ 2 | Minor (10–15%) synaptic | Cerebral cortex (layers V-VI), hippocampus, cerebellum, amygdala, brainstem (including raphe nuclei and locus coeruleus), spinal cord. |
| α 4 β n δ/γ | Minor (<10%) extrasynaptic | Hippocampus (dentate gyrus), thalamus, cortex. |
| α 5 β n γ 2 | Minor (<5%) extrasynaptic | Cerebral cortex, hippocampus, amygdala, hypothalamus, spinal cord. |
| α 6 β n γ2/δ | Minor (<5%)(extra) synaptic | Cerebellum. |
Thus, GABAA receptor subtypes probably possess diverging functional properties dependent on the subunit composition, contributing to the GABA signaling complexity [13]. Additionally, GABAA receptors are found synaptically as well as extrasynaptically. Synaptic receptors usually contain γ subunits and mediate fast phasic inhibition accompanied by transient high GABA concentrations [16]. By contrast, GABA has higher potency (at μM concentrations) at extrasynaptic GABAA receptors that usually contain a δ subunit, preferentially assemble with α 6 or α 4 subunits and have slow desensitization kinetics [20]. Also, α 5 subunits may be localized extrasynaptically [21]. Extrasynaptic tonic inhibition—which is not modified by benzodiazepines—is suggested to modulate excitability of neuronal networks throughout the brain.
2.2. Benzodiazepines from a Nonspecific towards a Subunit-Specific Pharmacology
Classical benzodiazepines allosterically modulate GABA-induced IPSPs by binding to the benzodiazepine site of GABAA receptors that contain an α 1, α 2, α 3, or α 5 subunit in combination with a β and a γ 2 subunit (Figure 1). The exact binding site of benzodiazepines at the GABAA receptor is located between the α and γ subunit. In contrast, benzodiazepines do not interact with GABAA receptors that contain an α 4- or α 6-subunit. In addition to benzodiazepines, other drug classes can bind to the GABAA receptor complex, including several anticonvulsants, ethanol, barbiturates, neurosteroids, and some anesthetics [15]. The fact that classical benzodiazepines non-selectively bind to different α subunits led to the hypothesis that the pharmacological profile with anxiolytic, sedative, anticonvulsive and myorelaxant properties may be further dissected. Both genetic and pharmacological approaches explored the hypothesis that α subunits differentially contribute to the different effects of classical benzodiazepines The genetic approach consisted of point mutations into specific α subunits (α 1(H101R), α 2(H101R), α 3(H126R), and α 5(H105R)), turning them functionally insensitive to benzodiazepines without altering their GABA sensitivity [22]. Pharmacological research on the GABAA receptor has focused on the development of compounds that show differential efficacy across the various α subunits [13]. Such drugs generally bind with equal affinity to all α subunits (i.e., α 1, α 2, α 3, and α 5 subunits), but selectively alter the capacity to increase GABA binding to one or more of them. Using this strategy, various efficacy-selective (and some affinity-selective) compounds have been developed with preferential agonistic activity at the α 1 (zolpidem and zaleplon), α 2/3 (TPA023, L838, 417, and SL651498), or inverse agonistic activity the α 5 subunit (α5IA, L-655,708, and MRK-016) (see also Table 2).
Table 2.
Summary of novel GABAA receptor subtype selective compounds.
| Target | Name | Efficacy (compared to a classical benzodiazepine) |
Affinity/Remarks | Ref |
|---|---|---|---|---|
| α 1 | Zolpidem | Comparable at α 1/α 2/α 3/α 5 | 5-10-fold higher affinity for α 1 versus α 2/3 > 1000 fold higher affinity for α 1 versus α 5 | [15] |
| α 2/3 | TPA023 | α 1 (0%), α 2 (11%), α 3 (21%), α 5 (5%) | Equivalent affinity | [8] |
| α 2/3 | TPA123 | α 1 (23%), α 2 (35%), α 3 (43%), α 5 (19%) | Equivalent affinity. Reinforcing efficacy and physiological dependence remained present | [8] |
| α 2/3 | L838,417 | α 1 (1.5%), α 2 (43%), α 3 (43%), α 5 (39%) | Equivalent affinity | [23] |
| α 2/3 | SL651498 |
α
1 (45%), α
2 (115%), α
3 (83%), α
5 (50%) Compared to zolpidem for α 1 efficacy |
5–10-fold increased affinity for α 2/3, 10–20 fold lower affinity for α 5 | [31] |
In line with a specific central localization and distribution of GABAergic subunits, these genetic and pharmacological approaches have demonstrated that different α subunits of the GABAA receptor mediate the distinct effects of benzodiazepines. Specifically, α 1-containing GABAA receptors probably mediate the sedative, amnesic, and anticonvulsant actions of classical benzodiazepines [13, 23]. In contrast, muscle relaxation and anxiety reduction after benzodiazepine administration was primarily ascribed to α 2 (and possibly α 3) subunit activation [24], whereas α 5 subunit-containing GABAA receptors appear to be involved in learning and memory [25, 26].
In light of the topic of this review, studies investigating the contribution of GABAergic subunits in benzodiazepine abuse liability, drug reinforcement, and tolerance development are of particular interest. Unfortunately, studies applying genetic and subtype-selective methodologies to examine the development of tolerance are scarce. One study using α subunit point mutation mice implicated a critical role for α 5 subunits together with α 1 subunits in the decreasing sedative efficacy of the classical benzodiazepine diazepam after chronic treatment [27]. We will discuss this finding in detail later in this paper. Studies on the background of physical dependence and abuse liability using subtype-selective GABAAR modulators are more abundant. Using self-administration studies, it was shown that efficacy at α 1-containing GABAA subtypes significantly contributed to the reinforcing effects and withdrawal symptoms of benzodiazepines [8, 28, 29]. Specifically, TPA123, which still possesses 23% intrinsic activity at α 1 subunits still led to benzodiazepine-like drug reinforcement and withdrawal symptoms, whereas TPA023 with 0% α 1 intrinsic activity did not, even at full GABAA receptor-binding capacity [8]. However, there is still the possibility that the lower α 2 and α 3 efficacy of TPA023 may have contributed to the absence of drug reinforcement and withdrawal. In support, L-838,417 also led to continued self-administration, even though it lacks efficacy for the α 1 subtype [30]. In any case, the α 5 subunit may not be directly involved in the abuse potential of classical benzodiazepines as the α 1-preferring hypnotic zolpidem with no affinity for the α 5 subunit still led to self-administration in primates [30]. This finding is surprising as it suggests that the α 5 subunit may be involved in tolerance development but not in drug reinforcement. Consequently, these processes could be independently mediated, even though they are both incorporated in the definition of benzodiazepine dependence.
2.3. GABA Metabolism
As benzodiazepines enhance the inhibitory effects of GABA and shift the GABA concentration-response curve to the left, the synaptic GABA concentration affects benzodiazepine efficacy. GABA is converted from glutamate by the enzyme glutamic acid decarboxylase (GAD) that maintains intracellular levels of GABA and exists in two independent isoforms (GAD65 and GAD67). In contrast to the localization of GAD67 in the neuronal body, GAD65 is primarily expressed in axon terminals, suggesting a role for GAD65 in synaptic neurotransmission and a more general role for GAD67 in regulating GABA synthesis [32]. Synaptic GABA is removed from the cleft into the presynaptic axon terminals by GABA transporters (GATs). So far, four GAT subtypes have been identified, with the highly expressed GAT1 and GAT4 being the most widely distributed [33].
3. The Development of Benzodiazepine Tolerance
Before examining the possible mechanisms underlying the development of benzodiazepine tolerance after long-term exposure, it is important to review its evidence and determine whether it is clinically relevant. Overall, there is little doubt that benzodiazepines are acutely effective in reducing anxiety, sleep latency and preventing convulsions. The tolerance that is eventually thought to develop appears to occur at different rates and to a different degree for each of the benzodiazepine effects [34]. Preclinical studies have shown that tolerance to the sedative and hypnotic effects occurs rather rapidly, followed by tolerance to the anticonvulsant effects, whereas tolerance to the anxiolytic effects of benzodiazepines are absent or partially develop after long-term treatment (for reviews, see [34–36]). As these preclinical studies have already been extensively reviewed, and novel preclinical studies on benzodiazepine tolerance have been limited in the last years to our knowledge, it is beyond this paper to reproduce all preclinical data on tolerance development. In general, preclinical studies are in agreement with the clinical divergent picture, even though in most preclinical studies, tolerance is not directly related to the applied dose, dosing interval, or the drug's plasma levels or half-life. Here, we will focus on the clinical evidence for (the rate of) tolerance development for each benzodiazepine action, even though we will also include preclinical studies when clinical studies are lacking or inconclusive.
3.1. Clinical Studies on Sedative and Hypnotic Tolerance
A study in low-dose benzodiazepine-dependent subjects showed a complete loss of hypnotic activity independent of the half-life of the prescribed benzodiazepine, even though a substantial suppression of REM sleep still occurred [37]. Also, other studies have shown that chronic users displayed no increase in sedation or motor impairment after the acute application of a benzodiazepine [38–40]. Moreover, tolerance to benzodiazepine-induced decreased reaction speed was shown after 10 days of alprazolam treatment [41]. Oral administration of triazolam, a short acting benzodiazepine, initially improved both sleep induction and maintenance, but latency to sleep and the number of awakenings were back to baseline values after two weeks of triazolam use [42]. Importantly, early-morning insomnia associated with short-acting benzodiazepines triazolam and midazolam markedly worsened after 7 days of treatment [43]. However, conflicting studies with triazolam exist that did not show any tolerance development [44, 45]. Another study applying the longer-acting benzodiazepine temazepam (15 or 30 mg) for either 26 or 54 nights in 7-8 subjects with chronic insomnia found no development of drug tolerance due to long-term temazepam administration [46]. Flurazepam, which has a relatively long elimination half-life, was shown to be effective for initiating and maintaining sleep with intermediate and long-term use (over 4 weeks), even though daytime sedation diminished during prolonged use [47]. Thus, even though tolerance to the sedative effects quickly emerges in most studies, these effects seem to be most prominent with benzodiazepines with a short half-life. Tolerance could thus depend on the half life of the applied benzodiazepine. However, this may be an overgeneralization, as a review showed that tolerance in human subjects only marginally emerged after chronic treatment with the short-acting drugs midazolam and zolpidem, even though the short-acting drug triazolam was associated with tolerance [48]. A limitation of most studies is their relatively short duration of exposure. Another issue is that convincing evidence for improved sleep after long-term use is lacking [49], yet this may not be the sole result of tolerance but could also be attributable to a generalized lack of efficacy. In support, in human subjects, discontinuation of benzodiazepines did not decrease sleep quality compared to a group that stayed on benzodiazepines up to 52 weeks after cessation [50], or even increased sleep quality and slow wave sleep after discontinuation in insomnia patients [51].
3.2. Clinical Studies on Anticonvulsant Tolerance
The use of benzodiazepines over a longer period of time in epilepsy is limited due to the development of tolerance [52]. In line with preclinical studies [53–55], tolerance develops during the first several months in 30–50% of epilepsy patients treated with either clobazam or clonazepam [56]. Thus, benzodiazepines are only prescribed in acute epileptic seizures or in a status epilepticus. However, in certain cases, intermittent use may be indicated, which may reduce the likelihood of tolerance [57]. Chronic treatment in rodents with the α 1-preferential compound CL218, 572 resulted in loss of picrotoxin-induced seizures [58]. In contrast to classical benzodiazepines, partial GABAA receptor PAMs including bretazenil did not result in anticonvulsant tolerance in several preclinical studies [54, 59, 60]. However, to our knowledge, these drugs have not been tested for (continuing) anticonvulsant activity in humans, precluding firm conclusions on their tolerance-inducing effects in epilepsy patients.
3.3. Clinical Studies on Amnesic Tolerance
Most studies have found continued short-term memory impairment after acute administration of benzodiazepines in chronically treated subjects [38, 39, 61]. Also, no tolerance for memory-impairing effects of alprazolam was found during a 10-day acute treatment [41]. However, another study reported tolerance to the acute amnesic effects of alprazolam after chronic use [40]. A major concern is that loss of memory associated with benzodiazepine use may be lasting, even after treatment discontinuation [62, 63], although other studies reported improved cognitive functioning after discontinuation with increased speed and accuracy of information processing, improved reaction time and working memory [50, 64–66]. Collectively, clinical data do not support the existence of tolerance to benzodiazepine-induced cognitive impairments.
3.4. Clinical Studies on Anxiolytic Tolerance
If developing al all, tolerance to the anxiolytic effects seems to develop more slowly compared to tolerance to the hypnotic effects. In patients with panic disorder, neither anxiolytic tolerance nor daily dose increase was observed after 8 weeks of alprazolam treatment with continued efficacy [67]. This was confirmed by another study in panic disorder patients who already chronically took alprazolam. Here, no differences were found in cortisol responsivity or anxiolytic efficacy compared to alprazolam-naïve patients, independent of disease severity [40]. Another double-blind study allocated 180 chronically anxious outpatients to diazepam (15 to 40 mg/day) and found that prolonged diazepam treatment (6–22 weeks) did not result in tolerance to the anxiolytic effects of diazepam [68]. Furthermore, additional studies all show a continuing anxiolytic effect, at least for panic disorder [69–72], generalized anxiety disorder [73], and social phobia [74–76]. Although a declining anxiolytic efficacy after long-term use of benzodiazepines cannot be clearly established, it is important to remember that other disadvantages prevent benzodiazepines to chronically treat anxiety symptoms, such as continued memory impairment, accident risk, hip fractures, and withdrawal symptoms [7, 77]. In conclusion, there is no solid evidence from the existing literature that anxiolytic efficacy declines following chronic benzodiazepine use in humans.
3.5. Clinical Studies on Drug Reinforcement Tolerance
The relevant topic of benzodiazepine tolerance to the reinforcing effects of benzodiazepines was already discussed by Licata and Rowlett [9]. They concluded that tolerance to reinforcing effects of benzodiazepines appears unlikely, supported by studies in nonhuman primates in which midazolam and zolpidem maintained stable self-injection and physical dependence under conditions of chronic continuous availability [78, 79]. Also, in humans, tolerance to drug reinforcement could lead to dose escalation that would maintain the vicious cycle of tolerance and dependence. In clinical practice, the majority of patients do not escalate their dose, suggesting that drug reinforcement tolerance may not emerge [80].
3.6. Conclusion
In conclusion, tolerance develops relatively quickly for the sedative, hypnotic, and anticonvulsant actions of benzodiazepines. Tolerance to anxiolytic and amnesic effects most probably does not appear at all. The fact that benzodiazepine dosage may be hard to reduce after chronic use can be ascribed to physical dependence to avoid withdrawal symptoms rather than the development of tolerance.
With diverging rates and varying completeness of tolerance development, it may be speculated that either (i) different tolerance mechanisms exist depending on the benzodiazepine effect, or that (ii) a uniform mechanism accounts for tolerance but revolves around the subunit composition of the targeted GABAA receptor subtype and the brain region involved. However, from the presented evidence it is difficult to conclude that benzodiazepines indeed produce a robust and reproducible tolerance for all (side) effects. It is clear however, that benzodiazepine tolerance is not a uniform process for all clinical effects and does not apply to all available benzodiazepines. However, it is not known which factors predict whether a certain benzodiazepine possesses the potential to produce tolerance. Unfortunately, many studies address the physical dependence of benzodiazepines and their abuse potential, but do not specifically investigate tolerance.
4. Mechanisms Underlying Tolerance
4.1. General
Decades of research into the molecular effects of long-term benzodiazepine treatment have already importantly advanced our understanding of tolerance and several excellent reviews on this topic have already been published [5, 11, 34, 77]. The general assumption is that chronic benzodiazepine use leads to compensating changes in the central nervous system. This way, the GABAA receptor may become less responsive to the continuing acute effects of benzodiazepines, either as a result of adaptations in the GABAA receptor itself, intracellular mechanisms, or changes in other neurotransmitter systems, such as the glutamatergic system. Although adaptive processes probably play an important role, it is important to realize that the development of tolerance is not uniform for all its actions, and differences between preclinical and clinical tolerance development may exist. Therefore, the possibility that not one but multiple adaptive mechanisms simultaneously coexist complicates research into benzodiazepine tolerance. Moreover, these adaptive changes could be limited to one or more specific brain areas. This makes it very challenging to single out one a priori unifying mechanism underlying tolerance. In support, a study in rats using 2-deoxyglucose quantitative autoradiography showed that during chronic diazepam treatment, heterogeneous tolerance to the diazepam-induced reduction of glucose utilization occurred in the brain, depending on treatment duration and brain region [6]. Whereas acute diazepam administration resulted in reductions in glucose utilization throughout the brain, 3 days of diazepam treatment led to tolerance in brain structures associated with sensory processing (parietal cortex, auditory cortex, cochlear nucleus) which was interpreted to correlate with reduced sedation. After 28-day diazepam treatment, tolerance to the depressant effect of diazepam on cerebral glucose occurred in the mamillary body, subiculum, and caudate nucleus, whereas changes in the frontal cortex approached significance. Of particular interest is the finding that none of the amygdaloid nuclei showed any blunting over time, in line with persistent anxiolytic effects of benzodiazepines.
Before taking a closer look at specific mechanisms that have been proposed to underlie benzodiazepine tolerance, it is important to note that pharmacokinetic factors probably do not play a major role in the development of tolerance [81]. In support, plasma levels after acute diazepam administration did not differ between chronically alprazolam-treated and untreated panic disorder patients, even though sedative and amnesic tolerance was observed [40]. The most obvious candidate to mediate the adaptive changes in cellular and synaptic function after chronic benzodiazepine treatment is the GABAA receptor. Therefore, we will first discuss the evidence supporting changes in the GABA system (including GABAA receptor coupling and GABA receptor expression) after chronic benzodiazepine exposure.
4.2. GABAA System Hypotheses
4.2.1. Mechanism 1: GABAA Receptor Uncoupling
One explanation for a loss of benzodiazepine function is a loss in GABAA receptor allosteric coupling. The GABAA receptor contains two GABA-binding sites and one benzodiazepine-binding site that are allosterically coupled, that is, binding to the benzodiazepine-binding site potentiates binding of GABA to the GABA-binding site (Figure 1). Benzodiazepines are generally referred to as positive allosteric modulators (PAMs) because their binding alters the GABAA receptor conformation with an increased capacity to bind GABA, leading to increased channel opening frequency, increased chloride influx, and, consequently, to hyperpolarization. GABAA receptor uncoupling is defined as a decreased ability of benzodiazepines to enhance GABA-induced IPSPs at the GABAA receptor. In terms of tolerance development, it has been hypothesized that chronic treatment affects the benzodiazepines' capacity to pharmacologically enhance the GABA response (i.e., tolerance leads to uncoupling). A decreased coupling may develop as a result of changed GABAA receptor subunit composition, alterations to the GABAA receptor itself (including phosphorylation) or its second messenger ligands, or any process affecting the conformational state of the GABAA receptor. The receptor uncoupling hypothesis is attractive as it does not assume any changes in subunit expression and ligand binding yet uses the knowledge on the specialized functions of the GABAA receptor and the different subunits. However, the uncoupling process is an aspecific process as it can be induced by exposure to different classes of GABAA receptor modulators acting at different modulatory sites, such as neurosteroids and barbiturates [82].
Already in 1984, an electrophysiological study indicated that allosteric coupling may play a role by showing a 50% decrease in the GABA enhancement of benzodiazepine-binding without significant changes in benzodiazepine-binding site density or affinity [83]. Also, more recent indications for reduced allosteric coupling were found after chronic treatment using transfected cell lines that express GABAA receptors or in neurons [84–94]. The mechanisms underlying possible differences in coupling remain poorly understood. If the GABAA receptor assembly process is modified, GABA receptor composition can be modified due to subunit replacements or altered expression in the receptor. This way, GABAA receptors with a different functionality could potentially possess reduced benzodiazepine sensitivity due to reduced GABAA receptor coupling. To our knowledge, no studies exist which have directly investigated GABAA receptor subunit composition after chronic exposure. Another mechanism to affect receptor coupling is GABAA receptor phosphorylation. GABAA receptors are phosphorylated by various protein kinases and dephosphorylated by phosphatases [95]. Dynamic functional alterations in GABAA receptor phosphorylation status may directly affect the inhibitory synaptic strength, with changes in channel openings (or indirectly influence receptor trafficking). However, the precise effects of phosphorylation on neuronal GABAA receptor function are complex, even though key residues within the intracellular loop of the GABAA receptor seem of particular importance. Using whole-cell patch-clamp recordings of GABAA receptor IPSCs in hippocampal neurons, brain-region-dependent effects of activation of cAMP-dependent protein kinase A (PKA) or Ca2+/phospholipid-dependent protein kinase C (PKC) were shown [96]. Also, PKA activity was found to be directly involved in changed GABAA receptor functioning in hippocampal pyramidal cells following chronic flurazepam treatment [97]. Probably, phosphorylation patterns rather than individual sites are of importance, supported by the finding that mutation to one PKA phosphorylation site is not involved in tolerance [90]. Using a point mutation genetic approach, transcriptional reduction was found in calcium-/calmodulin-dependent kinase IIα and MAP kinase phosphatase-1in control mice but not in α1(H101R) after acute administration of diazepam [98]. Unfortunately, no chronic treatment was included in these studies.
It remains to be seen whether changes in allosteric coupling are relevant to the development of tolerance in vivo. Because benzodiazepine tolerance gradually develops over days to weeks, this would suggest that structural changes take place, whereas posttranslational compensation would be expected to be directly manifest. In support, uncoupling seems to develop rapidly, with the classical benzodiazepine chlordiazepoxide (applied together with GABA) stimulating the rate and extent of desensitization produced in a single neuron within several seconds [99]. Also, the observed uncoupling after chronic benzodiazepine treatment is rapidly reversed by a brief exposure in vivo to the benzodiazepine antagonist flumazenil [83, 86].
4.2.2. Mechanism 2: Alterations in GABAA Receptor Subunit Expression
The most straightforward hypothesis to explain impaired sensitivity after chronic benzodiazepine exposure would be a general downregulation of GABAA receptors throughout the brain. Indeed, the process of tolerance requires GABAA receptors at least to some extent, as cell lines expressing one specific type of the GABAAR are susceptible to tolerance [86, 87, 90]. Because classical (nonselective) benzodiazepines bind to GABAA receptors that contain an α 1, α 2, α 3, or α 5 subunit, it could be expected that expression of receptors containing these α subunits (plus a γ 2 subunit) is changed. Of course, this would depend on the cellular and anatomical distribution of GABAA receptors. Already earlier in Section 2.1, the differentiated and unique distribution of GABAergic subunits in the CNS was discussed. With regard to the benzodiazepine-sensitive α subunits, the α 1 subunit is ubiquitously expressed in the entire brain, whereas the other α subunits (α 2, α 3 and α 5) display a more restricted pattern of expression (see Table 1). If receptor internalization simply downregulates GABAA receptor density, then a priori regional differentiation would be expected based on receptor distribution.
The processes that control the assembly, membrane trafficking, and synaptic accumulation of GABAA receptors are complex (for review, see [100]). In short, GABAA receptors are assembled from individual subunits out of the endoplasmic reticulum within minutes after their translation, with amino acid sequences in the N-terminus influencing the GABAA receptor subtype (Figure 2). Then, receptor trafficking to the plasma membrane takes place, facilitated by diverse helper GABAA receptor-associated proteins (among that GABARAP, BIG2, PRIP, gephyrin, and radixin). Ultimately, (clathrin-dependent) endocytosis occurs after receptor dephosphorylation, after which degradation or recycling may ensue (Figure 2). If prolonged activation of the GABA system leads to receptor downregulation, then this could be established by interfering at multiple steps of the dynamic GABAA receptor life cycle. These include decreased subunit mRNA transcription, subunit degradation in the endoplasmic reticulum (e.g., by ubiquitylation), decreased expression of GABAA receptor-associated helper proteins, and alterations in the endocytosis of specific GABAA receptor subtypes. The finding that the protein synthesis inhibitor cycloheximide and the RNA synthesis inhibitor actinomycin D blocked the effects of chronic diazepam exposure in recombinant cells expressing GABAA receptors indicates that GABAA receptor synthesis is of at least some importance [87].
Figure 2.
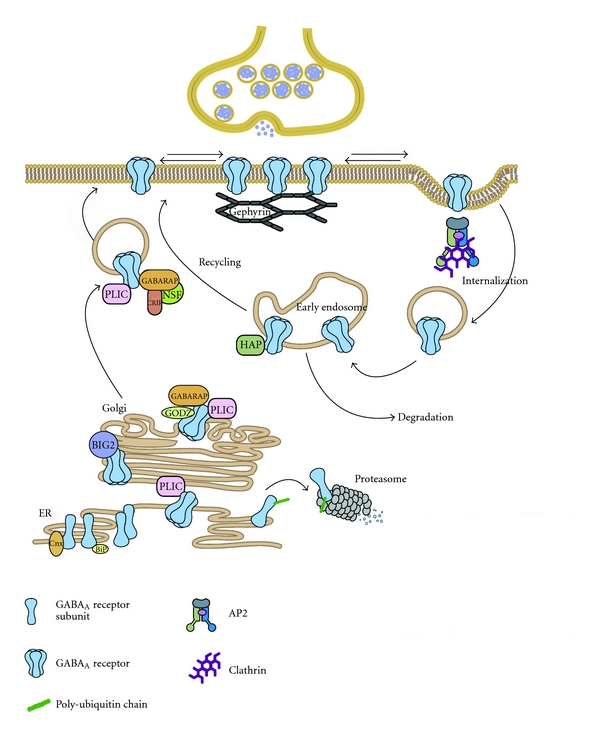
GABAA receptor trafficking and associated proteins. GABAA receptors are assembled from individual subunits in the endoplasmatic reticulum (ER) where the chaperones BiP and Calnexin assist in quality control. Unassembled GABAA receptor subunits that are to be targeted for ER-associated degradation are ubiquitinated and degraded in the proteasome. The ubiquitin-like protein PLIC can interact with GABAA receptors thereby inhibiting their targeting for proteasomal degradation. Assembled pentameric GABAA receptors exit the ER and bind the guanidine exchange factor brefeldin-A-inhibited GDP/GTP exchange factor 2 (BIG2) in the Golgi. Here they also interact with the palmitoylase transferase GODZ and Gamma-aminobutyric acid receptor-associated protein (GABARAP). GABARAP interacts with the NEM sensitive fusion (NSF) protein, as does the GABAA receptor β subunit, and this association may facilitate transport of the receptor complexes to the cell surface. GABAA receptors are inserted at extrasynaptic sites and can diffuse along the plasma membrane in and out of synaptic domains. At synapses they are stabilized by an interaction with the scaffolding protein Gephyrin. The interaction of the GABAA receptor intracellular loops with the μ2 subunit of the adaptin complex AP2 is important for GABAA receptor internalization. GABAA receptors are delivered by a clathrin-mediated pathway to early endosomes where they can be targeted for degradation in the lysosome or for recycling upon binding of Huntington-associated protein (HAP1). Reprinted by permission from Elsevier, reprinted from [101].
Up to now, a plethora of studies have tried to address whether chronic benzodiazepine treatment indeed affects GABAA receptor expression (and thus benzodiazepine binding sites) using compounds with different subtype selectivity profiles at different doses and varying treatment duration. A recent excellent review summarized all data on the regulation of GABAA receptor subunit after chronic benzodiazepine treatment that was mostly studied in rats [102]. It is beyond the scope of this review to repeat the meticulous work laid down in this paper. Of all subunits, α, β, and γ subunits have been mostly examined. This paper confirms that both for mRNA and protein subunit levels, the available evidence leads to a divergent and sometimes conflicting picture, although the majority of the studies essentially do not show any significant difference in subunit expression [102]. Furthermore, a lack of consistency appears for subunit changes in different specific brain areas. Moreover, the length and method of chronic treatment seem relevant since differences in GABAA receptor subunit mRNA levels after chronic diazepam treatment in rats can depend on whether diazepam is administered as daily systemic injections or via osmotic minipumps [103]. Binding studies also generally report no changes in benzodiazepine binding after chronic treatment [92, 93, 104]. Together, GABAA receptor expression (both mRNA and protein levels) is not consistently and robustly altered after various long-term treatment regimens. Thus, a general central downregulation or even consistent region-specific changes in GABAA receptor expression after chronic benzodiazepine use are not supported by the literature. Even though methodological differences (e.g., treatment regimen, species, route of administration, and applied drug) may account for some conflicting findings, the results seem overall inconsistent. Moreover, molecular results are often not combined with behavioral tests, preventing a direct correlation between behavioral tolerance and molecular changes. Clinical studies applying in vivo binding or postmortem GABAA receptor expression after chronic benzodiazepine treatment are to the knowledge of the authors lacking.
Changes in rates of GABAA receptor endocytosis, receptor membrane insertion, intracellular trafficking, and association with helper GABAA receptor-associated proteins could still play a role, leading to a reduction in membrane surface receptors without affecting overall subunit protein expression (e.g., see [105]). Another interesting suggestion is that a possible loss of synaptic function after chronic exposure could be due to a shift to a perisynaptic or even an extrasynaptic localization of GABAA receptors, away from clustering of GABAA receptors at synapses (Figure 2) [106]. At least in alcohol research, such dynamic changes in plasticity at inhibitory synapses have been shown [107]. Moreover, it cannot be excluded that particular subunits play a role in the development of tolerance after chronic treatment in the absence a direct up- or downregulation. Using the previously mentioned α subunit point mutation mice, acutely administered diazepam still reduced locomotor activity in α5 (H105R) mice even after chronic 8-day diazepam treatment at a combined daily dose of 15 mg/kg [27]. This suggests that the α 1 subunit that mediates the sedative effects remains responsive, indicating that simultaneous activation of the α 1 and α 5 subunit may be necessary for tolerance to the locomotor-reducing effects of classical benzodiazepines. Specifically, it was hypothesized that increased phasic signaling would alter extrasynaptic tonic inhibition mediated by α 5-containing GABAA receptors, whereas a decrease in hippocampal α 5-specific binding was reported in diazepam-tolerant mice. Also, in contrast to α 1-, α 2-, and α 3-containing receptors, α 5-containing GABAA receptors are located extrasynaptically at the base of dendritic spines where they can modulate excitatory glutamatergic input. However, α1(H101R) mice are not sensitive to the acute sedative benzodiazepine effects, making a comparison to isolated α 1 subunit activation not possible. Moreover, only tolerance to the sedative effects of diazepam was reported. Thus, it may still be possible that tolerance to other benzodiazepine effects is mediated by other subunits.
4.3. Glutamate System Hypotheses
4.3.1. General
From the previous sections, we conclude that compensatory changes solely arising from the GABA system may at most partially explain the tolerance arising following chronic treatment with benzodiazepines. Glutamate is an excitatory neurotransmitter acting on glutamate receptors. Together with the GABA system, they constitute the two fast-acting and opposing neurotransmitter systems that can modulate synaptic plasticity. In support, close neuroanatomical connections exist between GABAergic and glutamatergic neurons [108, 109]. With a presence in at least 30–50% of all synapses in the CNS, inhibitory GABA and excitatory glutamate together coordinate the balance in the brain's excitability. Therefore, it is not surprising that as these two opposing and fast-acting neurotransmitter systems form a delicate balance, chronic (increased) activation of the GABAergic system during benzodiazepine treatment may pertubate glutamatergic transmission. The basis of benzodiazepine tolerance could then lie in sensitization of the glutamatergic system—a putative process that could account for the withdrawal symptoms after chronic benzodiazepine discontinuation [5, 110]. Such sensitization is reminiscent to adaptive glutamatergic processes as seen in kindling experiments, although it should be noted that kindling only occurs with intermittent and not after continuous treatment [111]. Glutamatergic sensitization could thus play a role in the development of tolerance as well as withdrawal symptoms upon cessation of treatment. Glutamatergic changes after benzodiazepine withdrawal will not be discussed here, but there are indications that the glutamatergic system plays a role in withdrawal states with accompanying increases in anxiety and seizure activity (for review see [5]). However, glutamate receptor mRNA and protein changes may be dynamic during withdrawal, with unchanged levels during the early phase of withdrawal but changes occurring several days later [112]. This consequently complicates the interpretation of withdrawal studies and their significance for our understanding of benzodiazepine tolerance.
Similar to the GABAergic system, the glutamate system is diverse and complex, generally being divided into ionotropic and metabotropic receptor types. Ionotropic glutamate receptors form a class of heteromeric ligand-gated cation channels that potentiate the influx of K+, Na+, or Ca2+ ions following glutamate binding. Three classes of the ionotropic glutamate receptor occur in het central nervous system: the NMDA receptor (N-methyl-D-aspartate), the AMPA receptor (alpha-amino-3-hydroxy-5-methyl-4-isoxazole-4-propionic acid), and the kainate receptor (for a recent review see [113]). Functional NMDA receptors contain two obligatory GluN1 and two regulatory GluN2/3 subunits and are vital for synaptic plasticity (for review, see [114]). Each GluN subunit contains extracellular loops where coagonists glycine or D-serine (GluN1 and GluN3 subunits) and glutamate (GluN2 subunits) can bind [115]. Although the channel is blocked by Mg2+ ions, changes in membrane potential can make the channel permeable to Na+, Ca2+, and K+ ions. The central distribution of GluN2 subunits eventually ensures heterogeneity in the NMDA receptor system. AMPA receptors are widespread heterotetrameric ligand-gated ion channels composed of four types of subunits (GluR1–4), and are crucial to long-term synaptic plasticity such as long-term potentiation (for review see [116]). Although glutamate possesses lower affinity for the AMPA receptor compared to NMDA receptors, faster excitation-inducing kinetics are present at the AMPA receptor. Relevant to this review, a study showed that AMPA receptor desensitization was caused by a rupture of a domain interface which allowed the ion channel to close, providing a simple yet elegant explanation [117]. Kainate receptors are made up of four subunits, GluR5, GluR6, GluR7, KA1, and KA2, which are similar to AMPA and NMDA receptor subunits and can be arranged in different ways to form a functional tetramer (for review, see [118]). Compared to NMDA and AMPA receptors, synaptic kainate receptors exhibit slow rise and decay properties.
4.3.2. Mechanism 3: Role of Ionotropic Glutamatergic Receptors
Several studies have addressed the compensatory glutamate sensitization hypothesis during chronic benzodiazepine exposure to account for the development of tolerance (as reviewed by [5, 110]).
In rodents, the development of tolerance to the sedative effects of the classical benzodiazepines diazepam and chlordiazepoxide was prevented by coadministration of the NMDA receptor antagonists CPP, dizocilpine, MK-801, and ketamine [119–121]. Also, lorazepam-induced tolerance to its acute anticonvulsant effects was partially prevented with simultaneous CPP treatment [122]. In contrast, the development of tolerance to the anxiolytic effects of diazepam in a social interaction test was not blocked by concomitant administration of dizocilpine [123]. This suggests that the mechanism underlying tolerance to the anxiolytic effects of diazepam is different from that underlying tolerance to the sedative effects. Increases in cortical mRNA of NMDA NR1 and NR2B subunits have been reported in rats tolerant to diazepam [124, 125], which were prevented by concomitant treatment with the NMDA receptor antagonist MK-801 [126]. However, another study showed decreases in hippocampal NR2B subunits after chronic flurazepam treatment, even though the total amount of NMDA receptors was unchanged [127].
In support, after long-term (but not acute) lorazepam treatment, no differences were found in the affinity or density of NMDA receptors, even though increased in vitro glutamate release and NMDA-induced cGMP efflux in the hippocampus was reported [128]. Together, these data suggest that NMDA-dependent mechanisms contribute to the development of benzodiazepine tolerance. However, as anxiolytic tolerance was not blocked by NMDA receptor antagonism, the NMDA system could also play a differential role in tolerance depending on the specific behavioral effects [123]. Moreover, a straightforward glutamate sensitization may be an oversimplification, as tolerance to the sedative effects of lorazepam after 21-day treatment correlated with a decreased rather than an increased sensitivity for glutamate (using[(3)H] glutamate binding) [129].
Even though the AMPA receptor antagonist GYKI 52466 did not affect the development of tolerance to the sedative effects of diazepam [121], changes in AMPA receptor subunits have been reported to be altered after long-term benzodiazepine exposure [130]. Specifically, significant reductions of mGLuR1 (cortex and amygdala) and mGluR2 mRNA (amygdala) were reported in rats treated chronically with diazepam, even though the effects were complex and dependent on treatment route (subcutaneous or intraperitoneal injections). Adding to the complexity of the published data, another study did not show changes in hippocampal GluR1-3 subunit proteins following chronic flurazepam treatment, even though mEPSCs were found and nonspecific binding was increased using the AMPA receptor antagonist[(3)H] Ro48-8587 [131]. A genetic approach with GluR1 knockout mice showed that after subchronic flurazepam treatment, these mice developed a reduced and incomplete tolerance to the muscle relaxation and sedative effects of flurazepam, even though acute flurazepam effects were comparable between knockout and wild-type mice [132].
With regard to glutamatergic kainate receptors, we found no pharmacological or genetic studies investigating the development of tolerance.
Together, the evidence does not support a universal and replicable glutamatergic component, even though there are indications that NMDA receptor blockade can prevent tolerance to at least some behavioral benzodiazepine effects. However, molecular data are diverse and sometimes inconsistent, which are reminiscent of the molecular changes in the GABA system after chronic benzodiazepine treatment (see Section 4.2.2).
4.4. Other Mechanisms
4.4.1. Mechanism 4: Transcriptional and Neurotrophic Factors
Although the hypothesis that downstream signaling events adjust in response to chronic exposure to benzodiazepines seems plausible, a surprising paucity of data exist in this field. It is tempting to speculate on the expression of diverse helper GABAA receptor-associated proteins (including GABARAP, BIG2, PRIP, gephyrin, and radixin) after long-term benzodiazepine use (Figure 2). In addition, changes in intracellularly located cAMP-response-element-binding protein (CREB) or calcium, vital in various second messenger systems, could be altered, and prolonged GABA concentrations in a neuronal culture have been shown to affect voltage-gated calcium channels [133]. However, until further studies provide additional proof for chronic benzodiazepine-induced downstream intracellular changes, the evidence that this process plays a role is inconclusive.
Neurotrophic proteins support neuronal survival, synaptic growth, and differentiation throughout the brain via tyrosine kinase receptors (Trk) and, with lower affinity, via p75 receptors (p75NTRs) [134]. Neurotrophic factors that have discovered so far include brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and neurotrophin-4 (NT-4), and nerve growth factor (NGF). Since they act as potent factors in regulating fast synaptic inhibition, adaptations leading to tolerance following chronic benzodiazepine treatment could in part be mediated via these neurotrophic factors. In support, BDNF (and NT-4) was found to acutely reduce postsynaptic GABAA receptor immunoreactivity via activation of TrkB receptors [135–139], even though one study reported an increase [140], and another study reports that chronic BDNF treatment potentiates GABAergic inhibition [141]. This reduced immunoreactivity was hypothesized to be caused by a reduction in GABAA receptor surface expression and was accompanied by reduced postsynaptic responses with the direct GABAA receptor agonist muscimol [142]. Mechanistically, BDNF-induced suppression of GABAergic signaling was hypothesized to stem from altered GABAA receptor composition, increased GABAA receptor phosphorylation, decreased subunit synthesis, or increased postsynaptic receptor internalization or diffusion [139]. Interestingly, all these proposed mechanisms were already discussed in this paper. Thus, neurotrophin-induced changes may not be an independent mechanism, but be a player in a causal chain of events. Again, to our knowledge, no studies exist on the effects of chronic benzodiazepine treatment on neurotrophic expression and functionality.
4.4.2. Mechanism 5: Serotonin, Dopamine, Acetylcholine Systems
There is ample evidence that the serotonin, dopamine, and acetylcholine receptor systems can modulate the GABAA receptor functionality [143–146] (Figure 3). For example, the receptor for activated C kinase (RACK-1) potentiated PKC-dependent phosphorylation of GABAA receptors mediated by the activation of muscarinic acetylcholine receptors [145], and serotonergic neurotransmission inhibited GABAergic signaling via GABAA receptor PKC-dependent phosphorylation, again with involvement of RACK-1 [144]. Altogether, these neurotransmitter systems act via G-protein-coupled receptors to activate protein kinases (PKA and PKC) and scaffold proteins that may subsequently modulate GABAA receptor β and γ 2 subunit phosphorylation (Figure 3) [95].
Figure 3.
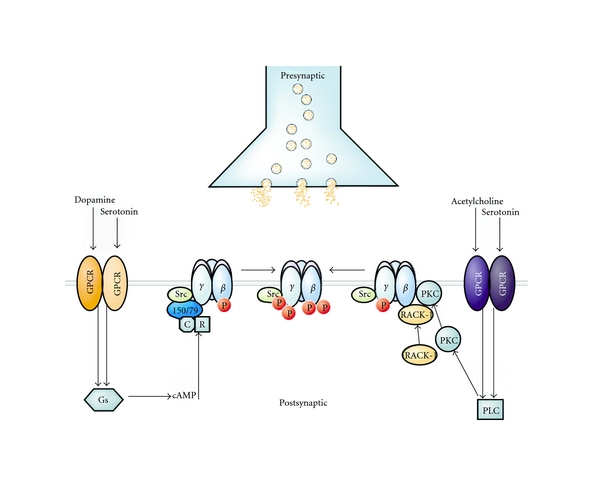
Functional crosstalk between G-protein coupled receptors (GPCRs) (which are present in the serotonin, dopamine, acetylcholine system) and GABAA receptors is facilitated through multiple protein kinases and scaffold proteins. GABAA receptor β and γ2 subunits are phosphorylated (P) by PKA and PKC upon the activation of individual GPCRs for dopamine and serotonin. PKA phosphorylation of GABAA receptor β1 and β3 subunits is dependent upon AKAP150/79, which directly interacts with these receptor subunits. AKAP150/79 also binds inactive PKA composed of regulatory (R) and catalytic (C) subunits. In addition, PKC phosphorylates the receptor β1–3 and γ2 subunits. Upon the activation of the appropriate GPCR, PKC-mediated phosphorylation is facilitated by the direct (but independent) interaction of the receptor for activated C kinase (RACK-1) and the β isoform of PKC with the GABAA receptor β1–3 subunits. RACK-1 facilitates functional regulation of GABAA receptors by controlling the activity of PKC associated with these proteins. The GABAA receptor γ2 subunit is also phosphorylated by Src, and this kinase is capable of binding to receptor β and γ2 subunits. Finally, the functional effects of phosphorylation are diverse and range from inhibitions to enhancements of GABAA receptor activity, dependent upon the receptor subunit composition. Reprinted by permission from Elsevier, reprinted from [95].
However, studies investigating the role of the serotonin, dopamine, and acetylcholine system in response to chronic benzodiazepine treatment are scarce. Three weeks of diazepam treatment (25 mg/day) in healthy male volunteers resulted in tolerance to the prolactin and growth hormone response induced by the 5-HT precursor L-tryptophan, even though sedative effects of L-tryptophan remained present [147]. Another study showed that chronic diazepam treatment resulted not only in diazepam tolerance but also in a very modest reduced efficacy of the 5-HT1A receptor agonist 8-OH-DPAT to induce flat body posture and forepaw treading [148]. In contrast, only acute but not chronic diazepam treatment decreased basal extracellular dopamine levels in rats, even though both acute and chronic treatment regimens could reverse the stress-induced rise of cortical dopamine levels [149].
4.4.3. Mechanism 6: Neurosteroids
There is ample and convincing evidence that neurosteroids are endogenous allosteric regulators that interact with GABAA receptors to modulate both tonic (extrasynaptic) and phasic (synaptic) inhibition (for reviews, see [150, 151]). Also, acute or chronic neurosteroid treatment may change GABAA receptor subunit expression, especially extrasynaptic α 4 and δ subunits [151]. In light of the plasticity-inducing actions of neurosteroids on inhibitory signaling, long-term enhancement of the GABA system with benzodiazepines may in turn evoke changes in the neurosteroids system such as changes in neurosteroid synthesis and metabolism, although classical benzodiazepines may differ in their potency to cause such changes [152]. In support, ovariectomy attenuated the development of tolerance to the anticonvulsant actions of diazepam [153]. Moreover, co-administration of the neurosteroids allopregnanolone or pregnenolone (but not dehydroepiandrosterone) prevented the development of tolerance after chronic treatment with either triazolam and diazepam [154]. Adding to the complexity of the putative involvement of neurosteroids in benzodiazepine tolerance, factors such as GABAA receptor subunit composition, phosphorylation mechanisms, and ((extra)synaptic) localization—which are all factors that were already found to be involved in tolerance development—influence the specific dynamics of neurosteroid activity.
4.4.4. Conclusion
From our review of the literature on the various mechanisms that may underlie benzodiazepine tolerance, it occurs that there is a considerable variance in the published data. The heterogeneity of the data lies in the application of different methodologies, species, treatment regimens, and benzodiazepines. Specifically, we have considered classical benzodiazepines as a homogenous drug class since they all lead to a nonspecific enhancement of GABAA receptors that contain an α 1, α 2, α 3, or α 5 subunit. However, in vivo pharmacodynamic potency and pharmacokinetic half-life differences could greatly impact on tolerance processes [7]. In support, subchronic treatment with different classical benzodiazepines lead to differential propensity for FG7142-induced seizures in mice, with triazolam, clonazepam, and diazepam producing around seizures in around 80% of the mice, whereas alprazolam and midazolam did so in 60% of the animals and lorazepam in 40% of the animals [155]. Surprisingly, chlordiazepoxide did not lead to any precipitated seizures, even though a comparable GABAA receptor occupancy was obtained. Therefore, the assumption that classical benzodiazepines act as a homogeneous class probably complicates the interpretation of the current literature.
Altogether, it appears that none of the proposed putative mechanisms can sufficiently explain tolerance development. Thus, multiple mechanisms may (synergistically) coexist, or an additional yet undiscovered mechanism may be present. However, the complex and adaptive nature of the GABA system and the existing heterogeneous literature on benzodiazepine tolerance suggest that one unifying tolerance mechanism may be a vast oversimplication. In any case, the proposed tolerance mechanisms are not completely independent, exemplified by the fact that neurotrophic factors and neurosteroids are influenced by GABAA receptor composition and phosphorylation status, which are themselves proposed to be involved in benzodiazepine tolerance. Unfortunately, the present literature does not consistently support a clear recommendation in terms of a pharmacological GABAA receptor profile (e.g., subunit preference) to aid in the development of novel and more selective benzodiazepines that lack tolerance development and are suitable for long-term treatment.
5. Tolerance to Novel Subtype-Selective Benzodiazepines
Here, we will review the evidence for tolerance development with novel GABAA receptor subtype selective compounds that provide the direct opportunity to evaluate their roles in tolerance. With the development of subunit-selective benzodiazepines, it has become possible to dissect the different effects of classical benzodiazepines (see Section 2.2 and Table 2). However, declining efficacy over time is a complex process which may not be easily attributed to one specific α subunit. Still, if novel drugs possess reduced propensity to lead to tolerance development, this will be greatly welcomed from a clinical perspective. Continuing efficacy with these drugs would advance the clinical use of drugs acting at the GABAA receptor benzodiazepine site. Unfortunately, not many studies have directly addressed tolerance development using these novel compounds. Recent data from our laboratory suggest that no tolerance develops to the acute hypothermic, anxiolytic, or sedative effect of diazepam in mice treated for 28 days with the GABAA-α 2/α 3 selective compound TPA023 (Table 2) [156], indicating that chronic activation of GABAA-α 2/α 3 receptors does not lead to anxiolytic tolerance after acute diazepam challenge (unpublished data). Also, in contrast to morphine, no analgesic tolerance occurred in rats after a 9-day treatment with the α 2/3 subtype GABAA receptor positive allosteric modulator L838,417 using a model of neuropathic pain [157]. From these data, it seems that tolerance development after chronic administration of GABAA-α 2/α 3 subtype selective drugs may not develop, or, alternatively, that tolerance to diazepam's sedative actions needs concomitant activation of GABAA-α 1/GABAA-α 5 receptors. In support of the latter hypothesis, ligands that do not bind to the α 5 subunit such as zolpidem have a reduced tendency to engender tolerance [158, 159], supported by studies in which chronic treatment with zolpidem (but not midazolam) did not produce any tolerance to sedative and anticonvulsant effects in mice and rats [160–162].
In addition to studies directly assessing tolerance, several studies have investigated the precipitated withdrawal after (sub) chronic treatment with subtype-selective compounds. Compounds with selective efficacy at α 2, α 3, and α 5 GABAA receptor subtypes were shown to lead to differential seizures susceptibility in mice in response to the inverse agonist FG-7142 [155]. Chronic treatment with zolpidem, as well as the selective compounds L-838,417 (partial agonist at α2 GABAA, α3GABAA, and α5GABAA receptors) and SL651498 (full agonist at α2GABAA and α3GABAA receptors, partial agonist at α1GABAA and α5GABAA receptors), did not result in seizures following FG-7142 administration [31, 155] (Table 2). Similarly, chronic treatment with TPA023 (partial agonist at α2GABAA, α3GABAA, and α5GABAA receptors) also did not result in FG-7142-induced seizures in mice [156]. However, because these studies do not specifically address tolerance development, the rather general conclusion from these studies is that partial or selective modulation of the GABAA receptor results in a reduced liability for physical dependence. Thus, it is important to note that, even though zolpidem does not seem to engender any obvious tolerance development, zolpidem can lead to withdrawal symptoms that are comparable to those seen after chronic classical benzodiazepine treatment [29, 77]. Thus, tolerance and withdrawal symptoms may constitute separate entities in benzodiazepine dependence. In support, one study demonstrated that marked withdrawal symptoms appeared upon abrupt discontinuation of chronic clorazepate treatment in dogs, even though tolerance was present to a rather limited extent [163].
Together, it can be concluded that so far, α 2/α 3 subtype selective compounds have neither been found to lead to tolerance nor withdrawal symptoms. This would constitute a significant improvement over currently used benzodiazepines, even though the anxiolytic profile of these compounds remains to be determined [164], and abuse liability may still be present [8]. However, interpretations should be made with caution since chronic treatment with nonselective partial positive allosteric modulators such as bretazenil did neither result in anticonvulsant tolerance [54, 59, 60] nor in FG-7142-precipitated seizures [155]. These studies implicate that the potency of classical and subtype-selective compounds, in addition to or despite subtype selectivity, may also be of importance in the development of tolerance. It could be also hypothesized that low efficacy at the α 1 subunit, rather than selectivity or reduced efficacy at α 2/α 3 subtypes, may be the causal mechanism preventing tolerance development. Also, the clinical anxiolytic efficacy of α 2/α 3 subtype selective compounds has not yet been established. In addition to a specific efficacy profile, tolerance development may also depend on a compound's affinity at certain GABAA receptor subtypes. This way, tolerance processes may be different with affinity-selective compounds such as zolpidem compared to efficacy-selective compounds such as TPA023. Circumstantial evidence stems from the fact that α 1-preferential affinity-selective compounds such as zolpidem produce physical dependence [165], even though the compound TPA123 that possesses 23% efficacy at the α 1 subunit (but is not affinity selective) did also result in physical dependence [8]. However, based on the currently available evidence, no definite conclusions can be drawn regarding the subtype involved in tolerance. Also, it is not possible to distinguish tolerance processes in selective binding (affinity) and selective activation (efficacy).
6. Conclusion
In the present paper, we summarized the rather inconsistent data regarding changes in several neurotransmitter systems to explain the development of tolerance. Specifically, we addressed possible changes at the level of (i) the GABAA receptor (subunit expression and receptor coupling), (ii) intracellular changes stemming from transcriptional and neurotrophic factors, (iii) ionotropic glutamate receptors, (iv) other neurotransmitters (serotonin, dopamine, and acetylcholine systems), and (v) the neurosteroid system. From the large variance in the studies, it appears that either different (simultaneous) tolerance mechanisms occur depending on the benzodiazepine effect, or that one tolerance-inducing mechanism depends on the activated GABAA receptor subtypes. This is not unlikely, given that tolerance is a heterogeneous process that occurs at different rates for the various effects and also depends on the profile of the (subtype selective) benzodiazepine. Adaptations could then occur on different time scales depending on the receptor subtype and brain region involved. In line with this hypothesis, tolerance develops relatively quickly for the sedative and anticonvulsant actions of benzodiazepines, whereas tolerance to anxiolytic and amnesic effects most probably do not develop at all. It is intriguing that anxiolytic effects of classical benzodiazepines may not decline during prolonged treatment. In addition to subtype selectivity, additional factors may be important for a (subtype-selective) benzodiazepine to cause tolerance, including GABAA receptor potency (efficacy) and in vivo receptor occupancy over time. The finding that partial agonists with an overall but comparable lower efficacy at all α subunits of the GABAA receptor such as bretazenil did not result in anticonvulsant tolerance raises the possibility that chronic clinical use of these compounds is associated with a lower tolerance.
An important question is how the development of tolerance of benzodiazepines could be reduced. One interesting suggestion could be—rather than intermittent use that can be defined by an individual—to develop benzodiazepine dosing schedules with varying daily doses including placebos. This could result in continued clinical efficacy (obviously depending on the indication) and utilize the placebo effect. The other possibility to reduce tolerance is the currently developing and promising body of literature on subtype-selective GABAA receptor PAMs. From the literature we reviewed, it appears that α 2/α 3 subtype selective compounds do not lead to tolerance or withdrawal symptoms. However, the underlying mechanism (reduced α 1 efficacy or a generally reduced efficacy profile) is unknown. Also, it is presently unclear whether this lack of tolerance also applies to α 1- and α 5-selective GABAergic positive allosteric modulators, although a broad and unspecific tolerance resulting from selective (and often low potency) compounds seems unlikely.
In conclusion, the development of tolerance following chronic benzodiazepine treatment is a complex process in which multiple processes may simultaneously act to cause varying rates of tolerance depending on the studied effect and the administered drug. There is no convincing evidence that subtype-selective compounds acting at the benzodiazepine site lead to tolerance at a level comparable to classical benzodiazepines. If this is indeed the case, one consequence may be that such subtype-selective compounds are unlikely to engender clinical tolerance, which would be a clinically significant improvement over classical benzodiazepines.
References
- 1.Lader M. History of benzodiazepine dependence. Journal of Substance Abuse Treatment. 1991;8(1-2):53–59. doi: 10.1016/0740-5472(91)90027-8. [DOI] [PubMed] [Google Scholar]
- 2.Owen RT, Tyrer P. Benzodiazepine dependence. A review of the evidence. Drugs. 1983;25(4):385–398. doi: 10.2165/00003495-198325040-00003. [DOI] [PubMed] [Google Scholar]
- 3.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV-TR. 2000. [Google Scholar]
- 4.Petursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455–1459. doi: 10.1111/j.1360-0443.1994.tb03743.x. [DOI] [PubMed] [Google Scholar]
- 5.Allison C, Pratt JA. Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence. Pharmacology and Therapeutics. 2003;98(2):171–195. doi: 10.1016/s0163-7258(03)00029-9. [DOI] [PubMed] [Google Scholar]
- 6.Pratt JA, Brett RR, Laurie DJ. Benzodiazepine dependence: from neural circuits to gene expression. Pharmacology Biochemistry and Behavior. 1998;59(4):925–934. doi: 10.1016/s0091-3057(97)00539-x. [DOI] [PubMed] [Google Scholar]
- 7.Woods JH, Katz JL, Winger G. Benzodiazepines: use, abuse, and consequences. Pharmacological Reviews. 1992;44(2):151–347. [PubMed] [Google Scholar]
- 8.Ator NA, Atack JR, Hargreaves RJ, Burns HD, Dawson GR. Reducing abuse liability of GABAA/benzodiazepine ligands via selective partial agonist efficacy at α 1 and α 2/3 subtypes. Journal of Pharmacology and Experimental Therapeutics. 2010;332(1):4–16. doi: 10.1124/jpet.109.158303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Licata SC, Rowlett JK. Abuse and dependence liability of benzodiazepine-type drugs: GABAA receptor modulation and beyond. Pharmacology Biochemistry and Behavior. 2008;90(1):74–89. doi: 10.1016/j.pbb.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan KR, Brown M, Labouébe G, et al. Neural bases for addictive properties of benzodiazepines. Nature. 2010;463(7282):769–774. doi: 10.1038/nature08758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wafford KA. GABAA receptor subtypes: any clues to the mechanism of benzodiazepine dependence? Current Opinion in Pharmacology. 2005;5(1):47–52. doi: 10.1016/j.coph.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Krystal JH, Staley J, Mason G, et al. γ-aminobutyric acid type A receptors and alcoholism: intoxication, dependence, vulnerability, and treatment. Archives of General Psychiatry. 2006;63(9):957–968. doi: 10.1001/archpsyc.63.9.957. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph U, Mohler H. GABA-based therapeutic approaches: GABAA receptor subtype functions. Current Opinion in Pharmacology. 2006;6(1):18–23. doi: 10.1016/j.coph.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 14.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends in Neurosciences. 1996;19(4):139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 15.Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacological Reviews. 1995;47(2):181–234. [PubMed] [Google Scholar]
- 16.Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Current Topics in Medicinal Chemistry. 2002;2(8):795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- 17.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyl P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378(6552):75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 18.Lewis DA, Cho RY, Carter CS, et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. American Journal of Psychiatry. 2008;165(12):1585–1593. doi: 10.1176/appi.ajp.2008.08030395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. Journal of Pharmacology and Experimental Therapeutics. 2002;300(1):2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 20.Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. Journal of Neuroscience. 2009;29(41):12757–12763. doi: 10.1523/JNEUROSCI.3340-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL. Synaptic and nonsynaptic localization of GABAA receptors containing the α5 subunit in the rat brain. Journal of Comparative Neurology. 2006;499(3):458–470. doi: 10.1002/cne.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature. 1999;401(6755):796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 23.McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nature Neuroscience. 2000;3(6):587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 24.Atack JR. GABAA receptor subtype-selective modulators. I. alpha2/alpha3-selective agonists as non-sedating anxiolytics. Current Topics in Medicinal Chemistry. 2010;2:331–360. doi: 10.2174/156802611795371350. [DOI] [PubMed] [Google Scholar]
- 25.Collinson N, Kuenzi FM, Jarolimek W, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the α5 subunit of the GABAA receptor. Journal of Neuroscience. 2002;22(13):5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawson GR, Maubach KA, Collinson N, et al. An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1335–1345. doi: 10.1124/jpet.105.092320. [DOI] [PubMed] [Google Scholar]
- 27.van Rijnsoever C, Täuber M, Choulli MK, et al. Requirement of α5-GABAA receptors for the development of tolerance to the sedative action of diazepam in mice. Journal of Neuroscience. 2004;24(30):6785–6790. doi: 10.1523/JNEUROSCI.1067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ator NA. Contributions of GABAA receptor subtype selectivity to abuse liability and dependence potential of pharmacological treatments for anxiety and sleep disorders. CNS Spectrums. 2005;10(1):31–39. doi: 10.1017/s1092852900009883. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths RR, Sannerud CA, Ator NA, Brady JV. Zolpidem behavioral pharmacology in baboons: self-injection, discrimination, tolerance and withdrawal. Journal of Pharmacology and Experimental Therapeutics. 1992;260(3):1199–1208. [PubMed] [Google Scholar]
- 30.Rowlett JK, Platt DM, Lelas S, Atack JR, Dawson GR. Different GABAA receptor subtypes mediate the anxiolytic, abuse-related, and motor effects of benzodiazepine-like drugs in primates. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(3):915–920. doi: 10.1073/pnas.0405621102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griebel G, Perrault G, Simiand J, et al. SL651498: an anxioselective compound with functional selectivity for alpha2- and alpha3-containing gamma-aminobutyric acidAGABAA receptors. Journal of Pharmacology and Experimental Therapeutics. 2001;298(2):753–768. [PubMed] [Google Scholar]
- 32.Benes FM, Todtenkopf MS, Logiotatos P, Williams M. Glutamate decarboxylase(65)-immunoreactive terminals in cingulate and prefrontal cortices of schizophrenic and bipolar brain. Journal of Chemical Neuroanatomy. 2000;20(3-4):259–269. doi: 10.1016/s0891-0618(00)00105-8. [DOI] [PubMed] [Google Scholar]
- 33.Schousboe A, Sarup A, Larsson OM, White HS. GABA transporters as drug targets for modulation of GABAergic activity. Biochemical Pharmacology. 2004;68(8):1557–1563. doi: 10.1016/j.bcp.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 34.Bateson AN. Basic pharmacologic mechanisms involved in benzodiazepine tolerance and withdrawal. Current Pharmaceutical Design. 2002;8(1):5–21. doi: 10.2174/1381612023396681. [DOI] [PubMed] [Google Scholar]
- 35.File SE. Tolerance to the behavioral actions of benzodiazepines. Neuroscience and Biobehavioral Reviews. 1985;9(1):113–121. doi: 10.1016/0149-7634(85)90037-5. [DOI] [PubMed] [Google Scholar]
- 36.File SE. The history of benzodiazepine dependence: a review of animal studies. Neuroscience and Biobehavioral Reviews. 1990;14(2):135–146. doi: 10.1016/s0149-7634(05)80214-3. [DOI] [PubMed] [Google Scholar]
- 37.Schneider-Helmert D. Why low-dose benzodiazepine-dependent insomniacs can’t escape their sleeping pills. Acta Psychiatrica Scandinavica. 1988;78(6):706–711. doi: 10.1111/j.1600-0447.1988.tb06408.x. [DOI] [PubMed] [Google Scholar]
- 38.Lucki I, Rickels K. The behavioral effects of benzodiazepines following long-term use. Psychopharmacology Bulletin. 1986;22(2):424–433. [PubMed] [Google Scholar]
- 39.Lucki I, Rickels K, Geller AM. Chronic use of benzodiazepines and psychomotor and cognitive test performance. Psychopharmacology. 1986;88(4):426–433. doi: 10.1007/BF00178503. [DOI] [PubMed] [Google Scholar]
- 40.Cowley DS, Roy-Byrne PP, Radant A, et al. Benzodiazepine sensitivity in panic disorder: effects of chronic alprazolam treatment. Neuropsychopharmacology. 1995;12(2):147–157. doi: 10.1016/0893-133X(94)00074-A. [DOI] [PubMed] [Google Scholar]
- 41.Allen D, Curran HV, Lader M. The effects of repeated doses of clomipramine and alprazolam on physiological, psychomotor and cognitive functions in normal subjects. European Journal of Clinical Pharmacology. 1991;40(4):355–362. doi: 10.1007/BF00265843. [DOI] [PubMed] [Google Scholar]
- 42.Kales A, Scharf MB, Kales JD. Rebound insomnia: a new clinical syndrome. Science. 1978;201(4360):1039–1041. doi: 10.1126/science.684426. [DOI] [PubMed] [Google Scholar]
- 43.Kales A, Soldatos CR, Bixler EO, Kales JD. Early morning insomnia with rapidly eliminated benzodiazepines. Science. 1983;220(4592):95–97. doi: 10.1126/science.6131538. [DOI] [PubMed] [Google Scholar]
- 44.Pegram V, Hyde P, Linton P. Chronic use of triazolam: the effects on the sleep patterns of insomniacs. Journal of International Medical Research. 1980;8(3):224–231. doi: 10.1177/030006058000800308. [DOI] [PubMed] [Google Scholar]
- 45.Roth T, Kramer M, Lutz T. Intermediate use of triazolam: a sleep laboratory study. Journal of International Medical Research. 1976;4(1):59–63. doi: 10.1177/030006057600400109. [DOI] [PubMed] [Google Scholar]
- 46.Mitler MM, Carskadon MA, Phillips RL, et al. Hypnotic efficacy of temazepam: a long-term sleep laboratory evaluation. British Journal of Clinical Pharmacology. 1979;8(1):63S–68S. doi: 10.1111/j.1365-2125.1979.tb00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kales A, Kales JD. Sleep laboratory studies of hypnotic drugs: efficacy and withdrawal effects. Journal of Clinical Psychopharmacology. 1983;3(2):140–150. doi: 10.1097/00004714-198304000-00038. [DOI] [PubMed] [Google Scholar]
- 48.Soldatos CR, Dikeos DG, Whitehead A. Tolerance and rebound insomnia with rapidly eliminated hypnotics: a meta-analysis of sleep laboratory studies. International Clinical Psychopharmacology. 1999;14(5):287–303. [PubMed] [Google Scholar]
- 49.Monane M, Glynn RJ, Avorn J. The impact of sedative-hypnotic use on sleep symptoms in elderly nursing home residents. Clinical Pharmacology and Therapeutics. 1996;59(1):83–92. doi: 10.1016/S0009-9236(96)90027-2. [DOI] [PubMed] [Google Scholar]
- 50.Curran HV, Collins R, Fletcher S, Kee SCY, Woods B, Iliffe S. Older adults and withdrawal from benzodiazepine hypnotics in general practice: effects on cognitive function, sleep, mood and quality of life. Psychological Medicine. 2003;33(7):1223–1237. doi: 10.1017/s0033291703008213. [DOI] [PubMed] [Google Scholar]
- 51.Poyares D, Guilleminault C, Ohayon MM, Tufik S. Chronic benzodiazepine usage and withdrawal in insomnia patients. Journal of Psychiatric Research. 2004;38(3):327–334. doi: 10.1016/j.jpsychires.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Browne TR, Penry JK. Benzodiazepines in the treatment of epilepsy: a review. Epilepsia. 1973;14(3):277–310. doi: 10.1111/j.1528-1157.1973.tb03965.x. [DOI] [PubMed] [Google Scholar]
- 53.Loscher W, Rundfeldt C, Honack D, Ebert U. Long-term studies on anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. I. Comparison of diazepam, clonazepam, clobazam and abecarnil. Journal of Pharmacology and Experimental Therapeutics. 1996;279(2):561–572. [PubMed] [Google Scholar]
- 54.Rundfeldt C, Wlaz P, Honack D, Loscher W. Anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. Comparison of diazepam, Bretazenil and Abecarnil. Journal of Pharmacology and Experimental Therapeutics. 1995;275(2):693–702. [PubMed] [Google Scholar]
- 55.Gonsalves SF, Gallager DW. Time course for development of anticonvulsant tolerance and GABAergic subsensitivity after chronic diazepam. Brain Research. 1987;405(1):94–99. doi: 10.1016/0006-8993(87)90993-0. [DOI] [PubMed] [Google Scholar]
- 56.Ko D, Rho JM, DeGiorgio CM, Sato S. Benzodiazpines. In: Engel JJ, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Philadelphia, Pa, USA: Lippincott-Raven; 1997. pp. 1475–1489. [Google Scholar]
- 57.Brodie MJ. Established anticonvulsants and treatment of refractory epilepsy. The Lancet. 1990;336(8711):350–354. doi: 10.1016/0140-6736(90)91886-f. [DOI] [PubMed] [Google Scholar]
- 58.File SE, Wilks L. Effects of acute and chronic treatment on the pro- and anti-convulsant actions of CL 218, 872, PK 8165 and PK 9084, putative ligands for the benzodiazepine receptor. Journal of Pharmacy and Pharmacology. 1985;37(4):252–256. doi: 10.1111/j.2042-7158.1985.tb05054.x. [DOI] [PubMed] [Google Scholar]
- 59.Feely M, Boyland P, Picardo A, Cox A, Gent JP. Lack of anticonvulsant tolerance with RU 32698 and Ro 17-1812. European Journal of Pharmacology. 1989;164(2):377–380. doi: 10.1016/0014-2999(89)90482-2. [DOI] [PubMed] [Google Scholar]
- 60.Loscher W, Rundfeldt C, Honack D, Ebert U. Long-term studies on anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. II. The novel imidazoquinazolines NNC 14-0185 and NNC 14-0189. Journal of Pharmacology and Experimental Therapeutics. 1996;279(2):573–581. [PubMed] [Google Scholar]
- 61.Curran HV, Bond A, O’Sullivan G, et al. Memory functions, alprazolam and exposure therapy: a controlled longitudinal study of agoraphobia with panic disorder. Psychological Medicine. 1994;24(4):969–976. doi: 10.1017/s0033291700029056. [DOI] [PubMed] [Google Scholar]
- 62.Tata PR, Rollings J, Collins M, Pickering A, Jacobson RR. Lack of cognitive recovery following withdrawal from long-term benzodiazepine use. Psychological Medicine. 1994;24(1):203–213. doi: 10.1017/s0033291700026969. [DOI] [PubMed] [Google Scholar]
- 63.Barker MJ, Greenwood KM, Jackson M, Crowe SF. Persistence of cognitive effects after withdrawal from long-term benzodiazepine use: a meta-analysis. Archives of Clinical Neuropsychology. 2004;19(3):437–454. doi: 10.1016/S0887-6177(03)00096-9. [DOI] [PubMed] [Google Scholar]
- 64.Tsunoda K, Uchida H, Suzuki T, Watanabe K, Yamashima T, Kashima H. Effects of discontinuing benzodiazepine-derivative hypnotics on postural sway and cognitive functions in the elderly. International Journal of Geriatric Psychiatry. 2010;25(12):1259–1265. doi: 10.1002/gps.2465. [DOI] [PubMed] [Google Scholar]
- 65.Tonne U, Hiltunen AJ, Vikander B, et al. Neuropsychological changes during steady-state drug use, withdrawal and abstinence in primary benzodiazepine-dependent patients. Acta Psychiatrica Scandinavica. 1995;91(5):299–304. doi: 10.1111/j.1600-0447.1995.tb09786.x. [DOI] [PubMed] [Google Scholar]
- 66.Rickels K, Lucki I, Schweizer E, García-España F, Case WG. Psychomotor performance of long-term benzodiazepine users before, during, and after benzodiazepine discontinuation. Journal of Clinical Psychopharmacology. 1999;19(2):107–113. doi: 10.1097/00004714-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Schweizer E, Rickels K, Weiss S, Zavodnick S. Maintenance drug treatment of panic disorder: I. Results of a prospective, placebo-controlled comparison of alprazolam and imipramine. Archives of General Psychiatry. 1993;50(1):51–60. doi: 10.1001/archpsyc.1993.01820130053009. [DOI] [PubMed] [Google Scholar]
- 68.Rickels K, Case G, Downing RW, Winokur A. Long-term diazepam therapy and clinical outcome. The Journal of the American Medical Association. 1983;250(6):767–771. [PubMed] [Google Scholar]
- 69.Charney DS, Woods SW. Benzodiazepine treatment of panic disorder: a comparison of alprazolam and lorazepam. Journal of Clinical Psychiatry. 1989;50(11):418–423. [PubMed] [Google Scholar]
- 70.Ballenger JC. Long-term pharmacologic treatment of panic disorder. Journal of Clinical Psychiatry. 1991;52(2, supplement):18–23. [PubMed] [Google Scholar]
- 71.Burrows GD, Judd FK, Norman TR. Long-term drug treatment of panic disorder. Journal of Psychiatric Research. 1993;27(supplement 1):111–125. doi: 10.1016/0022-3956(93)90022-t. [DOI] [PubMed] [Google Scholar]
- 72.Pollack MH, Tesar GE, Rosenbaum JF, Spier SA. Clonazepam in the treatment of panic disorder and agoraphobia: a one-year follow-up. Journal of Clinical Psychopharmacology. 1986;6(5):302–304. [PubMed] [Google Scholar]
- 73.Cohn JB, Wilcox CS. Long-term comparison of alprazolam, lorazepam and placebo in patients with an anxiety disorder. Pharmacotherapy. 1984;4(2):93–98. doi: 10.1002/j.1875-9114.1984.tb03327.x. [DOI] [PubMed] [Google Scholar]
- 74.Sutherland SM, Tupler LA, Colket JT, Davidson JRT. A 2-year follow-up of social phobia: status after a brief medication trial. Journal of Nervous and Mental Disease. 1996;184(12):731–738. doi: 10.1097/00005053-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 75.Davidson JRT, Ford SM, Smith RD, Potts NLS. Long-term treatment of social phobia with clonazepam. Journal of Clinical Psychiatry. 1991;52(11, supplement):16–20. [PubMed] [Google Scholar]
- 76.Otto MW, Pollack MH, Gould RA, et al. A comparison of the efficacy of clonazepam and cognitive-behavioral group therapy for the treatment of social phobia. Journal of Anxiety Disorders. 2000;14(4):345–358. doi: 10.1016/s0887-6185(00)00027-x. [DOI] [PubMed] [Google Scholar]
- 77.Griffiths RR, Weerts EM. Benzodiazepine self-administration in humans and laboratory animals—implications for problems of long-term use and abuse. Psychopharmacology. 1997;134(1):1–37. doi: 10.1007/s002130050422. [DOI] [PubMed] [Google Scholar]
- 78.Weerts EM, Griffiths RR. Zolpidem self-injection with concurrent physical dependence under conditions of long-term continuous availability in baboons. Behavioural Pharmacology. 1998;9(3):285–297. [PubMed] [Google Scholar]
- 79.Weerts EM, Kaminski BJ, Griffiths RR. Stable low-rate midazolam self-injection with concurrent physical dependence under conditions of long-term continuous availability in baboons. Psychopharmacology. 1998;135(1):70–81. doi: 10.1007/s002130050487. [DOI] [PubMed] [Google Scholar]
- 80.Soumerai SB, Simoni-Wastila L, Singer C, et al. Lack of relationship between long-term use of benzodiazepines and escalation to high dosages. Psychiatric Services. 2003;54(7):1006–1011. doi: 10.1176/appi.ps.54.7.1006. [DOI] [PubMed] [Google Scholar]
- 81.Fernandes C, File SE, Berry D. Evidence against oppositional and pharmacokinetic mechanisms of tolerance to diazepam’s sedative effects. Brain Research. 1996;734(1-2):236–242. [PubMed] [Google Scholar]
- 82.Friedman LK, Gibbs TT, Farb DH. γ-aminobutyric acidA receptor regulation: heterologous uncoupling of modulatory site interactions induced by chronic steroid, barbiturate, benzodiazepine, or GABA treatment in culture. Brain Research. 1996;707(1):100–109. doi: 10.1016/0006-8993(95)01226-5. [DOI] [PubMed] [Google Scholar]
- 83.Gallager DW, Lakoski JM, Gonsalves Rauch SFSL. Chronic benzodiazepine treatment decreases postsynaptic GABA sensitivity. Nature. 1984;308(5954):74–77. doi: 10.1038/308074a0. [DOI] [PubMed] [Google Scholar]
- 84.Wong G, Lyon T, Skolnick P. Chronic exposure to benzodiazepine receptor ligands uncouples the γ—aminobutyric acid type A receptor in WSS-1 cells. Molecular Pharmacology. 1994;46(6):1056–1062. [PubMed] [Google Scholar]
- 85.Tietz EI, Chiu TH, Rosenberg HC. Regional GABA/benzodiazepine receptor/chloride channel coupling after acute and chronic benzodiazepine treatment. European Journal of Pharmacology. 1989;167(1):57–65. doi: 10.1016/0014-2999(89)90747-4. [DOI] [PubMed] [Google Scholar]
- 86.Primus RJ, Yu J, Xu J, et al. Allosteric uncoupling after chronic benzodiazepine exposure of recombinant γ-aminobutyric acidA receptors expressed in Sf9 cells: ligand efficacy and subtype selectivity. Journal of Pharmacology and Experimental Therapeutics. 1996;276(3):882–890. [PubMed] [Google Scholar]
- 87.Pericic D, Strac DS, Jembrek MJ, Vlainic J. Allosteric uncoupling and up-regulation of benzodiazepine and GABA recognition sites following chronic diazepam treatment of HEK 293 cells stably transfected with α1β2γ2S subunits of GABAA receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2007;375(3):177–187. doi: 10.1007/s00210-007-0152-z. [DOI] [PubMed] [Google Scholar]
- 88.Klein RL, Harris RA. Regulation of GABAA receptor structure and function by chronic drug treatments in vivo and with stably transfected cells. Japanese Journal of Pharmacology. 1996;70(1):1–15. doi: 10.1254/jjp.70.1. [DOI] [PubMed] [Google Scholar]
- 89.Klein RL, Mascia MP, Harkness PC, Hadingham KL, Whiting PJ, Harris RA. Regulation of allosteric coupling and function of stably expressed γ-aminobutyric acid GABAA receptors by chronic treatment with GABAA and benzodiazepine agonists. Journal of Pharmacology and Experimental Therapeutics. 1995;274(3):1484–1492. [PubMed] [Google Scholar]
- 90.Ali NJ, Olsen RW. Chronic benzodiazepine treatment of cells expressing recombinant GABAA receptors uncouples allosteric binding: studies on possible mechanisms. Journal of Neurochemistry. 2001;79(5):1100–1108. doi: 10.1046/j.1471-4159.2001.00664.x. [DOI] [PubMed] [Google Scholar]
- 91.Itier V, Granger P, Perrault G, Depoortere H, Scatton B, Avenet P. Protracted treatment with diazepam reduces benzodiazepine1 receptor-mediated potentiation of gamma-aminobutyric acid-induced currents in dissociated rat hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics. 1996;279(3):1092–1099. [PubMed] [Google Scholar]
- 92.Heninger C, Gallager DW. Altered γ-aminobutyric acid/benzodiazepine interaction after chronic diazepam exposure. Neuropharmacology. 1988;27(10):1073–1076. doi: 10.1016/0028-3908(88)90070-6. [DOI] [PubMed] [Google Scholar]
- 93.Brett RR, Pratt JA. Changes in benzodiazepine-GABA receptor coupling in an accumbens-habenula circuit after chronic diazepam treatment. British Journal of Pharmacology. 1995;116(5):2375–2384. doi: 10.1111/j.1476-5381.1995.tb15083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roca DJ, Rozenberg I, Farrant M, Farb DH. Chronic agonist exposure induces down-regulation and allosteric uncoupling of the γ-aminobutyric acid/benzodiazepine receptor complex. Molecular Pharmacology. 1990;37(1):37–43. [PubMed] [Google Scholar]
- 95.Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Current Opinion in Neurobiology. 2003;13(3):341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 96.Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. Journal of Neuroscience. 1999;19(2):674–683. doi: 10.1523/JNEUROSCI.19-02-00674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lilly SM, Zeng XJ, Tietz EI. Role of protein kinase A in GABAA receptor dysfunction in CA1 pyramidal cells following chronic benzodiazepine treatment. Journal of Neurochemistry. 2003;85(4):988–998. doi: 10.1046/j.1471-4159.2003.01746.x. [DOI] [PubMed] [Google Scholar]
- 98.Huopaniemi, L, Keist R, Randolph A, Certa U, Rudolph U. Diazepam-induced adaptive plasticity revealed by alpha1 GABAA receptor-specific expression profiling. Journal of Neurochemistry. 2004;88(5):1059–1067. doi: 10.1046/j.1471-4159.2003.02216.x. [DOI] [PubMed] [Google Scholar]
- 99.Mierlak D, Farb DH. Modulation of neurotransmitter receptor desensitization chlordiazepoxide stimulates fading of the GABA response. Journal of Neuroscience. 1988;8(3):814–820. doi: 10.1523/JNEUROSCI.08-03-00814.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nature Reviews Neuroscience. 2008;9(5):331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Arancibia-Cárcamo IL, Kittler JT. Regulation of GABAA receptor membrane trafficking and synaptic localization. Pharmacology and Therapeutics. 2009;123(1):17–31. doi: 10.1016/j.pharmthera.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 102.Uusi-Oukari M, Korpi ER. Regulation of GABAA receptor subunit expression by pharmacological agents. Pharmacological Reviews. 2010;62(1):97–135. doi: 10.1124/pr.109.002063. [DOI] [PubMed] [Google Scholar]
- 103.Arnot MI, Davies M, Martin IL, Bateson AN. GABAA receptor gene expression in rat cortex: differential effects of two chronic diazepam treatment regimes. Journal of Neuroscience Research. 2001;64(6):617–625. doi: 10.1002/jnr.1115. [DOI] [PubMed] [Google Scholar]
- 104.Little HJ, Nutt DJ, Taylor SC. Bidirectional effects of chronic treatment with agonists and inverse agonists at the benzodiazepine receptor. Brain Research Bulletin. 1987;19(3):371–378. doi: 10.1016/0361-9230(87)90106-7. [DOI] [PubMed] [Google Scholar]
- 105.Tehrani MH, Barnes EM., Jr. Sequestration of gamma-aminobutyric acidA receptors on clathrin-coated vesicles during chronic benzodiazepine administration in vivo . Journal of Pharmacology and Experimental Therapeutics. 1997;283(1):384–390. [PubMed] [Google Scholar]
- 106.Barnes EM., Jr. Intracellular trafficking of GABAA receptors. Life Sciences. 2000;66(12):1063–1070. doi: 10.1016/s0024-3205(99)00469-5. [DOI] [PubMed] [Google Scholar]
- 107.Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. Journal of Neuroscience. 2006;26(6):1749–1758. doi: 10.1523/JNEUROSCI.4702-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herman JP, Mueller NK, Figueiredo H. Role of GABA and glutamate circuitry in hypothalamo-pituitary-adrenocortical stress integration. Annals of the New York Academy of Sciences. 2004;1018:35–45. doi: 10.1196/annals.1296.004. [DOI] [PubMed] [Google Scholar]
- 109.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. Journal of Neuroscience. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stephens DN. A glutamatergic hypothesis of drug dependence: extrapolations from benzodiazepine receptor ligands. Behavioural Pharmacology. 1995;6(5-6):425–446. [PubMed] [Google Scholar]
- 111.Marley RJ, Heninger C, Hernandez TD, Gallager DW. Chronic administration of β-carboline-3-carboxylic acid methylamide by continuous intraventricular infusion increases GABAergic function. Neuropharmacology. 1991;30(3):245–251. doi: 10.1016/0028-3908(91)90151-z. [DOI] [PubMed] [Google Scholar]
- 112.Izzo E, Auta J, Impagnatiello F, Pesold C, Guidotti A, Costa E. Glutamic acid decarboxylase and glutamate receptor changes during tolerance and dependence to benzodiazepines. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3483–3488. doi: 10.1073/pnas.051628698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bard L, Groc L. Glutamate receptor dynamics and protein interaction: lessons from the NMDA receptor. Molecular and Cellular Neuroscience. 2011;48(4):298–307. doi: 10.1016/j.mcn.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 115.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Current Opinion in Pharmacology. 2007;7(1):39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 116.Nakagawa T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Molecular Neurobiology. 2010;42(3):161–184. doi: 10.1007/s12035-010-8149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E. Measurement of conformational changes accompanying desensitization in an Ionotropic glutamate receptor. Cell. 2006;127(1):85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 118.Lerma J. Kainate receptor physiology. Current Opinion in Pharmacology. 2006;6(1):89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 119.Khanna JM, Chau A, Shah G. Effect of NMDA antagonists on rapid tolerance to benzodiazepines. Brain Research Bulletin. 1997;42(2):99–103. doi: 10.1016/s0361-9230(96)00219-5. [DOI] [PubMed] [Google Scholar]
- 120.File SE, Fernandes C. Dizocilpine prevents the development of tolerance to the sedative effects of diazepam in rats. Pharmacology Biochemistry and Behavior. 1994;47(4):823–826. doi: 10.1016/0091-3057(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 121.Steppuhn KG, Turski L. Diazepam dependence prevented by glutamate antagonists. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(14):6889–6893. doi: 10.1073/pnas.90.14.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koff JM, Pritchard GA, Greenblatt DJ, Miller LG. The NMDA receptor competitive antagonist CPP modulates benzodiazepine tolerance and discontinuation. Pharmacology. 1997;55(5):217–227. doi: 10.1159/000139531. [DOI] [PubMed] [Google Scholar]
- 123.Fernandes C, File SE. Dizocilpine does not prevent the development of tolerance to the anxiolytic effects of diazepam in rats. Brain Research. 1999;815(2):431–434. doi: 10.1016/s0006-8993(98)01160-3. [DOI] [PubMed] [Google Scholar]
- 124.Tsuda M, Shimizu N, Yajima Y, Suzuki T, Misawa M. Hypersusceptibility to DMCM-induced seizures during diazepam withdrawal in mice: evidence for upregulation of NMDA receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1998;357(3):309–315. doi: 10.1007/pl00005172. [DOI] [PubMed] [Google Scholar]
- 125.Perez MF, Salmiron R, Ramirez OA. NMDA-NR1 and -NR2B subunits mRNA expression in the hippocampus of rats tolerant to diazepam. Behavioural Brain Research. 2003;144(1-2):119–124. doi: 10.1016/s0166-4328(03)00072-x. [DOI] [PubMed] [Google Scholar]
- 126.Almiron RS, Perez MF, Ramirez OA. MK-801 prevents the increased NMDA-NR1 and NR2B subunits mRNA expression observed in the hippocampus of rats tolerant to diazepam. Brain Research. 2004;1008(1):54–60. doi: 10.1016/j.brainres.2004.01.080. [DOI] [PubMed] [Google Scholar]
- 127.Van Sickle BJ, Cox AS, Schak K, Greenfield LJ, Tietz EI. Chronic benzodiazepine administration alters hippocampal CA1 neuron excitability: NMDA receptor function and expression. Neuropharmacology. 2002;43(4):595–606. doi: 10.1016/s0028-3908(02)00152-1. [DOI] [PubMed] [Google Scholar]
- 128.Bonavita C, Ferrero A, Cereseto M, Velardez M, Rubio M, Wikinski S. Adaptive changes in the rat hippocampal glutamatergic neurotransmission are observed during long-term treatment with lorazepam. Psychopharmacology. 2003;166(2):163–167. doi: 10.1007/s00213-002-1373-y. [DOI] [PubMed] [Google Scholar]
- 129.Bonavita CD, Bisagno V, Bonelli CG, Acosta GB, Rubio MC, Wikinski SI. Tolerance to the sedative effect of lorazepam correlates with a diminution in cortical release and affinity for glutamate. Neuropharmacology. 2002;42(5):619–625. doi: 10.1016/s0028-3908(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 130.Allison C, Pratt JA. Differential effects of two chronic diazepam treatment regimes on withdrawal anxiety and AMPA receptor characteristics. Neuropsychopharmacology. 2006;31(3):602–619. doi: 10.1038/sj.npp.1300800. [DOI] [PubMed] [Google Scholar]
- 131.Van Sickle BJ, Tietz EI. Selective enhancement of AMPA receptor-mediated function in hippocampal CA1 neurons from chronic benzodiazepine-treated rats. Neuropharmacology. 2002;43(1):11–27. doi: 10.1016/s0028-3908(02)00065-5. [DOI] [PubMed] [Google Scholar]
- 132.Aitta-aho T, Vekovischeva OY, Neuvonen PJ, Korpi ER. Reduced benzodiazepine tolerance, but increased flumazenil-precipitated withdrawal in AMPA-receptor GluR-A subunit-deficient mice. Pharmacology Biochemistry and Behavior. 2009;92(2):283–290. doi: 10.1016/j.pbb.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 133.Lyons HR, Land MB, Gibbs TT, Farb DH. Distinct signal transduction pathways for GABA-induced GABAA receptor down-regulation and uncoupling in neuronal culture: a role for voltage-gated calcium channels. Journal of Neurochemistry. 2001;78(5):1114–1125. doi: 10.1046/j.1471-4159.2001.00501.x. [DOI] [PubMed] [Google Scholar]
- 134.Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nature Reviews Neuroscience. 2005;6(8):603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 135.Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. European Journal of Neuroscience. 2001;13(7):1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- 136.Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. Journal of Neuroscience. 2004;24(2):522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. Journal of Neuroscience. 1997;17(9):2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABAA receptor-mediated responses via postsynaptic mechanisms. Journal of Physiology. 2003;548(3):711–721. doi: 10.1113/jphysiol.2002.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Henneberger C, Jüttner R, Rothe T, Grantyn R. Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. Journal of Neurophysiology. 2002;88(2):595–603. doi: 10.1152/jn.2002.88.2.595. [DOI] [PubMed] [Google Scholar]
- 140.Mizoguchi Y, Kanematsu T, Hirata M, Nabekura J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. Journal of Biological Chemistry. 2003;278(45):44097–44102. doi: 10.1074/jbc.M305872200. [DOI] [PubMed] [Google Scholar]
- 141.Bolton MM, Pittman AJ, Lo DC, Lo Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. Journal of Neuroscience. 2000;20(9):3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hewitt SA, Bains JS. Brain-derived neurotrophic factor silences GABA synapses onto hypothalamic neuroendocrine cells through a postsynaptic dynamin-mediated mechanism. Journal of Neurophysiology. 2006;95(4):2193–2198. doi: 10.1152/jn.01135.2005. [DOI] [PubMed] [Google Scholar]
- 143.Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Molecular Neurobiology. 2002;26(2-3):203–216. doi: 10.1385/MN:26:2-3:203. [DOI] [PubMed] [Google Scholar]
- 144.Feng J, Cai X, Zhao J, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. Journal of Neuroscience. 2001;21(17):6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Brandon NJ, Jovanovic JN, Smart TG, Moss SJ. Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABAA receptors with the activation of G-protein-coupled receptors. Journal of Neuroscience. 2002;22(15):6353–6361. doi: 10.1523/JNEUROSCI.22-15-06353.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang X, Zhong P, Yan Z. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. Journal of Neuroscience. 2002;22(21):9185–9193. doi: 10.1523/JNEUROSCI.22-21-09185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nutt DJ, Cowen PJ, Franklin M. The effect of diazepam on indices of 5-HT function in man. Pharmacology Biochemistry and Behavior. 1986;24(5):1491–1495. doi: 10.1016/0091-3057(86)90217-0. [DOI] [PubMed] [Google Scholar]
- 148.Khan A, Haleem DJ. Tolerance in the anxiolytic profile following repeated administration of diazepam but not buspirone is associated with a decrease in the responsiveness of postsynaptic 5-HT-1A receptors. Acta Biologica Hungarica. 2007;58(4):345–357. doi: 10.1556/ABiol.58.2007.4.2. [DOI] [PubMed] [Google Scholar]
- 149.Hegarty AA, Vogel WH. The effect of acute and chronic diazepam treatment on stress-induced changes in cortical dopamine in the rat. Pharmacology Biochemistry and Behavior. 1995;52(4):771–778. doi: 10.1016/0091-3057(95)00177-x. [DOI] [PubMed] [Google Scholar]
- 150.Lambert JJ, Cooper MA, Simmons RDJ, Weir CJ, Belelli D. Neurosteroids: endogenous allosteric modulators of GABAA receptors. Psychoneuroendocrinology. 2009;34(supplement 1):S48–S58. doi: 10.1016/j.psyneuen.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 151.Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharmacology and Therapeutics. 2007;116(1):20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 152.Usami N, Yamamoto T, Shintani S, et al. Substrate specificity of human 3(20)α-hydroxysteroid dehydrogenase for neurosteroids and its inhibition by benzodiazepines. Biological and Pharmaceutical Bulletin. 2002;25(4):441–445. doi: 10.1248/bpb.25.441. [DOI] [PubMed] [Google Scholar]
- 153.Wilson MA, Biscardi R. Effects of gender and gonadectomy on responses to chronic benzodiazepine receptor agonist exposure in rats. European Journal of Pharmacology. 1992;215(1):99–107. doi: 10.1016/0014-2999(92)90614-a. [DOI] [PubMed] [Google Scholar]
- 154.Reddy DS, Kulkarni SK. Neurosteroid coadministration prevents development of tolerance and augments recovery from benzodiazepine withdrawal anxiety and hyperactivity in mice. Methods and Findings in Experimental and Clinical Pharmacology. 1997;19(6):395–405. [PubMed] [Google Scholar]
- 155.Mirza NR, Nielsen EO. Do subtype-selective gamma-aminobutyric acid A receptor modulators have a reduced propensity to induce physical dependence in mice? Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1378–1385. doi: 10.1124/jpet.105.094474. [DOI] [PubMed] [Google Scholar]
- 156.Atack JR, Wafford KA, Tye SJ, et al. TPA023 [7-(1, 1-dimethylethyl)-6-(2-ethyl-2H-1, 2, 4-triazol-3-ylmethoxy)-3-(2-fluor ophenyl)-1, 2, 4-triazolo[4, 3-b]pyridazine], an agonist selective for alpha2- and alpha3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. Journal of Pharmacology and Experimental Therapeutics. 2006;316(1):410–422. doi: 10.1124/jpet.105.089920. [DOI] [PubMed] [Google Scholar]
- 157.Knabl J, Witschi R, Hosl K, et al. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451(7176):330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- 158.Zammit G. Comparative tolerability of newer agents for insomnia. Drug Safety. 2009;32(9):735–748. doi: 10.2165/11312920-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 159.Ravishankar A, Carnwath T. Zolpidem tolerance and dependence—two case reports. Journal of Psychopharmacology. 1998;12(1):103–104. doi: 10.1177/026988119801200114. [DOI] [PubMed] [Google Scholar]
- 160.Sanger DJ, Zivkovic B. Differential development of tolerance to the depressant effects of benzodiazepine and non-benzodiazepine agonists at the omega (BZ) modulatory sites of GABAA receptors. Neuropharmacology. 1992;31(7):693–700. doi: 10.1016/0028-3908(92)90148-i. [DOI] [PubMed] [Google Scholar]
- 161.Sanger DJ, Zivkovic B. Investigation of the development of tolerance to the actions of zolpidem and midazolam. Neuropharmacology. 1987;26(10):1513–1518. doi: 10.1016/0028-3908(87)90172-9. [DOI] [PubMed] [Google Scholar]
- 162.Perrault G, Morel E, Sanger DJ, Zivkovic B. Lack of tolerance and physical dependence upon repeated treatment with the novel hypnotic zolpidem. Journal of Pharmacology and Experimental Therapeutics. 1992;263(1):298–303. [PubMed] [Google Scholar]
- 163.Scherkl R, Kurudi D, Frey HH. Clorazepate in dogs: tolerance to the anticonvulsant effect and signs of physical dependence. Epilepsy Research. 1989;3(2):144–150. doi: 10.1016/0920-1211(89)90042-9. [DOI] [PubMed] [Google Scholar]
- 164.Atack JR. Subtype-selective GABAA receptor modulation yields a novel pharmacological profile: the design and development of TPA023. Advances in Pharmacology. 2009;57:137–185. doi: 10.1016/S1054-3589(08)57004-9. [DOI] [PubMed] [Google Scholar]
- 165.Ator NA. Zaleplon and triazolam: drug discrimination, plasma levels, and self-administration in baboons. Drug and Alcohol Dependence. 2000;61(1):55–68. doi: 10.1016/s0376-8716(00)00123-x. [DOI] [PubMed] [Google Scholar]