

Abstract
Neurodegenerative tauopathies, such as Alzheimer's disease (AD), are characterized by insoluble deposits of hyperphosphorylated tau protein within brain neurons. Increased phosphorylation and decreased solubility has been proposed to diminish normal tau stabilization of microtubules (MTs), thereby leading to neuronal dysfunction. Earlier studies have provided evidence that small molecule MT-stabilizing drugs that are used in the treatment of cancer may have utility in the treatment of tauopathies. However, it has not been established whether treatment with a small molecule MT-stabilizing compound will provide benefit in a transgenic model with pre-existing tau pathology, as would be seen in human patients with clinical symptoms. Accordingly, we describe here an interventional study of the brain-penetrant MT-stabilizing agent, epothilone D (EpoD), in aged PS19 mice with existing tau pathology and related behavioral deficits. EpoD treatment reduced axonal dystrophy and increased axonal MT density in the aged PS19 mice, which led to improved fast axonal transport and cognitive performance. Moreover, the EpoD-treated PS19 mice had less forebrain tau pathology and increased hippocampal neuronal integrity, with no dose-limiting side effects. These data reveal that brain-penetrant MT-stabilizing drugs hold promise for the treatment of AD and related tauopathies, and that EpoD could be a candidate for clinical testing.
Introduction
Insoluble fibrillar inclusions comprised of hyperphosphorylated tau proteins are found in the brains of patients with Alzheimer's disease (AD) (Lee et al., 2001b; Ballatore et al., 2007; Brunden et al., 2009). The occurrence of tau aggregates in AD, frontotemporal lobar degeneration (FTLD), and several other “tauopathies” suggests that neurodegeneration is mechanistically linked to pathological tau. This theory is supported both by the discoveries that tau gene mutations cause inherited FTLD (Hong et al., 1998; Hutton et al., 1998) and by the correlation between tau pathology and the degree of AD dementia (Wilcock and Esiri, 1982; Arriagada et al., 1992; Gómez-Isla et al., 1997).
In healthy neurons, tau is enriched in axons and promotes microtubule (MT) stabilization (Cleveland et al., 1977a,b). The sequestration of tau into insoluble inclusions could result in disruption of normal tau function (Lee et al., 1994), which could alter MT stability and fast axonal transport (FAT). An approach to compensate for tau loss-of-function would be to use molecules that stabilize MTs (Lee et al., 1994, 2001b; Brunden et al., 2009). This strategy was first tested (Zhang et al., 2005) when the MT-stabilizing drug, paclitaxel, was administered to transgenic (Tg) mice that develop primarily brainstem and spinal cord tau pathology (Ishihara et al., 1999). The tau Tg mice had improved MT density, FAT, and motor performance after paclitaxel absorption at neuromuscular junctions. However, paclitaxel is not suitable for human tauopathies because of poor blood–brain barrier (BBB) permeability (Fellner et al., 2002; Brunden et al., 2011).
Subsequent studies demonstrated that intranasal delivery of the MT-stabilizing octapeptide, NAP, reduced tau and Aβ in the brains of 3X Tg mice (Matsuoka et al., 2007, 2008). This molecule (AL-108) is undergoing evaluation in patients with progressive supranuclear palsy (Gravitz, 2011). We more recently identified epothilone D (EpoD) as a preferred MT-stabilizing compound for the potential treatment of tauopathies (Brunden et al., 2010, 2011). In a preventative study, EpoD was administered weekly for 3 months to young PS19 tau Tg mice that initially lacked significant tau pathology. EpoD largely prevented the axonal MT loss and dystrophy, as well as spatial learning deficits, which manifested as these mice developed forebrain tau pathology with age (Yoshiyama et al., 2007; Brunden et al., 2010).
Left unresolved in the prophylactic study of EpoD was the important question of whether this agent would be efficacious in Tg mice with established tau pathology, which would more accurately model the human clinical setting where tauopathy patients are identified after the onset of cognitive deficits and tau pathology (Jack et al., 2010). We have now evaluated EpoD in aged PS19 mice with pre-existing tau pathology. EpoD treatment resulted in significant improvements of MT density, axonal integrity, FAT, and cognitive performance, without the onset of side effects. Moreover, EpoD treatment reduced hippocampal neuron and axon loss. Thus, EpoD safely corrected axonal defects in an interventional mouse model of AD-like tauopathy, suggesting that this agent has potential for the treatment of human tauopathies.
Materials and Methods
Preparation of EpoD.
EpoD was prepared as previously described (Lee et al., 2001a; Rivkin et al., 2004). The spectroscopic properties of EpoD were identical to those reported in the literature. Compound purity was >95% as demonstrated by liquid chromatography-mass spectrometry.
EpoD or vehicle treatment of PS19 and non-Tg WT mice.
A transgene encoding the human T34 tau isoform (1N4R) containing the P301S mutation found in FTDP-17 (Goedert and Jakes, 2005) and driven by the mouse PrP promoter was used to create tau Tg mice on a B6C3/F1 background as described previously (Yoshiyama et al., 2007). Groups (n = 12–13) of 9-month-old male PS19 tau Tg mice or 9-month-old non-Tg littermates were administered weekly intraperitoneal injections of 1 mg/kg EpoD or 0.3 mg/kg of EpoD, or vehicle (DMSO), for a total of 3 months. Male PS19 mice were chosen for these studies as they develop tau pathology more consistently than do females. Animals were monitored for signs of abnormal behavior or distress, and were weighed weekly. After 11 weeks of dosing, the mice underwent behavioral testing as described below. A subset of mice (n = 3) from each study were used for FAT studies (see below). The remaining animals were killed at study completion, with blood collected for complete blood cell counts and brain and optic nerve (ON) recovered for immunohistochemical and biochemical analyses. The mice from each group also underwent necropsy evaluation with organ weights recorded.
Electron microscopy evaluation of ON axonal dystrophy and MT density.
Transmission electron microscopy (EM) was performed on cross sections of ONs from vehicle- and drug-treated PS19 mice or non-Tg littermates. The ONs were removed and post-fixed in 2% glutaraldehyde and 2% paraformaldehyde, and then processed for standard EM as described previously (Zhang et al., 2005). Ultrathin sections from ON blocks were cut and mounted on EM grids and examined using a JEM1010 electron microscope (Jeol) at 80 kV. Cross sections of the entire ON were systematically sampled at 10,000× and 50,000× magnification, with dystrophic axons and MTs counted from multiple fields of 128 and 0.035 μm2, respectively, from at least 25 separate photos. The assessors were masked to the treatment group.
FAT studies.
Three mice from each treatment group underwent bilateral intravitreal injections (2 μl) of 0.5 mCi of [35S]methionine (PerkinElmer) with a 33 gauge needle connected to a 5 μl Hamilton syringe over a period of 2 min while under deep anesthesia (Brady et al., 1999; Kirkpatrick et al., 2001), in accordance with protocols approved by the University of Pennsylvania. Mice were killed 3 h after injection, with removal of ONs and dissection into seven consecutive 1-mm-long segments. Matched pairs of segments from each individual mouse were homogenized in 66 μl of BUST buffer (0.5% SDS, 8 m urea, 2% β-mercaptoethanol, 0.1% proteinase inhibitor mixture, and 0.1 m Tris HCl, pH 6.8) at room temperature, and then sonicated with 20 pulses of a tip-sonicator followed by centrifugation at 85,000 × g for 40 min at 30°C as previously described (Zhang et al., 2005). The extent of FAT was examined by comparing the proximal-to-distal distribution of a single 35S-labeled protein within each ON segment after SDS-PAGE. Aliquots of the ON segment homogenates containing equal volumes were resolved on 7.5% SDS-PAGE gels and transferred onto nitrocellulose membranes. The radioactive bands were detected and quantified with a Phosphor Imager (Molecular Dynamics/GE Healthcare) after 48 h exposure on Phosphor Imager plates. A mixed-effect model was used to examine the rate of proximal-to-distal decline of the percentage of the densitometric value of the protein band in the ON segments (Laird and Ware, 1982). The intercept and the regression coefficient for the distance were treated as random effects such that each sample has a unique intercept and regression coefficient for the distance. This statistical procedure accounts for the correlations that are due to the repeated measurements of outcomes in the same animals.
Immunohistochemical analyses.
Mice were perfused with 20 ml of PBS after being deeply anesthetized by an intraperitoneal injection of ketamine hydrochloride (1 mg/10 g) and xylazine (0.1 mg/10 g), in accordance with protocols approved by the University of Pennsylvania. The brains of mice were then removed and processed as described previously (Brunden et al., 2010). Paraffin-embedded tissue blocks were cut into 6 μm thick sections, and were stained with antibodies that recognize: tau phosphorylated at ser/thr 202/205 (AT8, 1:7500 dilution) (Goedert et al., 1995), misfolded tau (MC1, 1:2000 dilution; kind gift from Dr. Peter Davies) (Jicha et al., 1997), neurons (NeuN, 1:500 dilution; Millipore), synapses (synaptophysin, 1:500; Millipore), and axons [neurofilament light chain (NFL), 1:9000] (Tohyama et al., 1993). Semiquantitative analysis of tau pathology was performed using the AT8 and MC1 antibodies, utilizing previously published criteria adapted from the Braak staging system for AD that assesses both the intensity and spatial distribution of tau pathology in Tg mice (Hurtado et al., 2010). The area occupied by NeuN-positive neurons in each mouse was assessed from matched brain sections using 20× images of a defined portion of the hippocampal CA3 region (see Fig. 7). The NeuN-positive area was quantified using ImageJ software (National Institutes of Health). Synaptic density was assessed by determining the area of synaptophysin immunoreactivity using matched 20× images within the same CA3 area used for NeuN quantification, with free-hand demarcation of the synaptophysin-positive area as depicted in Figure 8, A and B. Quantification was with ImageJ software. Finally, hippocampal axonal integrity was evaluated in matched 20× images by determining the area occupied by mossy fibers after free-hand demarcation based on NFL immunostaining, as depicted in Figure 8, D and E. Quantification was with ImageJ software. All immunostained slides were assessed by a rater who was masked to the treatment group.
Figure 7.
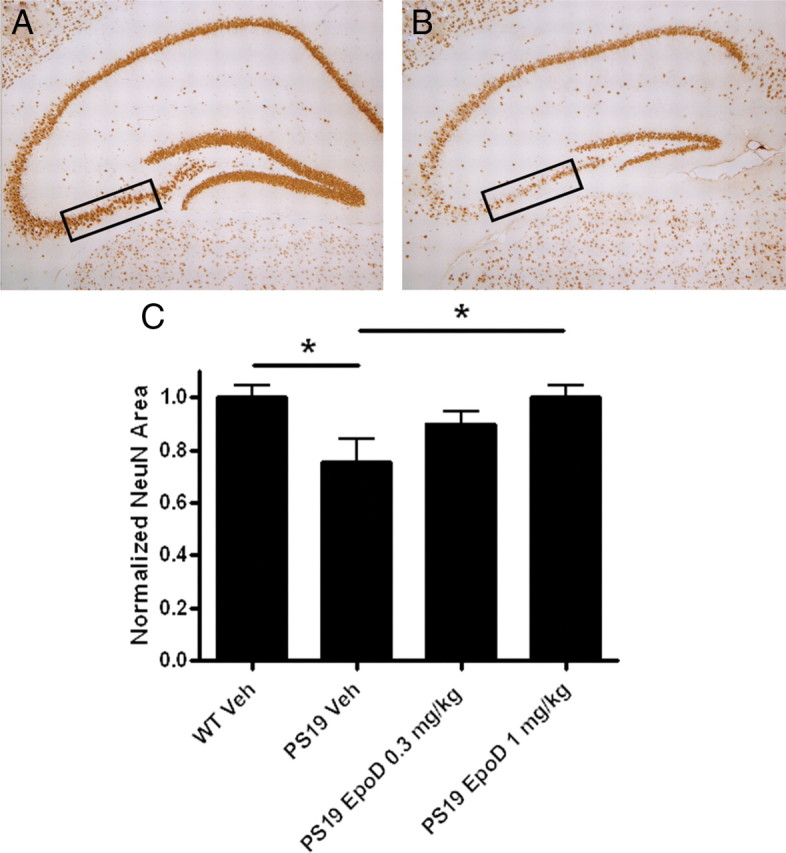
Assessment of hippocampal CA3 neurons in the brains of vehicle- or EpoD-treated 12-month-old mice PS19. A, B, Low-power representative images of hippocampal NeuN immunostaining from 12-month-old vehicle-treated WT (A) and PS19 (B) mice, with the regions sampled for quantification of 20× images denoted by boxes. C, Quantification of the CA3 NeuN area in WT or PS19 mice treated with vehicle or EpoD at the indicated dose. All data are normalized to the mean area obtained in the vehicle-treated WT mice. Error bars indicate SEM; *p < 0.05, as determined by one-way ANOVA and Tukey's multiple-comparison test, with n = 8–10 mice/group. Veh, Vehicle.
Figure 8.

Evaluation of hippocampal synaptic and axonal density in the brains of vehicle- or EpoD-treated 12-month-old mice PS19. A, B, Representative images (20×) of hippocampal CA3 synaptophysin immunostaining from 12-month-old vehicle-treated WT (A) and PS19 (B) mice, with the demarcated area quantified. C, Quantification of the CA3 synaptophysin-positive area in WT or PS19 mice treated with vehicle or EpoD at the indicated dose. D, E, Representative images (20×) of hippocampal NFL immunostaining from 12-month-old vehicle-treated WT (D) and PS19 (E) mice, with the demarcated area quantified. F, Quantification of the CA3 NFL-positive area in WT or PS19 mice treated with vehicle or EpoD at the indicated dose. All data are normalized to the mean area obtained in the vehicle-treated WT mice. Error bars indicate SEM; *p < 0.05, as determined by one-way ANOVA and Tukey's multiple-comparison test, with n = 8–10 mice/group.
Insoluble tau preparations.
Sequentially extracted brain homogenates were prepared as previously described (Yoshiyama et al., 2007). Whole-brain hemispheres were homogenized in 5 μl/mg tissue of cold high-salt (HS) buffer (0.1 m MES, 1 mm EGTA, 0.5 mm MgSO4, 0.75 m NaCl, 0.02 m NaF, 1 mm PMSF, and 0.1% protease inhibitor mixture), followed by centrifugation at 40,000 × g for 40 min at 4°C. Protein concentration within the supernatant fraction (HS soluble) was determined using a BCA assay. The HS-insoluble pellet was homogenized in HS buffer with 1 m sucrose and centrifuged at 85,000 × g for 20 min at 4°C to remove myelin and lipids. The resulting pellets were resuspended in a volume of RIPA (50 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40, and 5 mm EDTA) that was equal to the original HS buffer amount, and again centrifuged at 85,000 × g for 20 min at 4°C. The supernatants were removed and kept as the RIPA-soluble fraction, and the pellets were treated with 70% formic acid (FA) at a volume equal to 50% of the original HS buffer extract. The FA samples were sonicated and then centrifuged at 13,000 × g for 20 min at 4°C. The FA supernatant fractions were dried in a SpeedVac, and resuspended in RIPA containing 1% SDS at a volume equal to 50% of the original HS buffer extract. For ELISA measurements of total insoluble tau, equal fractions of the RIPA and FA samples from each brain homogenate were combined, and these were identically diluted over 1000-fold in EC buffer (0.02 m sodium phosphate, pH 7.0, 2 mm EDTA, 0.4 m NaCl, 0.2% BSA, 0.05% CHAPS, 0.4% Block Ace (AbD Serotec), and 0.05% NaN3 for analysis in a tau ELISA.
Tau ELISA.
Measurements of tau were made with a sandwich ELISA that is similar to that which has recently been described (Yamada et al., 2011). Plates (384-well) were coated with Tau5 antibody (5 μg/ml) in 30 μl of cold 0.1 m bicarbonate buffer. After overnight incubation at 4°C, the plates were washed with cold PBS containing 0.05% Tween (PBST) followed by the addition of 100 μl of Block Ace solution and overnight incubation at 4°C. The blocking solution was removed and the wells were washed with cold PBST. Aliquots (30 μl) of Tau40 (Goedert et al., 1989) standards (37.5–1200 pg/ml) diluted in EC buffer or diluted brain homogenate RIPA + FA samples were added to the wells, and were allowed to incubate overnight at 4°C. The plates were subsequently aspirated and washed with cold PBST, followed by addition of 30 μl/well of reporting antibodies BT2 and HT7 (Thermo Scientific) diluted to 125 ng/ml in C buffer (0.02 m sodium phosphate, pH 7.0, 2 mm EDTA, 0.4 m NaCl, 1% BSA, and 0.05% thimerosal). The plates were incubated overnight at 4°C, followed by washing with cold PBST and addition of 30 μl/well of streptavidin-HRP (ThermoFisher) that was diluted 1:8000 in C buffer. After 1 h incubation at 25°C, the plates were washed with PBST, followed by addition of peroxidase substrate solution (30 μl). The reaction was quenched after 5–10 min by addition of 30 μl of 10% phosphoric acid. Plates were read on a SpectraMax M5 plate reader at 450 nm. The amount of insoluble tau in the RIPA + FA samples was determined from the Tau40 standard curve, and normalized to the average amount of insoluble tau found in the samples from the vehicle-treated PS19 mice.
Complete blood counts.
Blood from PS19 or non-Tg mice was analyzed for complete blood cell counts as previously described (Brunden et al., 2010).
Behavioral testing.
All mice were tested for working memory using spontaneous alternation behavior (SAB) in a Y-maze, as previously described (Rosario et al., 2006), one week before killing. Briefly, each animal was placed in the maze and allowed to explore ad libitum for 8 min. SAB score was calculated as the proportion of alternations (an arm choice differing from the previous two choices) to the total number of alternation opportunities (total arm entries minus 2) (King and Arendash, 2002). To assess spatial learning and memory, mice were tested on the Barnes maze (San Diego Instruments; model 7001–0235) essentially as previously described (Brunden et al., 2010). Mice were randomly assigned to different escape compartments and were allowed to habituate to the testing room 1 h before testing, and to the maze for 30 s before each trial in a start compartment. On five consecutive testing days, mice were exposed to the Barnes maze for three 2.5 min trials that were 15 min apart. Performance was not scored during the first trial, and mice who did not find the hidden escape compartment were gently guided there and allowed to remain for 30 s. Mice were tested for their ability to learn and remember the fixed position of this escape compartment during Trials 2 and 3, and their performance was scored for success rate and the time taken before finding the correct compartment (Harrison et al., 2009; Patil et al., 2009). The maze was cleaned with 70% ethanol in between each animal to minimize odor cues. Statistical analyses of Barnes maze data were as previously described (Brunden et al., 2010).
Peripheral nerve testing.
To assess potential treatment effects on peripheral sensory nerve function, mice were evaluated for tactile allodynia by the von Frey Test (Pitcher et al., 1999; Smith et al., 2004). von Frey filaments of increasing diameters ranging from 0.008 to 300 g of force (Bioseb) were placed underneath the hindpaw of mice standing on a wire mesh grid to determine the minimum pressure needed to elicit a withdrawal response. Values were determined from the average of five trials.
Results
EpoD treatment reduces axonal dystrophy and increases MT stabilization in aged PS19 tau Tg mice
Prior pharmacokinetic studies revealed that EpoD has a significantly longer residence time in the brain than in plasma of mice (Brunden et al., 2010, 2011), with the compound being detectable in brain for at least 1 week after a single 3 mg/kg administration. Moreover, a measurable pharmacodynamic effect, as evidenced by increased levels of a marker of stable MTs, acetyl-tubulin (Black et al., 1989; Laferrière et al., 1997), was apparent 1 week after a single 3 mg/kg dose of EpoD to normal mice (Brunden et al., 2011). These characteristics led to the selection of EpoD as a preferred MT-stabilizing agent for testing in a Tg mouse model of tauopathy (Brunden et al., 2010). Here we have assessed EpoD in an interventional study to address the important question of whether EpoD might improve efficacy outcomes in aged PS19 mice with pre-existing tau pathology, a paradigm that more closely approximates the clinical setting in which AD and other tauopathy patients will first display cognitive symptoms after the onset of tau pathology. In this study, groups of 9-month-old male PS19 mice with existing tau pathology (Yoshiyama et al., 2007; Brunden et al., 2010; Hurtado et al., 2010) were administered once-weekly intraperitoneal doses of either vehicle, 0.3 mg/kg of EpoD or 1.0 mg/kg of EpoD for 3 months, followed by the evaluation of a series of efficacy and safety endpoints. In addition, vehicle-treated, 9-month-old non-Tg WT littermates were included in the study as a control group.
PS19 mice show an age-dependent increase in axonal dystrophy that is detectable at 3 months of age and becomes very apparent by 6 months (Brunden et al., 2010). In agreement with these previous results, a comparison of the axonal dystrophy within the ONs of vehicle-treated WT and PS19 mice revealed a significant increase in dystrophic axons in the tau Tg mice (Fig. 1). Not surprisingly, the number of dystrophic axons per unit area was increased in the 12-month-old PS19 mice relative to the value previously reported for PS19 mice at 6 months of age (Brunden et al., 2010). Interestingly, the incidence of abnormal axons in the 12-month-old WT mice also appeared to be increased relative to that reported in 6-month-old WT mice (Brunden et al., 2010), suggesting an age-related deterioration. Notably, the 12-month-old PS19 mice treated with either dose of EpoD showed a significant decrease in axonal dystrophy, such that the density of abnormal axons was similar to that observed in the vehicle-treated WT mice (Fig. 1B). This decrease in the number of degenerating axons presumably resulted from EpoD-mediated effects on MTs, and an evaluation of MT density in the ON (Fig. 2) revealed an increase of MTs in the higher dose EpoD-treated PS19 mice that was significantly greater than vehicle-treated PS19 mice (Fig. 2C). Although the average MT density in the vehicle-treated, 12-month-old PS19 mice was reduced relative to that in WT mice (Fig. 2C), this difference did not reach statistical significance. The MT density of the 12-month-old, vehicle-treated PS19 mice is nearly identical to that reported for 6-month-old PS19 mice (Brunden et al., 2010); however, the MT number per area in the 12-month-old WT mice is somewhat diminished relative to what was observed in 6-month-old WT mice. This age-dependent reduction of MT density in the WT mice may cause the increase in axonal dystrophy that was observed when comparing 12- and 6-month-old WT mice, and may explain why the difference in MT density between the vehicle-treated PS19 and WT mice did not reach statistical significance.
Figure 1.
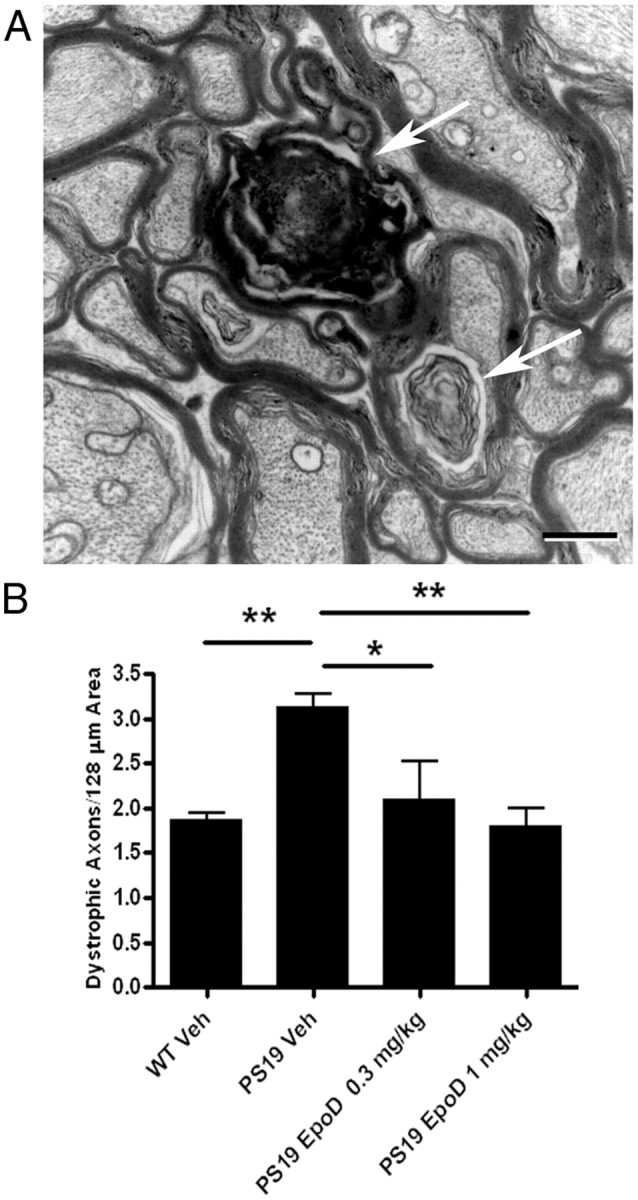
Comparison of axonal dystrophy in vehicle- or EpoD-treated 12-month-old PS19 and WT mice. The number of dystrophic axons per unit area of ON was determined for WT or PS19 mice that had received administration of either vehicle or EpoD from 9 to 12 months of age. A, A representative micrograph showing examples of dystrophic axons (arrows). Scale bar, 0.5 μm. B, Quantification of the number of dystrophic axons per area for the various treatment groups. Error bars indicate SEM; *p < 0.05, **p < 0.01, as determined by one-way ANOVA and Tukey's multiple-comparison test, with n = 8–10 mice evaluated/group. Veh, Vehicle.
Figure 2.

Assessment of ON MT density in vehicle- or EpoD-treated 12-month-old mice PS19 and WT mice. A, Cross sections of the ON were imaged at 50,000× (scale bar, 500 nm), and hexagonal fields of 0.035 μm2 were arrayed over the ON images. B, An expanded image, with MTs identified by arrows in one hexagon. To avoid repeat counting of MTs that reside on a hexagon border, only those MTs that were on three of the six borders of each hexagon, as indicated, were included in the counts. C, ON MT density in WT or PS19 mice treated with vehicle or EpoD at the indicated dose. Error bars indicate SEM; *p < 0.05, as determined by one-way ANOVA and Tukey's multiple-comparison test, with n = 8–10 mice/group. Veh, Vehicle.
EpoD treatment improves FAT and cognitive performance in aged PS19 tau Tg mice
The increase of MT stabilization and the decrement in dystrophic axons observed in the EpoD-treated PS19 mice suggest that axonal function might also be improved. To examine this, an assessment of FAT was made in ON samples from mice in each treatment group. Briefly, [35S]methionine was injected into the vitreous of each eye, and each ON was harvested 3 h later. The extent of FAT was determined by quantifying the proximal-to-distal distribution of a single chosen 35S-labeled protein as quantified by densitometric analysis after SDS-PAGE and autoradiography (Zhang et al., 2005) (Fig. 3A). As depicted in Figure 3B, a greater percentage of the total 35S-labeled protein was found in the most proximal segment, and a correspondingly smaller fraction had progressed to the more distal segments, in the vehicle-treated PS19 mice compared with the other cohorts. This reveals an impairment of FAT in the vehicle-treated PS19 mice relative to the WT mice. Importantly, the proximal-to-distal distribution in the PS19 mice treated with either dose of EpoD was nearly indistinguishable from the WT vehicle-treated mice. Thus, the reduction in ON axon damage observed upon EpoD treatment of PS19 mice translated into an improvement of axonal function.
Figure 3.

Comparison of ON FAT in vehicle- or EpoD-treated 12-month-old mice PS19 and WT mice. The extent of proximal-to-distal FAT along the ONs of WT or PS19 mice that had been dosed with vehicle or EpoD from 9 to 12 months of age as judged by [35S]methionine labeling of newly synthesized ON proteins and evaluation of the proximal-to-distal distribution of a transported protein as quantified by densitometric analysis of autoradiographs. A, SDS-PAGE autoradiographs of 35S-labeled proteins from ON segments of vehicle- and EpoD-treated PS19 mice. Each gel contains ON segments from a single mouse, with the lanes corresponding to individual 1 mm ON segments. The arrows in each gel depict the band that was quantified, with the migration of molecular markers indicated. B, Mean values for the proximal-to-distal distribution of the selected protein band. Error bars indicate SEM; *p < 0.05, ***p < 0.001, as determined using a mixed effect statistical model as described in Materials and Methods, with n = 3 mice evaluated/group.
PS19 mice show deficits in spatial learning by 6 months of age (Brunden et al., 2010), and thus the 9-month-old PS19 mice that first received EpoD treatment in this study are cognitively impaired. It was therefore of interest to determine whether the behavioral performance of the aged PS19 mice would be affected by 3 months of EpoD treatment. Working memory was analyzed using a Y-maze test (Hughes, 2004; Carroll et al., 2007) in which performance was assessed by determining whether the mice remembered which of the three arms of the maze they previously explored. The vehicle-treated PS19 mice performed more poorly in this task, demonstrating lower spontaneous alterations, than did the corresponding WT mice (Fig. 4A). The PS19 mice that received EpoD had improved performance relative to those that received vehicle, with the 1 mg/kg treatment group reaching statistical significance. To examine further the cognitive abilities of the treatment cohorts, the mice also underwent spatial learning analysis in the Barnes maze (Harrison et al., 2009; Brunden et al., 2010). Again, the vehicle-treated PS19 mice showed deficits compared with WT mice, both in their success at correctly identifying the target escape box, as well as in the time required to find the target (Fig. 4B,C). The EpoD-treated PS19 mice showed improved spatial learning when compared with the vehicle-treated PS19 mice, with the 1 mg/kg dose group reaching significance in the success endpoint and both EpoD dose groups showing significant improvement in the latency measure (Fig. 4B,C). The Y-maze and Barnes maze data reveal that administration of EpoD to aged PS19 mice with existing cognitive deficits can lead to improvements in working and spatial memory, such that overall performance approaches that of comparably aged WT mice.
Figure 4.

Cognitive evaluation of vehicle- or EpoD-treated 12-month-old PS19 and WT mice. A–C, Y-maze testing of spontaneous alternation (SA) behavior (A), and Barnes maze performance as judged by the percentage of trials that were successfully completed (B) and the time taken to identify the correct target (C) with WT or PS19 mice that had received vehicle or EpoD from 9 to 12 months of age. Error bars indicate SEM; *p < 0.05, **p < 0.01, ***p < 0.001, as determined for (A) by one-way ANOVA and Tukey's multiple-comparison test, and for (B, C) by a repeated measures statistical analysis, with n = 10–12 mice/group. veh, Vehicle.
EpoD reduces tau pathology in aged PS19 tau Tg mice
The enhancement of MT density and axonal function observed upon EpoD treatment of the aged PS19 mice would not be expected, a priori, to affect tau misfolding and aggregation unless tau pathology is exacerbated by axonal dysfunction. A modest decrease in forebrain phospho-tau (pTau) immunostaining was previously observed when young PS19 mice were treated with EpoD, although this change was not statistically significant (Brunden et al., 2010). An analysis of pTau (pS202/pT205) in the aged PS19 mice, using an immunohistochemical rating scale to monitor both the intensity and distribution of immunostaining (Hurtado et al., 2010), revealed a decrease of pTau in EpoD-treated PS19 mice relative to those receiving vehicle (Fig. 5A), which reached significance at the higher EpoD dose (Fig. 5C). To confirm that there was a reduction of pathological tau in the EpoD-treated PS19 mice, immunohistochemistry was also performed with the MC1 antibody that recognizes misfolded tau (Jicha et al., 1997). Again, both EpoD groups demonstrated less staining than did the vehicle group (Fig. 5B), with the higher dose of EpoD reaching statistical significance (Fig. 5D). Finally, the amount of pathological tau was also evaluated in the vehicle- and EpoD-treated PS19 mice by ELISA measurement of HS buffer-insoluble tau in hemi-brain homogenates. In accordance with the immunohistochemistry results, the amount of HS buffer-insoluble tau that was detected with the ELISA, which measures both phosphorylated and nonphosphorylated tau, was found to be decreased in the PS19 mice receiving EpoD, with the 1 mg/kg treatment result being statistically significant (Fig. 5E). The correspondence between the immunohistochemical and biochemical analyses of pathologic tau was further confirmed by comparing for each mouse the semiquantitative scores assigned from the immunostaining methodologies with the relative amount of insoluble tau as determined by ELISA. As depicted in Figure 6, there was a strong and highly significant correlation between the immunohistochemical ratings and the insoluble tau level for each animal. Thus, these data indicate that EpoD treatment of the PS19 mice resulted in diminished tau pathology.
Figure 5.

Assessment of tau pathology in the brains of vehicle- or EpoD-treated 12-month-old mice PS19. A, Low-power and higher power images of AT8 phospho-tau immunostaining, with the regions sampled for the 20× images denoted by boxes. The immunohistochemical (IHC) scores assigned to the vehicle- and EpoD-treated PS19 mice in these examples were 4 and 2, respectively. B, Low-power and higher power images of MC1 misfolded-tau immunostaining, with the regions sampled for the higher power images denoted by boxes. The IHC scores assigned to the vehicle- and EpoD-treated PS19 mice in these examples were 4 and 2, respectively. Scale bars, 1 mm in the low-power images, and 0.1 mm in the higher power images. C, D, Quantification of the extent of phospho-tau (C) and misfolded tau (D) in the forebrains of PS19 mice that had received vehicle or EpoD from 9 to 12 months of age, as determined by a semiquantitative rating of immunohistochemical staining using AT8 or MC1 monoclonal antibodies, respectively. E, The amount of insoluble tau in hemi-brain homogenates from vehicle- or EpoD-treated 12-month-old PS19 mice as determined with a tau ELISA. Error bars indicate SEM; *p < 0.05 as determined by a nonparametric Kruskal–Wallis analysis and Dunn's multiple-comparison test (C, D), or ANOVA and Tukey's multiple-comparison test (E); n = 9–10 mice/group.
Figure 6.

A, B, Comparison of MC1 (A) and AT8 (B) immunohistochemical (IHC) scores with relative insoluble tau amount as determined by ELISA analysis of brain homogenates for each vehicle- and EpoD-treated PS19 mouse. The line represents the best linear fit of the data; r = correlation coefficient.
The observed reduction of tau pathology in the PS19 mice receiving EpoD, coupled with the improvement of axonal function, suggested that these mice might have less hippocampal neuron loss than is normally seen in these mice (Yoshiyama et al., 2007). An example of this hippocampal neuron death can be observed by comparing NeuN-stained sections from 12-month-old WT (Fig. 7A) and PS19 mice (Fig. 7B). Quantification of NeuN area in the CA3 region of vehicle-treated WT and PS19 mice revealed a significant reduction of neurons in the latter group (Fig. 7C). The PS19 mice that received EpoD demonstrated a dose-dependent increase of CA3 neurons relative to the vehicle-treated group which reached statistical significance at the 1 mg/kg dose (Fig. 7C). A similar comparison of CA3 synaptic density, as measured by synaptophysin immunostaining, revealed a trend toward a loss of synapses in the 12-month-old PS19 mice (Fig. 8A–C), and both doses of EpoD caused a significant increase of synaptic area in the PS19 mice when compared with those receiving vehicle (Fig. 8C). Finally, hippocampal axonal density was assessed by determining the area within a defined CA3 region occupied by mossy fibers, as visualized by NFL immunostaining (Fig. 8D,E). A loss of mossy fibers was observed in the vehicle-treated PS19 mice relative to WT littermates, and EpoD treatment prevented this axonal loss, with the higher compound dose reaching significance. Thus, EpoD treatment reduced several measures of neurotoxicity in the 12-month-old PS19 mice.
No adverse effects observed in EpoD-treated mice
The data described above reveal that EpoD can significantly improve functional outcomes, reduce tau pathology, and prevent neurotoxicity in tau Tg mice when they are treated in an interventional setting. This suggests that EpoD, which has previously advanced to Phase II clinical testing for oncology, might have utility for the treatment of AD and related tauopathies. However, MT-stabilizing agents are known to induce side effects when used at the doses required for the treatment of cancer. Hence, it is important to note that the aged PS19 mice in this study received EpoD doses that were 30- and 100-fold less, on a milligrams per square meter basis, than was previously used in human testing (Beer et al., 2007). Thus, the substantially lower EpoD doses used here could attenuate the adverse complications associated with MT-stabilizing drugs, such as immune cell depletion and the onset of peripheral neuropathy (Cortes and Baselga, 2007; Cheng et al., 2008; Bedard et al., 2010). The PS19 mice receiving EpoD in the present study had body weights at the end of the dosing period that were indistinguishable from vehicle-treated PS19 or WT mice (Fig. 9A), indicating an absence of gross toxicities. Moreover, there were no significant differences in major organ weights among the study cohorts, including brain weight (Fig. 9B). An evaluation of complete blood cell counts from the different treatment groups demonstrated that there was no reduction of total white blood cells (WBCs) in the EpoD-treated PS19 mice relative to either the PS19 or WT mice that received vehicle (Fig. 9C). Similarly, there were no alterations in the neutrophil content among WBCs in the EpoD-treated mice (Fig. 9C). Thus, there is no evidence of immune suppression or neutropenia in the mice that received either of the EpoD doses. Finally, peripheral nerve sensory function was evaluated using the von Frey test to measure the sensitivity of the mouse footpad to sensory stimuli by determining the force required to elicit a withdrawal response (Smith et al., 2004). As summarized in Figure 9D, neither of the EpoD-treated cohorts showed any change in footpad sensitivity relative to the vehicle-treated PS19 mice. Therefore, it appears that the low doses of EpoD used in this study, which were sufficient to improve efficacy outcomes, elicited none of the adverse events that are observed when higher doses of MT-stabilizing agents are used for the treatment of cancer.
Figure 9.

Toxicological evaluation of vehicle- and EpoD-treated 12-month-old PS19 mice. A–D, WT or PS19 mice that had received vehicle or EpoD from 9 to 12 months of age were evaluated for final body weights (A), organ weights (B), WBC counts and percentage of WBCs that are neutrophils (C), and sensory nerve function using the von Frey test of footpad sensitivity (D). There were no statistically significant changes among any of the groups within these measures as determined by one-way ANOVA, with n = 8–10 mice/group. Veh/veh, Vehicle.
Discussion
There is growing evidence that hyperphosphorylation and misfolding of tau contributes to neurodegenerative processes in AD and other tauopathies through both gain-of-function and loss-of-function toxicities (Lee et al., 2001b; Ballatore et al., 2007; Brunden et al., 2009). Although there may be multiple mechanisms of tau-mediated neuron damage, there is evidence in AD patients of a decline of normal tau function. This includes data which demonstrate a reduction in the number and length of MTs in neurons within the AD brain (Cash et al., 2003), decreased levels of MT-binding competent tau in affected regions of AD brains (Bramblett et al., 1992), and diminished acetylated tubulin in AD neurons (Hempen and Brion, 1996). Moreover, decreased MT density has been observed in Tg mouse models of tauopathy (Ishihara et al., 1999; Zhang et al., 2005), including the PS19 mice that were used in the study presented here (Brunden et al., 2010). Tau Tg mouse models have proven to be valuable in testing the hypothesis that MT-stabilizing drugs that are used in the treatment of cancer can compensate for an alteration of tau-mediated stabilization of MTs. For example, paclitaxel improved MT density, axonal function, and motor performance after absorption at neuromuscular junctions in another tau Tg mouse model that develops tau inclusions primarily within the brainstem and spinal cord (Zhang et al., 2005). Because paclitaxel does not effectively cross the BBB (Fellner et al., 2002; Brunden et al., 2011), it is not suitable for the treatment of human tauopathies where forebrain tau inclusions predominate. More recently, we demonstrated that prophylactic administration of the brain-penetrant MT-stabilizing agent, EpoD, alleviated the onset of MT deficits and axonal dystrophy in young PS19 mice, resulting in improvements of cognitive performance (Brunden et al., 2010).
Here, we have confirmed and substantially extended these previous studies by examining the safety and efficacy of EpoD in an interventional treatment paradigm, using aged PS19 mice with existing tau pathology. This study design was critical for demonstrating that a BBB-permeable MT-stabilizing agent could decrease disease in a tauopathy model that more closely approximates the symptomatic patients who are candidates for therapeutic treatment. Although significant advances are being made in the ability to diagnose patients before the onset of advanced disease, it is likely that those at the prodromal stages of disease progression (i.e., mild cognitive impairment in AD patients) already have substantial tau pathology (Jack et al., 2010). Therefore, the determination that very low EpoD doses can safely and effectively mitigate axonal deficits and improve cognitive performance in aged PS19 mice with pre-existing tau pathology provides critical evidence that such a therapeutic strategy might ameliorate disease in human tauopathy patients.
Although EpoD was demonstrated to increase MT density in aged PS19 mice, the possibility exists that the drug-induced improvements of FAT, cognitive performance, tau pathology, and neuronal health also resulted from effects that are not directly attributable to tau loss-of-function. For example, tau overexpression in cultured neurons has been shown to reduce the engagement of the kinesin motor proteins required for anterograde FAT (Stamer et al., 2002; Vershinin et al., 2007; Dixit et al., 2008). Since there is evidence that tau binding to MTs is decreased by paclitaxel (Kar et al., 2003; Samsonov et al., 2004), and because EpoD shares a common MT-binding site with paclitaxel (Bollag et al., 1995), it is conceivable that EpoD may increase FAT in the PS19 mice by decreasing tau binding to MTs and thus enhance kinesin interaction with MTs. It is also possible that tau overexpression in the PS19 mice could result in MT overstabilization, with EpoD normalizing MT stability by decreasing tau binding to MTs. However, it should be noted that the modest decrease in MT density observed in the PS19 mice relative to WT mice (Brunden et al., 2010; present study) would argue that the extent of tau bound to MTs in the Tg mice is below normal levels, and that compensation for tau loss-of-function is the most parsimonious explanation for the beneficial effects of EpoD.
A somewhat unexpected finding from our study was the reduction of tau pathology after EpoD treatment, as evidenced by decreased immunohistochemical staining with antibodies that recognize a major phospho-tau epitope (pS202/pT205) and misfolded tau, as well as by a reduction of insoluble tau. How a change of MT stabilization or an enhancement of FAT might reduce the amount of hyperphosphorylated and misfolded tau in the PS19 mice is unclear, although a similar reduction of tau pathology was observed after intranasal administration of the pleiotropic peptidic MT-stabilizing agent, NAP (Matsuoka et al., 2007, 2008). We have examined whether EpoD has a direct effect on tau fibrilization, using a well characterized in vitro assay (Crowe et al., 2009), and find that the compound has essentially no effect on tau assembly (data not shown). Although it is impossible to definitively exclude the possibility of off-target effects of EpoD that might lead to increased degradation of misfolded tau, it is interesting to note that kinesin-1-deficient mice develop dystrophic axons with increased amounts of hyperphosphorylated tau (Falzone et al., 2009). Moreover, JNPL3 tau Tg mice crossed to kinesin light chain knock-out mice have an exacerbation of tau pathology (Falzone et al., 2010). Thus, it appears that impaired FAT can lead to an enhancement of tau pathology, perhaps through JNK pathway activation (Falzone et al., 2009). These findings suggest that a vicious cycle may develop in tauopathies, whereby tau loss-of-function results both in MT destabilization and FAT deficits that can then further enhance tau pathology. This may explain why EpoD treatment prevents the development of additional tau pathology in the aged PS19 mice, as the compound improves FAT, and thus attenuates this possible feedforward mechanism.
In addition to reducing tau pathology, EpoD treatment also prevented the loss of hippocampal neurons, axons, and synapses in the PS19 mice. The improvements in neuronal measures, along with increased axonal transport, are consistent with the enhanced cognitive performance of the EpoD-treated PS19 mice. It is difficult to separate the absolute contributions of improved axonal transport, increased synaptic density, and greater neuronal survival to the observed learning and memory improvements in the EpoD-treated mice, as all of these parameters may be interrelated.
The efficacious properties of EpoD in this interventional study suggest that this compound, which progressed to Phase II clinical testing in cancer (Beer et al., 2007), might be a possible clinical candidate for the treatment of tauopathies. However, MT-stabilizing agents have been plagued by side effects in oncology settings (Bedard et al., 2010), and there is a concern that such drugs would not be tolerated when dosed on a chronic basis, as would likely be required for neurodegenerative diseases. For this reason, we have carefully monitored the mice in this study for potential drug-related adverse events. Notably, we did not observe any side effects after 3 months of weekly EpoD administration to aged PS19 mice, with the compound-treated mice showing normal body weight gain and no signs of organ toxicity. Moreover, the mice receiving EpoD did not show alterations of neutrophil counts or evidence of peripheral neuropathy, complications that are typical of this class of drug in cancer patients (Cortes and Baselga, 2007; Cheng et al., 2008; Bedard et al., 2010). The absence of EpoD-related side effects in this study presumably relates to the low doses that were used, which are 30–100× less than used in cancer patients (Beer et al., 2007). To the extent that the PS19 mice accurately model human tauopathy, our data would suggest that much lower concentrations of EpoD are required to overcome tau loss-of-function than are needed to inhibit the growth of rapidly dividing cancer cells. We should also note that the unique pharmacokinetic and tissue distribution properties of EpoD likely contribute to the favorable efficacy-to-safety margin (i.e., therapeutic index). In particular, the retention of EpoD in the brain long after the compound has been cleared from the blood (Brunden et al., 2010, 2011) appears to provide for prolonged MT stabilization in the brain while minimizing peripheral side effects. As a comparison, we have also examined in PS19 mice the drug ixabepilone (Ixempra), which is another member of the epothilone family of MT-stabilizing drugs that is currently approved for the treatment of metastatic breast cancer (Hunt, 2009). Although brain penetrant, the plasma levels of ixabepilone are higher than those of EpoD at the same drug dose (Brunden et al., 2011). A subset of PS19 mice receiving a weekly 4 mg/kg dose of this drug began to show significant body weight loss after 6–8 weeks of treatment, with some animals dying (data not shown). In contrast, we previously demonstrated that a weekly 3 mg/kg EpoD dose administered over a 12 week period was well tolerated (Brunden et al., 2010).
The combined safety and efficacy of EpoD in a mouse model with pre-existing tau pathology suggests that this compound merits consideration as a candidate for clinical testing in tauopathy patients. The results obtained with EpoD also provide impetus to identify additional BBB-permeable MT-stabilizing agents that might have an even greater therapeutic index. Finally, EpoD and related drug candidates might also have utility in other neurodegenerative conditions, particularly in Parkinson's disease (PD) where tau pathology can be found (Kalaitzakis and Pearce, 2009) and where genome-wide association studies have identified a significant association between PD and the tau gene locus (Simón-Sánchez et al., 2009; Edwards et al., 2010).
Footnotes
These studies were supported by National Institutes of Health Grant U01AG029213, and in part by the Marian S. Ware Alzheimer Program, the Karen Cohen Segal and Christopher S. Segal Alzheimer Drug Discovery Initiative Fund, the Paula C. Schmerler Fund for Alzheimer's Research, the Barrist Neurodegenerative Disease Research Fund, the Eleanor Margaret Kurtz Endowed Fund, the Mary Rasmus Endowed Fund for Alzheimer's Research, Gloria J. Miller, and Dr. Arthur Peck. We thank Biao Zuo of the Biomedical Imaging Core Facility of the University of Pennsylvania for his assistance. We appreciate Nabil Darwich's assistance in quantitative EM studies. Drs. Brunden, Lee, and Trojanowski collaborate with Bristol-Myers Squibb, which has certain intellectual property rights to epothilone D, on unrelated research studies.
References
- Arriagada et al., 1992.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimers disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Ballatore et al., 2007.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bedard et al., 2010.Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol. 2010;7:22–36. doi: 10.1038/nrclinonc.2009.186. [DOI] [PubMed] [Google Scholar]
- Beer et al., 2007.Beer TM, Higano CS, Saleh M, Dreicer R, Hudes G, Picus J, Rarick M, Fehrenbacher L, Hannah AL. Phase II study of KOS-862 in patients with metastatic androgen independent prostate cancer previously treated with docetaxel. Invest New Drugs. 2007;25:565–570. doi: 10.1007/s10637-007-9068-1. [DOI] [PubMed] [Google Scholar]
- Black et al., 1989.Black MM, Baas PW, Humphries S. Dynamics of alpha-tubulin deacetylation in intact neurons. J Neurosci. 1989;9:358–368. doi: 10.1523/JNEUROSCI.09-01-00358.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag et al., 1995.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- Brady et al., 1999.Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM. Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci. 1999;19:7278–7288. doi: 10.1523/JNEUROSCI.19-17-07278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramblett et al., 1992.Bramblett GT, Trojanowski JQ, Lee VM. Regions with abundant neurofibrillary pathology in human brain exhibit a selective reduction in levels of binding-competent-tau and accumulation of abnormal tau-isoforms (A68 proteins) Lab Invest. 1992;66:212–222. [PubMed] [Google Scholar]
- Brunden et al., 2009.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden et al., 2010.Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, 3rd, Lee VM, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden et al., 2011.Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan AM, Trojanowski JQ, Smith AB, 3rd, Lee VM. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer's disease and related tauopathies. Pharmacol Res. 2011;63:341–351. doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll et al., 2007.Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27:13357–13365. doi: 10.1523/JNEUROSCI.2718-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash et al., 2003.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avíla J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng et al., 2008.Cheng KL, Bradley T, Budman DR. Novel microtubule-targeting agents—the epothilones. Biologics. 2008;2:789–811. doi: 10.2147/btt.s3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland et al., 1977a.Cleveland DW, Connolly JA, Kalnins VI, Spiegelman BM, Kirschner MW. Physical properties and cellular localization of tau, a microtubule-associated protein which induces assembly of purified tubulin. J Cell Biol. 1977a;75:A283. [Google Scholar]
- Cleveland et al., 1977b.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and role of tau in microtubule assembly. J Mol Biol. 1977b;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- Cortes and Baselga, 2007.Cortes J, Baselga J. Targeting the microtubules in breast cancer beyond taxanes: the epothilones. Oncologist. 2007;12:271–280. doi: 10.1634/theoncologist.12-3-271. [DOI] [PubMed] [Google Scholar]
- Crowe et al., 2009.Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AM, Huang R, Wichterman J, McCoy J, Huryn D, Auld DS, Smith AB, 3rd, Inglese J, Trojanowski JQ, Austin CP, Brunden KR, Lee VM. The identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry. 2009;48:7732–7745. doi: 10.1021/bi9006435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit et al., 2008.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards et al., 2010.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Züchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone et al., 2009.Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS. Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci. 2009;29:5758–5767. doi: 10.1523/JNEUROSCI.0780-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone et al., 2010.Falzone TL, Gunawardena S, McCleary D, Reis GF, Goldstein LS. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum Mol Genet. 2010;19:4399–4408. doi: 10.1093/hmg/ddq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner et al., 2002.Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhänel M, Spruss T, Bernhardt G, Graeff C, Färber L, Gschaidmeier H, Buschauer A, Fricker G. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. doi: 10.1172/JCI15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert and Jakes, 2005.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Goedert et al., 1989.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cdna-encoding an isoform of microtubule-associated protein tau containing 4 tandem repeats: differential expression of tau protein messenger-rnas in human-brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert et al., 1995.Goedert M, Jakes R, Vanmechelen E. Monoclonal-antibody At8 recognizes tau-protein phosphorylated at both serine-202 and threonine-205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla et al., 1997.Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gravitz, 2011.Gravitz L. A tangled web of targets. Nature. 2011;475:S9–S11. doi: 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- Harrison et al., 2009.Harrison FE, Hosseini AH, McDonald MP. Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav Brain Res. 2009;198:247–251. doi: 10.1016/j.bbr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempen and Brion, 1996.Hempen B, Brion JP. Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer's disease. J Neuropathol Exp Neurol. 1996;55:964–972. doi: 10.1097/00005072-199609000-00003. [DOI] [PubMed] [Google Scholar]
- Hong et al., 1998.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Hughes, 2004.Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Hunt, 2009.Hunt JT. Discovery of ixabepilone. Mol Cancer Ther. 2009;8:275–281. doi: 10.1158/1535-7163.MCT-08-0999. [DOI] [PubMed] [Google Scholar]
- Hurtado et al., 2010.Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, Lee VM. A beta accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010;177:1977–1988. doi: 10.2353/ajpath.2010.100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton et al., 1998.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakravarty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ishihara et al., 1999.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Jack et al., 2010.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha et al., 1997.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Kalaitzakis and Pearce, 2009.Kalaitzakis ME, Pearce RK. The morbid anatomy of dementia in Parkinson's disease. Acta Neuropathol. 2009;118:587–598. doi: 10.1007/s00401-009-0597-x. [DOI] [PubMed] [Google Scholar]
- Kar et al., 2003.Kar S, Fan J, Smith MJ, Goedert M, Amos LA. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003;22:70–77. doi: 10.1093/emboj/cdg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King and Arendash, 2002.King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol Behav. 2002;75:627–642. doi: 10.1016/s0031-9384(02)00639-x. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick et al., 2001.Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST. Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci. 2001;21:2288–2297. doi: 10.1523/JNEUROSCI.21-07-02288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferrière et al., 1997.Laferrière NB, MacRae TH, Brown DL. Tubulin synthesis and assembly in differentiating neurons. Biochem Cell Biol. 1997;75:103–117. [PubMed] [Google Scholar]
- Laird and Ware, 1982.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38:963–974. [PubMed] [Google Scholar]
- Lee et al., 2001a.Lee CB, Wu Z, Zhang F, Chappell MD, Stachel SJ, Chou TC, Guan Y, Danishefsky SJ. Insights into long-range structural effects on the stereochemistry of aldol condensations: a practical total synthesis of desoxyepothilone F. J Am Chem Soc. 2001a;123:5249–5259. doi: 10.1021/ja010039j. [DOI] [PubMed] [Google Scholar]
- Lee et al., 1994.Lee VM, Daughenbaugh R, Trojanowski JQ. Microtubule stabilizing drugs for the treatment of Alzheimers-disease. Neurobiol Aging. 1994;15:S87–S89. doi: 10.1016/0197-4580(94)90179-1. [DOI] [PubMed] [Google Scholar]
- Lee et al., 2001b.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001b;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Matsuoka et al., 2007.Matsuoka Y, Gray AJ, Hirata-Fukae C, Minami SS, Waterhouse EG, Mattson MP, LaFerla FM, Gozes I, Aisen PS. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer's disease at early pathological stage. J Mol Neurosci. 2007;31:165–170. doi: 10.1385/jmn/31:02:165. [DOI] [PubMed] [Google Scholar]
- Matsuoka et al., 2008.Matsuoka Y, Jouroukhin Y, Gray AJ, Ma L, Hirata-Fukae C, Li HF, Feng L, Lecanu L, Walker BR, Planel E, Arancio O, Gozes I, Aisen PS. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2008;325:146–153. doi: 10.1124/jpet.107.130526. [DOI] [PubMed] [Google Scholar]
- Patil et al., 2009.Patil SS, Sunyer B, Höger H, Lubec G. Evaluation of spatial memory of C57BL/6J and CD1 mice in the Barnes maze, the multiple T-maze and in the Morris water maze. Behav Brain Res. 2009;198:58–68. doi: 10.1016/j.bbr.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Pitcher et al., 1999.Pitcher GM, Ritchie J, Henry JL. Paw withdrawal threshold in the von Frey hair test is influenced by the surface on which the rat stands. J Neurosci Methods. 1999;87:185–193. doi: 10.1016/s0165-0270(99)00004-7. [DOI] [PubMed] [Google Scholar]
- Rivkin et al., 2004.Rivkin A, Yoshimura F, Gabarda AE, Cho YS, Chou TC, Dong H, Danishefsky SJ. Discovery of (E)-9,10-dehydroepothilones through chemical synthesis: on the emergence of 26-trifluoro-(E)-9,10-dehydro-12,13-desoxyepothilone B as a promising anticancer drug candidate. J Am Chem Soc. 2004;126:10913–10922. doi: 10.1021/ja046992g. [DOI] [PubMed] [Google Scholar]
- Rosario et al., 2006.Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006;26:13384–13389. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonov et al., 2004.Samsonov A, Yu JZ, Rasenick M, Popov SV. Tau interaction with microtubules in vivo. J Cell Sci. 2004;117:6129–6141. doi: 10.1242/jcs.01531. [DOI] [PubMed] [Google Scholar]
- Simón-Sánchez et al., 2009.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith et al., 2004.Smith SB, Crager SE, Mogil JS. Paclitaxel-induced neuropathic hypersensitivity in mice: responses in 10 inbred mouse strains. Life Sci. 2004;74:2593–2604. doi: 10.1016/j.lfs.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Stamer et al., 2002.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlhyama et al., 1993.Tlhyama T, Lee VM, Trojanowski JQ. Coexpression of low-molecular-weight neurofilament protein and glial fibrillary acidic protein in established human glioma cell-lines. Am J Pathol. 1993;142:883–892. [PMC free article] [PubMed] [Google Scholar]
- Vershinin et al., 2007.Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. Multiple-motor based transport and its regulation by tau. Proc Natl Acad Sci U S A. 2007;104:87–92. doi: 10.1073/pnas.0607919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock and Esiri, 1982.Wilcock GK, Esiri MM. Plaques, tangles and dementia-a quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- Yamada et al., 2011.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama et al., 2007.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]