

Abstract
Highly porous calcium phosphate (CaP) scaffolds for bone-tissue engineering were fabricated by combining a robocasting process with a sol–gel synthesis that mixed Calcium Nitrate Tetrahydrate and Triethyl Phosphite precursors in an aqueous medium. The resulting gels were used to print scaffolds by robocasting without the use of binder to increase the viscosity of the paste. X-ray diffraction analysis confirmed that the process yielded hydroxyapatite and β-tricalcium phosphate biphasic composite powders. Thus, the scaffold composition after crystallization of the amorphous structure could be easily modified by varying the initial Ca/P ratio during synthesis. The compressive strengths of the scaffolds are ~6 MPa, which is in the range of human cancellous bone (2–12 MPa). These highly porous scaffolds (~73 vol% porosity) are composed of macro-pores of ~260 μm in size; such porosity is expected to enable bone ingrowth into the scaffold for bone repair applications. The chemistry, porosity, and surface topography of such scaffolds can also be modified by the process parameters to favor bone formation. The studied sol–gel process can be used to coat these scaffolds by dip-coating, which induces a significant enhancement of mechanical properties. This can adjust scaffold properties such as composition and surface morphology, which consequently may improve their performances.
1 Introduction
Calcium phosphate (CaP) materials, especially biphasic calcium phosphate (BCP) composed of hydroxyapatite (HA, Ca10(PO4)6(OH)2) and β-tricalcium phosphate (β-TCP, Ca3(PO4)2), are widely used as bone-substitute material and in various tissue-engineering applications [1–8]. Interest is growing in BCP because of the faster dissolution rate of β-TCP, which enhances the formation of new bone inside the implanted material [3, 4]. However, in vitro tests indicate that fast dissolution can decrease the mechanical strength of CaP materials [9]. Thus, the HA/β-TCP ratio is a key parameter controlling the performance of the scaffold for bone repair applications, since it determines both biodegradation speed and mechanical properties of the material [10].
Various liquid-solution techniques have been used to prepare HA and β-TCP powders, including sol–gel synthesis, hydrothermal techniques, microemulsion, gas phase reactions and precipitation [11, 12]. Among these techniques, the sol–gel method offers considerable advantages because of its low cost, simplicity, high versatility, homogeneous molecular composition, and low synthesis temperature [11–13]. Because of its homogenous composition and controllable Ca/P ratio, the sol–gel method constitutes an ideal processing technique for fabricating BCP materials with various compositions. Sol–gel synthesis technique is well established, and can produce nanocrystalline powders, amorphous monolithic solids, and thin films, thus opening a wide field of applications [11–21]. Especially, there have been numerous studies of deposition of CaP coatings by sol–gel for biomedical applications [16–21]. Besides, several reports on the fabrication of porous scaffolds using sol–gel technique are found in the literature [22–28]. However, these studies are primarily focused on bioactive SiO2-CaO-P2O5 glasses and derived hybrid compositions. No systematic studies on the use of sol–gel processing of BCP scaffolds have been reported. Thus, the combination of sol–gel and solid free-form fabrication techniques may result in new constructs with adjustable chemical and structural properties.
Scaffolds for bone regeneration require interconnected large (>50 μm) and small macro-pores (1–50 μm) that create favorable conditions for cell seeding, proliferation and vascularization, and new bone formation [29]. An optimal three-dimensional (3-D) scaffold must be strong enough to replace the bone, at least temporarily, while providing a substrate for cell attachment and proliferation. It also must dissolve gradually as new tissue grows [30]. A variety of techniques has been used to fabricate porous ceramic scaffolds for tissue engineering, including: replica templates [31, 32], emulsions [33], the use of porogens [34], freeze-casting [35], and solid freeform fabrications which include robocasting as a rapid prototyping technique [9, 31, 36–38]. Compared to these methods, the solid freeform fabrication system offers better control of the macro-porosity and facilitates the complex shape formation required for bone scaffolds [9, 31, 36–38]. The robocasting method applied in this study is a printing process that builds 3-D structures layer-by-layer, extruding a continuous filament through a needle guided by a computer-assisted positioning system [9, 31, 36–38].
In this study, we offer an original approach by combining this robocasting technique with a sol–gel process to fabricate sol–gel CaP scaffolds for bone regeneration. The first objective is to correlate the Ca/P ratio of the sol–gel synthesis with the final composition of the BCP materials. Then, a given Ca/P ratio was selected to produce amorphous gels which were used to print porous scaffolds using the robocasting technique. The structure, composition, total porosity, and compressive strength of these scaffolds are subsequently characterized. Finally, the effects on the scaffold properties of a sol–gel coating derived from the same sol–gel process are analyzed.
2 Experimental
2.1 Preparation of amorphous CaP solutions and gels
A flowchart summarizing the sol–gel process used in this work is presented in Fig. 1. The first solution was prepared by dissolving Calcium Nitrate Tetrahydrate (CNT) in absolute ethanol under vigorous stirring. Calcium concentration in the solution was 3 M, and since the CNT is a tetrahydrate, the H2O/Ca molar ratio was 4. In parallel, the Triethyl Phosphite (P(OEt)3) precursor was mixed with deionized water to get a H2O/P molar ratio of 4; and with absolute ethanol, to fix the P(OEt)3 concentration at 3 M. This mixture was sealed in a glass beaker immediately after solvent addition, and then stirred vigorously. Because of the immiscibility between the phosphite and water, the mixture initially appears opaque since the light is scattered by the emulsion phase. However, after approximately two hours of mixing, the emulsion clarified indicating that the phosphite was completely hydrolyzed in agreement with the literature data [16, 17]. Both solutions were aged for two days at room temperature before use. During that time, the hydrolysis reactions took place in the ethanol–water media leading to the formation of P(OEt)3−x(OH)x phosphoric esters and Ca(NO3)2−y(OEt)y following reactions (1) and (2), for P(OEt)3 and CNT precursors respectively [16].
Fig 1.
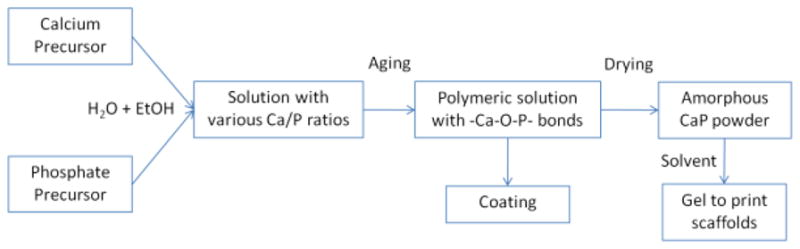
Sol-gel procedure to obtain calcium phosphate gels and solutions used, respectively, to print and to coat scaffolds
| (1) |
| (2) |
Then, the CNT and P(OEt)3 hydrolysed sols were mixed using a Ca/P ratio ranging from 1.5 to 1.67. Water was added to fix the H2O/P molar ratio at 20, which induces a final concentration of reactants at ~2.5 M. The solution was again aged for two days while hetero-condensation reactions occurred, yielding a polymeric solution composed of oligomeric derivatives containing -Ca–O–P-bonds following reaction (3) [16].
| (3) |
Evaporating the solvent from the solution at 40°C allows acceleration of the poly-condensation reactions, resulting in polymeric amorphous particles containing -(Ca–O–P)n- oligomers. These particles were dried at 110°C, and then they were carefully crushed with a mortar and sieved through a 105 μm mesh to obtain an amorphous CaP powder. SEM images of these amorphous particles are shown in Fig. 2. Next, the powders were mixed with a solvent, either water or isopropanol (IPA), to create gels. In the first case, 6 g of powder were mixed with 1 mL of water to get the printable amorphous gel. In the second case, amorphous sol–gel powder was mixed with IPA to obtain liquid slurry, which was ball-milled overnight. The printable gel was subsequently obtained after the partial evaporation of the IPA. Regardless of the solvent used, the gel was dried at 50°C few hours until it had the proper viscosity for printing by robocasting. Finally, prior to fabrication of the 3-D porous scaffolds, the gel was sieved though a 75 μm mesh to avoid agglomerates that could block the gel extrusion tip during printing. The untreated amorphous powder was also crystallized at 1,085°C to study the HA/β-TCP composition resulting from the sol–gel process before printing. As indicated in Fig. 1, the polymeric solution of the process may also be used for dip-coating to modify and adjust the properties of the scaffolds. Here, the dip-coating experiments were performed on sintered scaffolds with an immersion and withdrawal speed of 1 mm/s; and with complete immersion for 30 s. Coated samples were then heat treated at 1,085°C for two hours.
Fig 2.

SEM images of an amorphous sol–gel powder (Ca/P of 1.58) after the drying and sieving steps. The image on the right shows that the powder consists of micrometer-sized agglomerates; the image to the left demonstrates that the agglomerates themselves are composed of particles of several tens of nanometers
2.2 Scaffold printing
The gel was used to print HA/β-TCP ceramic grids (7.2 × 7.2 × 7.2 mm3) using a robotic deposition device (Robocad 3.0, 3-D Inks, Stillwater, OK) [9, 31, 36, 37]. The diameter of the printing nozzles controlling the diameter of the scaffold rods was 250 μm (EFD precision tips, EFD, East Providence, RI). The width between rods and the layer height were fixed at 330 and 180 μm, respectively. The scaffolds were printed over a mirror-polished silicon wafer (0.6 mm thick) and inside a reservoir of non-wetting oil (Lamplight, Menomonee Falls, WI) to prevent the scaffolds from drying during the printing process. The scaffold remains stable during printing because the silicon surface is hydrophilic, promoting adhesion with the first printed line. Moreover, the inert mirror-polished silicon surface allows easy removal of the samples after fabrication, which facilitates handling and prevents deformations due to uneven shrinkage during drying and sintering. After printing, the samples were dried and sintered on the silicon wafer. First, the plates were tilted to drain the excess oil. After drying for two days at room temperature, the scaffolds were fired at 600°C (2°C/min heating rate) for 2 h to evaporate the organics, followed by sintering for 2 h at 1,085°C with a heating and cooling rate of 5°C/min. The temperature of 1,085°C was chosen to avoid the formation of α-TCP crystals during the sintering [36]. Micro-cracks, which would decrease the mechanical performance of the scaffolds, can be generated as result of volume changes during the phase transformation [36, 39].
2.3 Characterization
X-ray diffraction (XRD) (Siemens D-500, Cu Kα1 = 1.5406 Å) was used to identify and quantify the HA and β-TCP phases. The patterns were compared with reference HA and β-TCP patterns (JCPDS 09-0432 and 09-0169 respectively) matching the peak intensities of each phase. As a result, the percentage of HA and β-TCP phases in the powders and scaffolds were quantified, and the crystalline Ca/P ratio can be calculated. Such a procedure was previously calibrated on a 60HA/40β-TCP industrial powder. Analyses of the scaffolds were made after grinding them to powder. The β-Di Calcium Pyrophosphate (β-DCPP, Ca2O7P2) phase matches the JCPDS pattern 09–0346.
Images of the scaffolds were taken with scanning electron microscopy (SEM) (Hitachi FE-SEM-4300EN) to visualize the quality of the scaffolds and to identify the grain and pore sizes of the scaffold rods.
The density of the scaffolds was calculated from the mass and dimensions of at least three samples with regular shapes. The theoretical densities of HA (3.16 g/cm3) and β-TCP (3.14 g/cm3) were used as references to calculate the total volume fraction of porosity. Also, the theoretical density of the β-DCPP phase has been reported to be 3.09 g/cm3 [40].
The compressive strength of the scaffolds was measured on a servo-hydraulic testing machine (MTS810, MTS Systems, Eden Prairie, MN), with a crosshead speed of 0.2 mm/min, for at least three samples. The sizes of the tested scaffolds are about 6 × 6 × 6 mm3 as discussed further in Sect. 3.2.
3 Results and discussion
3.1 Composition of sol–gel powders after sintering
The sol–gel powders were analyzed following the drying step described in the experimental section (Fig. 1) for different initial Ca/P ratios, after sintering at 1,085°C. As illustrated in Fig. 2 by SEM images, the powders consisted of particles of several tens of nanometers compacted into micrometer-sized agglomerates similar to previous studies on sol–gel syntheses [11, 12, 14]. Figure 3 shows the XRD patterns obtained for powders with initial Ca/P ratios of 1.5, 1.53, 1.58 and 1.67. Pure HA, Ca10(PO4)6(OH)2, and β-TCP, Ca3(PO4)2, phases give Ca/P ratio values of 1.67 and 1.5, respectively. The position of HA and β-TCP peaks are also indicated in this figure based on JCPDS patterns 09–0432 and 09–0169. For every Ca/P ratio in the range 1.5–1.67, the sol–gel process leads to HA/β-TCP biphasic powders after sintering, as presented in Fig. 3. When the initial Ca/P ratio increases from 1.5 to 1.67, the figure also shows that an increase in HA is associated with a decrease of the β-TCP phase. Matching the XRD peak intensities of each phase with the JCPDS patterns makes it possible to evaluate approximately the HA/β-TCP composition. Thus, our sol–gel process produces HA/β-TCP biphasic materials with weight compositions from 15HA/85β-TCP to 70HA/30β-TCP. The percentage of HA and β-TCP phases in the biphasic powders allows us to calculate the Ca/P ratio after sintering. Figure 4 presents the Ca/P ratio of the powder after crystallization as a function of the initial Ca/P ratio of the sol–gel solution. It clearly shows that the Ca/P ratio differs from the initial ratio of the sol–gel solution. Shifts to higher and smaller Ca/P ratio are observed when the initial Ca/P ratio is near 1.5 and 1.67. In other words, the system tends to produce biphasic materials instead of single phase materials. This is most likely the result of incomplete hydrolysis and polycondensation reactions during the sol–gel synthesis. Similar results have been observed in previous work, and it was shown that single phase HA or β-TCP can be obtained by adjusting pH and the temperature parameters during the sol–gel synthesis [14]. In summary, BCP powders were successfully produced by the sol–gel process, enabling the preparation of inks with different HA/β-TCP composition for robocasting.
Fig 3.

XRD patterns of sol–gel powders with initial Ca/P ratios of (a) 1.5, (b) 1.53, (c) 1.58 and (d) 1.67 after crystallization at 1,085°C. HA and β-TCP peaks are identified according to the JCPDS patterns 09–0432 and 09–0169, respectively
Fig 4.

Ca/P ratio of the sol–gel powder after sintering at 1,085°C as a function of Ca/P ratio of the initial sol–gel solution
3.2 Robocast scaffolds
Three-dimensional scaffolds were successfully printed by extruding gels through a 250 μm cone tip (Fig. 5). The gels were obtained by mixing a solvent with the amorphous sol–gel powder derived from the solution with an initial Ca/P ratio of 1.58. This composition should lead to the same Ca/P ratio after sintering of the scaffolds, i.e. a composition of ~50HA/50β-TCP, as shown in Fig. 4 and described in the previous section. Two solvents, water and isopropanol (IPA), have been studied here following the respective experimental steps described in the experimental section. For both solvents, the respective steps yielded a gel with a high viscosity which enables the printing of scaffolds by robocasting without any additional components. Neither hydrogel nor polymer as a binder is required to increase the viscosity of the gel in this work, unlike procedures frequently reported in the robotic deposition literature [9, 31, 36–38]. Indeed, the amorphous powder, consisting of an inorganic polymer network formed by -(Ca–O–P)n- oligomer chains, entraps water and IPA solvents to form a viscous gel. Formation of such a network was possible due to the 3- or 5-coordinated phosphate atoms yielding a 3D cross-linked network. Moreover, we observed that this network presents a transition state at around 30–35°C, transforming from a solid at low temperatures to a viscous gel at higher temperatures. This transition makes necessary careful control of the temperature and hygrometry during the drying step that follows the printing of the scaffolds. The oil and solvent inside the scaffolds from the printing process have to be dried for several days, in a dry atmosphere, at a temperature below 30°C prior to sintering. If these conditions are not carefully followed, the inorganic network, soft during the sintering because of the temperature increase, can be partially dissolved by the remaining solvent. That would further decrease the gel viscosity, resulting in a deformation of the scaffold shape. Moreover, since evaporation of IPA is faster than that of water, it is easier to manage scaffolds derived from IPA gel.
Fig 5.

SEM pictures of sol–gel scaffolds built with water and isopropanol (IPA) as solvent, and derived from the sol–gel synthesis with an initial Ca/P of 1.58. General view of the scaffolds made with both solvents (a). Corner (b), top (c) and side (d) views of an IPA scaffold. Cross-sections of scaffolds derived from IPA (e) and water (f) gels
A general view of the scaffolds is shown in Fig. 5, as well as SEM micrographs from the corner (Fig. 5b), the top (Fig. 5c) and the side (Fig. 5d). Also shown are cross-section views (Figs. 5e–f) for scaffolds derived from water-gel and IPA-gel after sintering at 1,085°C. The dimensions of the water-processed scaffolds are approximately 6.4 × 6.4 × 6.0 mm3 on average, which represent shrinkages of ~11% in the x and y directions and ~17% in the z direction (direction orthogonal to the substrate) after sintering. For the IPA scaffolds, the dimensions are ~6.15 × 6.15 × 5.8 mm3, which represent shrinkages of ~14% in the x–y directions and ~19% in the z direction. The higher shrinkage in the direction perpendicular to the substrate could probably be due to the effect of gravity from the weight of the scaffold during the drying and sintering steps. Different sintering for IPA and water-based inks can be related to different initial solid contents. Sintered scaffolds contain two characteristic dimensions of large macro-pores, the first of ~260 μm, resulting from the space between the rods in the x–y plan (Fig. 5b, c, e, f), and the second ~80 μm, induced by the space between the layers in the z direction (Fig. 5d, e, f). Such porosities should promote cell seeding, proliferation and vascularisation, and bone formation inside the scaffold as required for tissue engineering applications. A survey of the literature indicates that pores in the range of 100–400 μm are optimal for the bone ingrowth [41–45]. This macro-porosity must also provide transport pathways for nutrients, oxygen, and wastes necessary to maintain living cells within the scaffold. Since the macro-porosity results from the designed structure of the scaffold, the pore size can be easily controlled to fit the applications of the scaffold.
Water and IPA scaffold compositions were studied by XRD. Corresponding patterns are illustrated in Fig. 6, in which peaks are identified according to the CaP JCPDS patterns. Figure 6 shows that the printing of the water-gel yields the expected HA/β-TCP biphasic scaffolds (in agreement with Fig. 4). However, following the JCPDS patterns, the IPA-gel yields β-TCP/β-DCPP (β-DiCalcium Pyrophosphate, Ca2O7P2) biphasic scaffolds. As previously discussed, compositions of the scaffolds have been quantified from the XRD patterns, however, the β-TCP and β-DCPP peak overlaps made such quantification more difficult and less accurate. The scaffold compositions, as well as the scaffold Ca/P ratios, are reported in Table 1. The synthesis of β-TCP/β-DCPP biphasic material for the IPA scaffolds is related to a significant decrease of the Ca/P ratio from the initial amount of 1.58, while the Ca/P ratio stays almost constant when water is used as the solvent. This observation could be explained by (1) an unexpected effect of the IPA on the sintering condition of the amorphous gel which would modify the final composition of the scaffold after sintering; and/or (2) a slight evaporation of unreacted calcium nitrate precursors during the IPA evaporation step following the ball-milling performed for the IPA-gel preparation (see experimental section). In other words, the solvent itself, and/or different experimental steps used to prepare the gels, could significantly affect the CaP composition of the scaffolds after sintering of the amorphous gel structure. As a result, a wide range of CaP scaffold compositions can be obtained by controlling carefully the process parameters and/or varying the initial CaP ratio (see Sect. 3.1).
Fig 6.

XRD patterns of robocast scaffolds after sintering at 1,085°C and printed from a water gel (a) and an IPA gel (b), both with an initial Ca/P ratio of 1.58. The spectrum of an IPA scaffold coated by a sol–gel solution with a Ca/P ratio of 1.67 is also represented (c). HA, β-TCP and β-DiCalcium Pyrophosphate (β-DCPP) peaks are identified according to the JCPDS patterns 09–0432, 09–0169 and 09–0346, respectively
Table 1.
Composition and Ca/P ratio, after sintering at 1,085°C, for the scaffolds printed from water and IPA gels and for the coated IPA scaffolds
| Scaffold reference | Scaffold composition | Scaffold Ca/P ratio | Total porosity | Compressive strength |
|---|---|---|---|---|
| Water | HA/β-TCP 51/49 | 1.59 | 72.3 vol% ± 0.7 | 5.3 MPa ± 2.9 |
| IPA | β-TCP/β-DCPP 72/28 | 1.36 | 73.3 vol% ± 0.8 | 6.4 MPa ± 2.2 |
| IPA coated | β-TCP/β-DCPP 82/18 | 1.41 | 66.0 vol% ± 1.1 | 12.0 MPa ± 0.6 |
Total volume porosity (vol%) and compressive strength of the scaffolds are also reported
Images of the scaffold rods tested in this work before and after fracture are illustrated in Fig. 7. The rod surfaces of the IPA scaffolds exhibit a homogeneous roughness full of pores of ~1–2 μm (Fig. 7c). In addition to large pores from robocasting, surfaces with small pores could also promote a fast bone formation since it is believed that small pores are favorable for cell adhesion and crystallization of hydroxycarbonate apatite [26, 46]. In contrast, surfaces of scaffolds printed from a water-gel exhibit an inhomogeneous roughness composed of two pore ranges, a small one of ~1 μm and a second one ~5–30 μm (Figs. 5f, 7a). In fact, pores of 5–50 μm could also help to favor bone formation, since previous studies indicated that 5–20 μm is an optimum pore size for neovascularisation and fibroblast ingrowth [42, 44, 47]. Between these pores, which are dispersed randomly with a substantial distance between them, the surfaces of scaffolds using gels processed with water look very smooth. Cross-section (Fig. 5f) and fracture (Fig. 7b) images of water scaffolds show that both types of pores also appear inside the rods. The presence of large pores in the water scaffold could be explained by (1) an incomplete miscibility of water in the amorphous gel, which created water droplets inside the gel that evaporated during the sintering and/or (2) too-high viscosity of the gel, which did not allow the removal of air bubbles entrapped in the gel prior to printing. The fracture images also show a larger particle size for the IPA scaffold than for the water scaffold, ~1 μm compared to ~0.5 μm (Fig. 7b, d). This observation could be explained by an easier sintering of the IPA-gel structure, in agreement with the higher shrinkage value of the IPA scaffold, which could be related either by the chemistry or process variations. To conclude, depending of the sol–gel parameter, the sol–gel process can provide different kinds of surface morphology and rod structures that could be selected for different purposes.
Fig 7.

Surface (left) and fracture (right) SEM micrographs of robocast scaffold internal rods for sol–gel scaffolds derived from water (a and b) and isopropanol (c and d) gels, and derived from the isopropanol gel after sol–gel coating (e and f)
Finally, the total volume porosity and the compressive strength of the scaffolds have been measured and reported in Table 1. Regardless of which solvent is used, both scaffolds have an overall porosity of ~73 vol%. This porosity results from the addition of large macro-pores designed by the scaffold printing and the pores included into the printed rods. Since it is easy to control the scaffold design, i.e. the space between the rods and the layers, it is also easy to modify the volume porosity and the corresponding mechanical strength. Mechanical tests on these scaffolds, in the direction perpendicular to the printing plane, indicate a compressive strength of ~6 MPa for both solvents, which matches the range of compressive strength of human cancellous, also called trabecular, bone (2–12 MPa) [48]. Moreover, early studies indicate that, for these types of robocast scaffolds, compressive strength should be almost the same in all directions (parallel and perpendicular to the printing plane) [37]. In agreement with a recent review of CaP scaffolds [49], this compressive strength is similar to values reported in the literature for HA and β-TCP scaffolds with volume porosity in the same range. For instance, Cordell reported that robocast BCP scaffolds with a porosity of 67 vol% have compressive strength of ~8 MPa [50]. Bignon, on the other hand, described BCP scaffolds with a compressive strength of ~1.5 MPa for a porosity of 73 vol% [51]. Nevertheless, the obtained compressive strength values are sufficient for scaffold handling during in vitro or in vivo experiments. In summary, the sol–gel process developed here, combined with robocasting, yields CaP scaffolds with variable chemistry and with properties required for bone ingrowth. Moreover, the porosity and surface morphology characteristics can be modified by varying the experimental conditions.
3.3 Coated scaffolds
Coating derived from the polymeric solution of the studied sol–gel process (Fig. 1) was also analyzed in this work. IPA scaffolds were dip-coated into a sol–gel solution with an initial Ca/P ratio of 1.67. The choice of this ratio was made to increase the Ca/P ratio of the β-TCP/β-DCPP structure derived from the IPA-gel. After the dip-coating process, the samples were sintered at 1,085°C for two hours to crystallize the polymeric film. The coated scaffolds were characterized by their surface morphology, chemistry, porosity, and compressive strength. These traits were then compared with the properties of uncoated IPA scaffolds. SEM micrographs of the IPA-scaffold surfaces, with and without sol–gel coating, are shown in Fig. 7c, e. The images show an inhomogeneous film on the surface of the printed rods as a result of the applied sol–gel coating (Fig. 7e). The coating introduces a supplementary range of porosity ~5–20 μm on the top of the scaffold surface (Fig. 7e). This porosity could be induced by a dewetting effect of the sol–gel solution on the top of the rough surface after the scaffold dipping. As discussed in the previous section, this additional porosity might favor bone formation via faster neovascularisation and fibroblast ingrowth [42, 44, 47]. It should be noted that the macro-porosity of ~260 μm, necessary for bone ingrowth, is not modified by the coating process. Porosity and compressive strength properties of the coated IPA scaffolds are reported in Table 1. Compared with uncoated scaffolds, the data show a decrease of the volume porosity, from ~73 to ~66 vol%, which results from the deposition of the coating being on rod surfaces and from the infiltration of the sol–gel solution into the porous structure.
Figure 7d, f shows the images of the fractured scaffold rods with and without coating. As previously mentioned, the images confirm that the coating process decreases the pore volume due to sol–gel infiltration in the porous rods. Because the dipping process is followed by a heat-treatment step, the sol–gel matter sintered on the scaffold particles leading to a slight increase of the grain size (Fig. 7d, f). Also, significant increases in compressive strength occurred after the coating process, from ~6 to ~12 MPa. Because the smallest pores and cracks of the CaP structure are being filled by the sol–gel solution, denser scaffolds with increased mechanical properties are produced. Since macro-porosity does not vary after coating, both the porosity decrease and mechanical strength increase are related to the decrease of micro-porosity after sintering of the infiltrated solution. It was also verified that the additional heat treatment at 1,085°C on uncoated scaffolds does not affect the scaffold properties. Finally, the chemistry of the coated scaffold has been characterized by XRD. A comparison between uncoated and coated patterns of IPA scaffolds is showed in Fig. 6. Both analyses show a β-TCP/β-DCPP structure. However, matching the peak with the JCPDS patterns underscores a variation in β-TCP/β-DCPP ratio. Compositions and Ca/P ratios of uncoated and coated IPA scaffolds are reported in Table 1. The coating induces a variation of the β-TCP/β-DCPP ratio from 72/28 to 82/18 which is related to an increase of Ca/P ratio from 1.36 to 1.41. This significant increase of Ca/P ratio can be explained by the presence of solid matter from the coating process, with an initial Ca/P ratio of 1.67, which represents more than 20% of the coated-scaffold volume (in agreement to the total porosity measurement of Table 1). In conclusion, controlling the sol–gel coating appears to be a good way to manage scaffold properties such as surface morphology, porosity, chemistry, and mechanical strength. This provides a pathway to improving their bone regeneration performance.
4 Conclusion
A sol–gel process was developed to fabricate HA/β-TCP composites from amorphous polymeric solutions and powders. In particular, highly porous biphasic CaP scaffolds have been successfully produced by combining sol–gel synthesis with robocasting. The scaffolds exhibit large fractions of porosity with macro-pores ~260 μm, which should enable bone ingrowth for bone repair applications. The internal scaffold surfaces show morphologies with various pore sizes which could influence the in vivo bone formation. Also, with good control of process parameters, a large range of CaP compositions for these scaffolds should be easily accessible for further studies. The sol–gel process allows the coating of scaffolds by dip-coating in an amorphous polymeric solution. Such a coating can not only improve significantly the mechanical properties of the structure, but also modify the topography and the chemistry of the internal scaffold surface to favor bone formation. The entire process allows varying the scaffold chemistry, porosity, and surface morphology parameters that are key factors affecting scaffold performance. As sol–gel technique is quite versatile, these factors can be monitored during and after the scaffold fabrication, opening a wide field for other highly promising applications in bone-tissue engineering.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Dental and Craniofacial Research Grant No. 1 R01 DE015633.
Contributor Information
Manuel Houmard, Email: manuel.houmard@wanadoo.fr, Materials Science Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Qiang Fu, Materials Science Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Eduardo Saiz, Centre for Advanced Structural Materials, Department of Materials, Imperial College, London, UK.
Antoni P. Tomsia, Materials Science Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
References
- 1.Hench LL. Bioceramics. J Am Ceram Soc. 1998;81:1705. [Google Scholar]
- 2.Ramay HRR, Zhang M. Biphasic calcium phosphate nanocomposite porous scaffolds for load-bearing bone tissue engineering. Biomaterials. 2004;25:5171. doi: 10.1016/j.biomaterials.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 3.LeGeros RZ, Lin S, Rohanizadeh R, Mijares D, LeGeros JP. Biphasic calcium phosphate bioceramics: Preparation, properties and applications. J Mater Sci Med. 2003;14:201. doi: 10.1023/a:1022872421333. [DOI] [PubMed] [Google Scholar]
- 4.Daculsi G, LeGeros RZ, Nery E, Lynch K, Kerebel B. Transformation of biphasic calcium phosphate ceramics in vivo: ultrastructural and physicochemical characterization. J Biomed Mater Res. 1989;23(8):883. doi: 10.1002/jbm.820230806. [DOI] [PubMed] [Google Scholar]
- 5.LeGeros RZ, Daculsi G, LeGeros JP. Bioactive Bioceramics. In: Pietrzak WS, editor. Orthopedic Biology and Medicine: Musculoskeletal Tissue Regeneration: Biological Materials and Methods. Chapter 8. New Jersey: Humana Press Inc; 2008. pp. 153–81. [Google Scholar]
- 6.LeGeros RZ, LeGeros JP. Hydroxyapatite. In: Kokubo T, editor. Bioceramics and their clinical applications. Cambridge: Wood-head Publishing Ltd; 2008. pp. 367–94. [Google Scholar]
- 7.Malard O, Guicheux J, Bouler JM, Gauthier O, Beauvillain de Montreuil C, Aguado E, Pilet P, LeGeros RZ, Daculsi G. Calcium phosphate scaffold and bone marrow for bone reconstruction in irradiated area: a dog study. Bone. 2005;36:323. doi: 10.1016/j.bone.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 8.Arinzeh TL, Peter SJ, Archambault MP, Van Den Bos C, Gordon S, Kraus K, Smith A, Kadiyala S. Allogeneic mesynchymal stem cells rgenerate bone in a critical sized canine segmental defect. J Bone Jt Surg Am. 2003;85A:1927. doi: 10.2106/00004623-200310000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Miranda P, Pajares A, Saiz E, Tomsia AP, Guiberteau F. Mechanical properties of calcium phosphate scaffolds fabricated by robocasting. J Biomed Mater Res A. 2008;85A:218. doi: 10.1002/jbm.a.31587. [DOI] [PubMed] [Google Scholar]
- 10.Daculsi G, Passuti N, Martin S, Deudon C, Legeros RZ, Raher S. Macroporous calcium phosphate ceramic for long bone surgery in humans and dogs: Clinical and histological study. J Biomed Mater Res. 1990;24(3):379. doi: 10.1002/jbm.820240309. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Shaw LL. Synthesis of high purity hydroxyapatite nanopowder via sol–gel combustion process. J Mater Sci. 2009;20:1223. doi: 10.1007/s10856-008-3685-x. [DOI] [PubMed] [Google Scholar]
- 12.Sanosh KP, Chu MC, Balakrishnan A, Kim TN, Cho SJ. Sol–gel synthesis of pure nano sized β-tricalcium phosphate crystalline powders. Curr Appl Phys. 2010;10:68. [Google Scholar]
- 13.Gupta R, Kumar A. Bioactive materials for biomedical applications using sol–gel technology. Biomed Mater. 2008;3:034005. doi: 10.1088/1748-6041/3/3/034005. [DOI] [PubMed] [Google Scholar]
- 14.Raynaud S, Champion E, Bernache-Assollant D, Thomas P. Calcium phosphate apatites with variable Ca/P atomic ratio I. Synthesis, characterisation and thermal stability of powders. Biomaterials. 2002;23:1065. doi: 10.1016/s0142-9612(01)00218-6. [DOI] [PubMed] [Google Scholar]
- 15.Fellah BH, Layrolle P. Sol–gel synthesis and characterization of macroporous calcium phosphate bioceramics containing micro-porosity. Acta Biomater. 2009;5:735. doi: 10.1016/j.actbio.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Liu DM, Troczynski T, Tseng WJ. Water-based sol–gel synthesis of hydroxyapatite: process development. Biomaterials. 2001;22:1721. doi: 10.1016/s0142-9612(00)00332-x. [DOI] [PubMed] [Google Scholar]
- 17.Liu DM, Yang Q, Troczynski T. Sol–gel hydroxyapatite coatings on stainless steel substrates. Biomaterials. 2002;23:691. doi: 10.1016/s0142-9612(01)00157-0. [DOI] [PubMed] [Google Scholar]
- 18.Manso M, Langlet M, Jiménez C, Martínez-Duart JM. Hydroxyapatite coatings obtained by the thermal activation of polymeric sols. Int J Inorg Mater. 2001;3:1153. [Google Scholar]
- 19.Manso M, Langlet M, Jiménez C, Martínez-Duart JM. Microstructural study of aerosol–gel derived hydroxyapatite coatings. Biomol Eng. 2002;19:63. doi: 10.1016/s1389-0344(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 20.Manso-Silván M, Langlet M, Jiménez C, Fernández M, Martínez-Duart JM. Calcium phosphate coatings prepared by aerosol-gel. J Eur Ceram Soc. 2003;23:243. [Google Scholar]
- 21.Stoch A, Jastrzębski W, Długoń E, Lejda W, Trybalska B, Stoch GJ, Adamczyk A. Sol–gel derived hydroxyapatite coatings on titanium and its alloy Ti6Al4 V. J Mol Struct. 2005;744–747:633. [Google Scholar]
- 22.Chen QZ, Thompson ID, Boccaccini AR. 45S5 Bioglass®-derived glass–ceramic scaffolds for bone tissue engineering. Biomaterials. 2006;27:2414. doi: 10.1016/j.biomaterials.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 23.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27:3413. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 24.Jones JR, Ehrenfried LM, Hench LL. Optimising bioactive glass scaffolds for bone tissue engineering. Biomaterials. 2006;27:964. doi: 10.1016/j.biomaterials.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Jones JR, Tsigkou O, Coates EE, Stevens MM, Polak JM, Hench LL. Extracellular matrix formation and mineralization on a phosphate-free porous bioactive glass scaffold using primary human osteoblast (HOB) cells. Biomaterials. 2007;28:1653. doi: 10.1016/j.biomaterials.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Almeida RM, Gama A, Vueva Y. Bioactive sol–gel scaffolds with dual porosity for tissue engineering. J Sol-Gel Sci Technol. 2011;57:336. [Google Scholar]
- 27.Marques AC, Jain H, Kiely C, Song K, Kiely CJ, Almeida RM. Nano/macroporous monolithic scaffolds prepared by the sol–gel method. J Sol-Gel Sci Technol. 2009;51:42. [Google Scholar]
- 28.Peña J, Román J, Cabañas MV, Vallet-Regí M. An alternative technique to shape scaffolds with hierarchical porosity at physiological temperature. Acta Biomater. 2010;6:1288. doi: 10.1016/j.actbio.2009.10.049. [DOI] [PubMed] [Google Scholar]
- 29.Hulbert SF, Morrison SJ, Klawitte JJ. Tissue reaction to three ceramics of porous and non-porous structures. J Biomed Mater Res. 1972;6:347. doi: 10.1002/jbm.820060505. [DOI] [PubMed] [Google Scholar]
- 30.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 31.Saiz E, Gremillard L, Menendez G, Miranda P, Gryn K, Tomsia AP. Preparation of porous hydroxyapatite scaffolds. Mater Sci Eng C. 2007;27:546. [Google Scholar]
- 32.Ben-Nissan B. Natural bioceramics: from coral to bone and beyond. Curr Opin Solid State Mater Sci. 2003;7:283. [Google Scholar]
- 33.Bohner M, Van Lenthe GH, Grunenfelder S, Hirsiger W, Evison R, Muller R. Synthesis and characterization of porous beta-tri-calcium phosphate blocks. Biomaterials. 2005;26:6099. doi: 10.1016/j.biomaterials.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 34.Sous M, Bareille R, Rouais F, Clement D, Amedee J, Dupuy B, Baquey Ch. Cellular biocompatibility and resistance to compression of macroporous beta-tricalcium phosphate ceramics. Biomaterials. 1998;19:2147. doi: 10.1016/s0142-9612(98)00118-5. [DOI] [PubMed] [Google Scholar]
- 35.Deville S, Saiz E, Tomsia AP. Freeze casting of hydroxyapatite scaffolds for bone tissue engineering. Biomaterials. 2006;27:5480. doi: 10.1016/j.biomaterials.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 36.Franco J, Hunger P, Launey ME, Tomsia AP, Saiz E. Direct write assembly of calcium phosphate scaffolds using a water-based hydrogel. Acta Biomater. 2010;6:218. doi: 10.1016/j.actbio.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miranda P, Saiz E, Gryn K, Tomsia AP. Sintering and robo-casting of β-tricalcium phosphate scaffolds for orthopaedic applications. Acta Biomater. 2006;2:457. doi: 10.1016/j.actbio.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Duoss EB, Twardowski M, Lewis JA. Sol–gel inks for direct-write assembly of functional oxides. Adv Mater. 2007;19:3485. [Google Scholar]
- 39.Case ED, Smith IO, Baumann MJ. Microcracking and porosity in calcium phosphates and the implications for bone tissue engineering. Mater Sci Eng A. 2005;390:246. [Google Scholar]
- 40.Wang JW, Hon MH. Sugar-mediated chitosan/poly(ethylene glycol)-β-dicalcium pyrophosphate composite: Mechanical and microstructural properties. J Biomed Mater Res A. 2003;64A:262. doi: 10.1002/jbm.a.10358. [DOI] [PubMed] [Google Scholar]
- 41.Murphy CM, Haugh MG, O’Brien FJ. The effect of mean pore size on cell attachment, proliferation and migration in collagen–glycosaminoglycan scaffolds for bone tissue engineering. Biomaterials. 2010;31:461. doi: 10.1016/j.biomaterials.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 42.Oh SH, Park IK, Kim JM, Lee JH. In vitro and in vivo characteristics of PCL scaffolds with pore size gradient fabricated by a centrifugation method. Biomaterials. 2007;28:1664. doi: 10.1016/j.biomaterials.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 43.Karageorgiou V, Kaplan D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials. 2005;26:5474. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Petite H, Viateau V, Bensaïd W, Meunier A, De Pollak C, Bourguignon M, Oudina K, Sedel L, Guillemin G. Tissue-engineered bone regeneration. Nat Biotechnol. 2000;18:959. doi: 10.1038/79449. [DOI] [PubMed] [Google Scholar]
- 45.Pineda LM, Büsing M, Meinig RP, Gogolewski S. Bone regeneration with resorbable polymeric membranes. III. Effect of poly(L-lactide) membrane pore size on the bone healing process in large defects. J Biomed Mater Res. 1996;31:385. doi: 10.1002/(SICI)1097-4636(199607)31:3<385::AID-JBM13>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 46.Campbell CE, Von Recum AF. Microtopography and soft tissue response. J Invest Surg. 1989;2:51. doi: 10.3109/08941938909016503. [DOI] [PubMed] [Google Scholar]
- 47.Yang S, Leong KF, Du Z, Chua CK. The design of scaffolds for use in tissue engineering. Part I. Traditional factors. Tissue Eng. 2001;7:679. doi: 10.1089/107632701753337645. [DOI] [PubMed] [Google Scholar]
- 48.Carter DR, Hayes WC. Bone compressive strength: the influence of density and strain rate. Science. 1976;194:1174. doi: 10.1126/science.996549. [DOI] [PubMed] [Google Scholar]
- 49.Wagoner Johnson AJ, Herschler BA. A review of the mechanical behavior of CaP and CaP/polymer composites for applications in bone replacement and repair. Acta Biomater. 2011;7:16. doi: 10.1016/j.actbio.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 50.Cordell JM, Vogl ML, Wagoner Johnson AJ. The influence of micropore size on the mechanical properties of bulk hydroxyapatite and hydroxyapatite scaffolds. J Mech Behav Biomed Mater. 2009;2:560. doi: 10.1016/j.jmbbm.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 51.Bignon A, Chouteau J, Chevalier J, Fantozzi G, Carret J, Chavassieux P, Boivin G, Melin M, Hartmann D. Effect of micro-and macroporosity of bone substitutes on their mechanical properties and cellular response. J Mater Sci Mater Med. 2003;14:1089. doi: 10.1023/b:jmsm.0000004006.90399.b4. [DOI] [PubMed] [Google Scholar]