

Abstract
Resistance to existing HIV therapies is an increasing problem, and alternative treatments are urgently needed. RNA interference (RNAi), an innate mechanism for sequence-specific gene silencing, can be harnessed therapeutically to treat viral infections, yet viral resistance can still emerge. Here, we demonstrate that HIV can develop indirect resistance to individual and combinatorial RNAi-targeting protein-coding regions up to 5,500 nucleotides (nt) downstream of the viral promoter. We identify several variants harboring mutations in the HIV promoter, and not within the RNAi targets, that produce more fully elongated transcripts. Furthermore, these variants are resistant to the RNAi, potentially by stoichiometrically overwhelming this cellular mechanism. Alarmingly, virus resistant to one short hairpin RNA (shRNA) also exhibits cross-resistance to a different shRNA, which targets a distinct and spatially distant region to which the virus has not been previously exposed. To our knowledge, this is the first example of HIV “cross-resistance” to viral inhibitors targeting different loci. Finally, combining anti-HIV RNAi with a small molecule enhancer of RNAi can inhibit the replication of an indirectly resistant mutant. These results suggest that indirect resistance to RNAi is a general mechanism that should be considered when investigating viral resistance and designing combinatorial RNAi therapies.
Introduction
Thirty years after the identification of AIDS, over 34 million people are living with HIV. Significant strides have been made in controlling the epidemic, including improved access to highly active antiretroviral therapies that potently suppress viral replication and have thereby extended the lives of millions of patients.1 However, HIV strains resistant to first-line therapies have been emerging,2 and new antiviral treatments are thus needed.
RNA interference (RNAi) is a mechanism that can potentially be harnessed for antiviral therapy, due to its ease of design and exquisite potential to differentiate between the host and pathogen. Specifically, the cellular enzyme Argonaute can be primed with ~21 nucleotide (nt) short-interfering RNAs to degrade HIV RNAs in a sequence-specific manner. However, due to this very potential for high sequence specificity, RNAi faces a similar challenge as other antiviral drugs: rapid viral sequence evolution and the subsequent emergence of resistance.3
It is well recognized that mutation within a RNAi target can yield resistance, and mutation near the target can also result in the structural rearrangement of viral mRNA to render it less susceptible to RNAi.4,5 To address these escape mechanisms, several highly conserved targets can be used in combination to delay resistance.6,7 However, we recently proposed a distinct mechanism for resistance in which HIV acquires mutations within the long terminal repeat (LTR), which contains the viral promoter, and thereby transcriptionally upregulates viral gene expression to stoichiometrically overwhelm cellular RNAi mechanisms.8 If general, this mechanism would be challenging to study and address since it involves sequence fluctuations in viral regions that may lie far away from a RNAi target.
There are a number of open questions about such indirect RNAi evasion via viral transcriptional upregulation. Our previous study directed RNAi against HIV's TAR element, an indispensible component of the HIV promoter and regulator of viral gene expression, and it is unclear whether RNAi directed against viral protein-coding sequences currently under clinical investigation9 could also favor modifications of the HIV promoter. Second, a troubling implication not considered in the original work is that transcriptional upregulation may be a general mechanism that enables a virus to evade not only RNAi directed against the original target, but also RNAi subsequently directed against other regions. The analogous situation for highly active antiretroviral therapies would be if resistance to a reverse transcriptase inhibitor also conferred resistance to a protease inhibitor. Finally, the original study was conducted with an attenuated viral strain, and it is unclear whether the indirect mechanism generalizes to full length HIV.10
Here, we assess the ability of full length HIV to evade RNAi directed against a single target or a combination of two targets within viral protein-coding regions. Using this combinatorial RNAi system, we investigate the hypothesis that resistance to one short hairpin (shRNA) could additionally confer resistance to a second shRNA targeting a distinct viral sequence. We demonstrate that mutation of the HIV LTR confers the virus with resistance to RNAi targeting protein-coding regions up to 5,500 nts downstream of the HIV promoter. Moreover, two mutants exhibited cross-resistance to shRNAs that they had not previously encountered, indicating that such a mechanism for indirect evasion that is independent of target sequence has the potential to significantly compromise combinatorial RNAi strategies. To our knowledge, this is the first demonstration of HIV cross-resistance for viral inhibitors targeting independent genes. Taken together, these data suggest that combinatorial therapies should be tailored to address this potential for indirect resistance.
Results
Validation of RNAi targets within protein-coding regions
A standard lentiviral expression system11 was used to generate cell lines expressing shRNAs against previously reported highly conserved viral targets.7,12 For each region of interest, several RNAi targets that overlap the previously tested 19 nt regions7,12 was tested, as the pSLIK vector is predicted to yield a 21 nt shRNA.11 In all, 14 shRNAs that target highly conserved regions of the packaging signal (psi)/gag, pol, vif, and tat/rev (Figure 1a) were analyzed, and shRNAs that effectively reduced the replication of wild-type (WT) NL4-3 HIV were identified for each target region except vif (Figure 1b). Notably, minor shifts of just 1 nt had substantial impacts on RNAi efficacy, consistent with previous results.7
Figure 1.

Inhibition of HIV with combinatorial RNA interference (RNAi) targeting protein-coding regions. (a) A list of previously tested, highly conserved RNAi targets up to 5,500 nucleotides (nts) downstream of the HIV promoter and targeting psi/gag, pol, vif, and tat/rev is shown. For each target gene of interest, several short hairpin RNAs (shRNAs) that encompass the previously tested 19 nt regions were tested, as the pSLIK vector is predicted to yield a 21 nt short-interfering RNA (siRNA).11 Nucleotide positions are based on NL4-3 RNA. (b) shRNAs targeting regions in a were tested for their ability to inhibit HIV replication after 8 days. A shRNA targeting firefly luciferase (Luc) was used as a negative control. Cells were infected with wild-type (WT) virus at a multiplicity of infection (MOI) of 0.0005. Experiments were conducted in biological triplicate, and error bars represent 1 SD. *A significantly different titer from unprotected cultures (P < 0.05). (c) Whisker plots were used to plot the diversity of the 21 nt Ldr3 and TatB2 targets for 956 subtype B HIV sequences from the Los Alamos National Lab database. Upper and lower boundaries of boxes represent upper and lower quartiles (respectively), and whiskers represent data within 1.5-fold of the upper and lower quartiles. *A significantly different median number of variations (P < 0.05, Mann–Whitney U-test). (d) The frequency of variation at each nucleotide position of the 21 nt Ldr3 and TatB2 targets is shown for the 956 HIV sequences analyzed in c. (e) Combinatorial RNAi can be equally distributed such that all protected cells (solid line) contain both shRNAs (gray), or compartmentalized, such that each protected cell contains one component of the combination (white and black). Also, unprotected cells (dashed line) will be present in vivo due to delivery limitations.
Because they provided similarly potent inhibition of HIV replication in the short-term assay, long-term studies were initiated with shRNAs Ldr3 and TatB2, which differ in several respects. Ldr3 spans the noncoding structural feature psi and the beginning of the late structural gene gag, whereas TatB2 targets two overlapping reading frames for early genes, tat and rev (Figure 1a and Supplementary Figure S1). Additionally, while both shRNAs target essential and highly conserved regions of the HIV genome, Ldr3 is significantly more conserved than TatB2 based on an analysis of 956 sequences from the Los Alamos National Lab HIV database (Figure 1c). Furthermore, variability in the TatB2 target is biased towards the 3′ end (Figure 1d), a key consideration for escape from RNAi as this represents the “seed” region most important in determining RNAi efficacy.13 We were thus also interested in observing how such variation in the seed region might affect long-term evolution of HIV using these two targets.
HIV eventually recovers from long-term inhibition
Several scenarios were considered in the experimental design for long-term cultures. First, it has been computationally predicted and experimentally confirmed that using multiple shRNAs reduces the likelihood of escape.6,7,14,15 We thus considered not only shRNA “monotherapies” but also two strategies for combinatorial shRNA delivery: equally distributed delivery of both shRNAs to all cells in a population versus compartmentalized delivery of individual shRNAs to a fraction or subpopulation of the cells within a population (Figure 1e). While the former is the strategy explored to date, our recent in silico analysis of compartmentalized delivery indicates that it may provide similar therapeutic benefits.16
In addition, due to delivery limitations, any clinical application of gene therapy will likely include a mix of gene-transduced and unmodified cells, and the latter could serve as a reservoir for viral replication and mutation.8,15 To more accurately reflect this heterogeneous environment, which has not been considered by others,8 we mixed Ldr3- or TatB2-protected cells with unmodified or unprotected cells at a ratio of 1:1. Furthermore, to account for the stochastic nature of long-term evolution studies, each mixture of unprotected and protected cells was tested in replicates of n = 12, over 32 days. Cultures whose infectious titer exceeded 1,000 IU/ml at any point were considered to have escaped, since cultures reaching this threshold consistently proceeded to higher titers. Although both shRNAs suppressed HIV replication to similar extents in a short-term trial (Figure 1b), Ldr3 enabled markedly more effective long-term suppression (Figure 2a). Moreover, HIV replication was suppressed even in the presence of high proportions of unprotected cells (up to 50%).
Figure 2.

HIV recovers from long-term RNA interference (RNAi)-mediated inhibition. HIV was propagated in the presence of a (a) Ldr3, TatB2, (b) compartmentalized, or equally distributed RNAi selective pressure for 32 days. In each sample, 4 × 105 cells of a given RNAi composition were infected at a multiplicity of infection (MOI) of 0.0005. Infectious titers were measured every 2 days, and every eighth day viral supernatant was transferred to a new culture with a composition identical to the original, for a total of 32 days. Cultures that exceeded 1,000 IU/ml were deemed to have “escaped.”
We also tested the shRNAs in combination. In the compartmentalized combination, two cell populations, each expressing a single shRNA, were mixed with unprotected cells at a ratio of 1:1:1. In the equally distributed combination, Ldr3 and TatB2 coexpressing cells were mixed with unprotected cells at a ratio of 2:1. To improve the capacity to identify trends in an inherently stochastic process, replicates of n = 48 were used for each condition. Interestingly, we found nearly identical frequencies of escape for the two combinatorial strategies over 32 days (Figure 2b), suggesting that at least for these shRNAs the exact combination strategy may not be a major factor in determining the efficacy of long-term inhibition in vitro, confirming our in silico predictions.16 We next analyzed potential mechanisms of escape for each RNAi strategy.
U3 is a hotspot for mutation
We sequenced the Ldr3 and TatB2 targets and flanking regions of HIV (Supplementary Figure S1) from cultures that generated high titers for three Ldr3-protected cultures, three TatB2-protected cultures, five compartmentalized cultures, and four equally distributed cultures. We also sequenced HIV propagated in unprotected cells to survey the natural diversity that arises from in vitro propagation without external RNAi-mediated selective pressure. Interestingly, sequence analysis of the Ldr3 region for 204 clones and the TatB2 region for 187 clones resulted in only 8 and 13 clones containing mutations within their respective target sequences, and no point mutations, insertions, or deletions became fixed (present in more than 50% of clones from any single culture) within the viral quasispecies for either target region (Table 1). While mutation of both RNAi target regions was likely in cultures that included both shRNAs, our sequencing methodology did not provide us with information on linkage between the Ldr3 and TatB2 sequences (Supplementary Figure S1). Importantly, no mutations occurred in the Ldr3 or TatB2 target in cultures that did not express a shRNA targeting that region, confirming that both regions are highly conserved and appropriate RNAi targets.
Table 1. Summary of mutationsa within Ldr3 and TatB2.
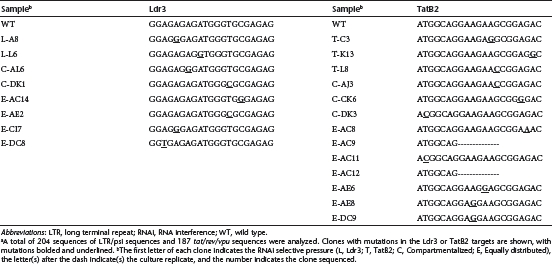
Since direct target mutation was not a dominant mechanism of resistance, we investigated whether, as we observed for RNAi targeting the TAR element,8 compensatory mutations may be contributing to indirect resistance. Two regions were analyzed using sequencing data adjacent to the RNAi targets: the LTR and psi, and exon 1 of tat and rev, and a portion of vpu (Supplementary Figure S1).
We first considered the sequence diversity of virus exposed to each selective pressure by quantifying the distribution of the number of mutations per clone within functionally relevant subsections of sequenced regions. For the LTR and psi, these regions include the untranscribed U3 enhancer region and the transcribed but noncoding R, U5, and psi regions. Since the LTR and psi are partially noncoding, their sequences are not suitable for an analysis of the ratio of nonsynonymous to synonymous substitutions as an indicator of positive selection, and alternative analysis for positive selection in promoters17 is difficult in a 9,000-nt genome with few noncoding regions. However, Kruskal–Wallis rank sum analysis of the median number of mutations per clone revealed that the U3 region from Ldr3 cultures and the equally distributed combination cultures have significantly higher median diversity compared to unprotected cultures (Figure 3a). The U3 region from compartmentalized combination cultures also has a higher median diversity than unprotected cultures, though this difference is not statistically significant once the Holm–Bonferroni correction for multiple comparisons is applied (P = 0.013). In contrast to Ldr3, the U3 region from TatB2 cultures—shown to be less suppressive of viral replication (Figure 2a)—does not show an elevated median diversity compared to virus from unprotected cultures. Likewise, the transcribed R, U5, and psi regions did not show any significant differences in median diversity for HIV clones from any RNAi-protected cultures (Figure 3b). Finally, the tat and rev exon 1 coding sequences and the vpu coding sequence that did not overlap with env did not exhibit significant differences in median sequence diversity in virus from unprotected vs. any RNAi-protected culture (Figure 3c,d).
Figure 3.

Exposure to a RNA interference (RNAi) selective pressure is associated with mutations in the U3 region. Whisker plots of the numbers of mutations per clone in the (a) U3; (b) R, U5, and psi; (c) tat/rev exon 1; and (d) vpu regions are shown for virus isolated from each selective pressure (UP, unprotected; Comp., compartmentalized; Equally, equally distributed). Upper and lower boundaries of boxes represent upper and lower quartiles, respectively, and whiskers represent data within 1.5-fold of the upper and lower quartiles. *A significantly different median number of mutations between a pair of conditions (P < 0.05, Kruskal–Wallis and Mann–Whitney U-tests with Holm–Bonferroni correction).
The significantly higher median diversity of the U3 region for HIV exposed to a RNAi selective pressure (Figure 3a) indicates that enhancer mutations may have contributed to the capacity for replication in the presence of antiviral RNAi. Analysis of substitution frequency at each nt revealed that several U3 mutations were fixed within particular cultures and unique to HIV exposed to RNAi (Figure 4a), including several mutations in a NFAT binding site (nts −230 to −220) and a mutation in YY1/LSF and LBP-1 sites just upstream of TAR (at nt −1). Furthermore, though not fixed within any particular culture, mutations in transcription factor binding sites for nuclear factor-κB, Sp1, AP4, and YY1/LSF (Supplementary Table S1) appeared at frequencies similar to the mutations within the Ldr3 and TatB2 target regions. By contrast, within tat/rev and vpu there were a couple frequent mutations, but none were unique to virus exposed to RNAi (Figure 4b).
Figure 4.

Mutations in the U3 are frequent and unique to RNA interference (RNAi)-exposed virus. The frequency of mutation at each nucleotide (nt) position was calculated for the (a) long-terminal repeat (LTR) (nts −240 to +182) and psi (nts +243 to +353) and (b) tat (nts +5,376 to +5,591), rev (nts +5,516 to + 5,591), and vpu (nts +5,608 to +5,768) regions for each selective pressure (UP, unprotected; Comp., Compartmentalized; Equally, equally distributed). Several mutations in the U3 (a, nts −240 to 0) are present at high frequencies in virus exposed to RNAi. Mutations in tat exon 1 (b) are present at high frequencies, but not unique to RNAi-exposed virus. Nucleotides are numbered based on NL4-3 RNA. Ldr3 and TatB2 targets are located at nts +329 to +349 and +5,516 to +5,536, respectively.
Mutations enhancing transcription confer viruses with cross-resistance to RNAi
We analyzed the replication of 12 variants that were chosen from cultures that escaped different RNAi strategies, represented both fixed and low frequency mutations (present in up to 25% of clones in any single culture), and encompassed mutations located in previously documented transcription factor-binding sites18 (Supplementary Table S1). Importantly, none of these variants included mutations in the Ldr3 or TatB2 targets. For quantitative comparison of viral replication dynamics in unprotected and RNAi-protected cultures, we calculated the “burst size” or cumulative viral replication for each viral strain by integrating the replication curves over the 10-day time course. Four of the twelve mutants exhibited significantly enhanced cumulative viral production in the presence of several RNAi strategies compared to the WT strain (Figure 5a and Supplementary Figure S2). For example, mutants T-C19 and C-CK1, which emerged from TatB2 and compartmentalized combination cultures, respectively, had significantly larger burst sizes compared to the WT virus in the presence of the TatB2 shRNA. Likewise, mutants L-K7 and L-L2, which were isolated from Ldr3 cultures, demonstrated enhanced cumulative viral replication compared to WT virus in the presence of the Ldr3 shRNA.
Figure 5.

Mutants develop cross-resistance to RNA interference (RNAi) and increased transcriptional activity. (a) Cells were infected with wild-type (WT) or mutant virus at a multiplicity of infection (MOI) of 0.005, and titers were measured every 2 days for 10 days. Viral burst size or cumulative viral replication was calculated by integrating 10-day replication curves (Supplementary Figure S3) and normalized to WT virus in unprotected (UP) cells. Experiments were conducted in biological triplicate, and error bars represent 1 SD. *A significantly different burst size compared to WT virus with the same inhibitory pressure (P < 0.05). (b) Cells were infected with WT or mutant virus at a MOI of 0.025 in the presence of 1 µmol/l saquinivir. Elongated viral transcripts were quantified in technical triplicate via reverse transcriptase quantitative PCR (RT-QPCR) and normalized to β-actin, and the resulting values were normalized to WT virus. Experiments were conducted in biological triplicate, and error bars represent 1 SD. *A significantly higher number of fully elongated transcripts compared to WT virus with the same inhibitory pressure (P < 0.05).
Strikingly, L-K7 and L-L2 also had significantly larger burst sizes compared to the WT virus in the presence of the TatB2 shRNA, and thus exhibited cross-resistance to TatB2, even though they were never previously exposed to this shRNA. Additionally, the mutant showing the strongest resistance to Ldr3 and TatB2 individually, L-K7, also exhibited cross-resistance to the equally distributed combination. Finally, the four mutants that demonstrated resistance to one or more RNAi strategies also showed altered replication dynamics in unprotected cells compared to the WT strain (Supplementary Figure S3). Accelerated peaks and subsequent drops in infectious titer (due to cell death induced by HIV replication) resulted in lower burst sizes than the WT strain in unprotected cells. Interestingly, we previously observed such accelerated replication dynamics and lower replicative fitness in unprotected cells for variants with enhanced transcriptional activity.8
To address whether their evasion of RNAi was due to transcriptional upregulation, we quantified transcriptional elongation in cells infected with the four resistant mutants (Figure 5b). Mutant L-L2 produced 1.5-fold more fully elongated transcripts compared to WT HIV, though this increase did not reach statistical significance (P = 0.14). However, mutants L-K7 and C-CK1 produced 2.3- and 1.7-fold more fully elongated transcripts, respectively, compared to WT HIV (P < 0.05).
Enoxacin acts synergistically with RNAi to inhibit a mutant indirectly resistant to RNAi alone
If the RNAi pathway becomes overwhelmed by large numbers of HIV transcripts, improving the flux of HIV RNAs flowing through this pathway should inhibit such indirectly resistant mutants. It was recently shown that enoxacin could enhance RNAi activity.19 We thus investigated if enoxacin would act synergistically with RNAi to inhibit indirectly resistant mutants.
While enoxacin inhibited WT and mutant virus replication in a RNAi-independent manner (Figure 6a), as previously reported,20,21 we found that enoxacin combined with RNAi inhibited replication of mutant LK-7 even when RNAi alone did not. If one knows the individual normalized burst sizes (i.e., the burst in the presence divided by the burst in the absence of the intervention) of two separate interventions, and the two interventions function independently, then the expected burst size of a combination of the two interventions is the product of their two individual normalized burst sizes.22,23 However, we found that the actual burst size for the combination of enoxacin and the Ldr3 shRNA was 2.3-fold lower than an expected burst size that assumes independent effects (P < 0.05; Figure 6b). Thus, enoxacin acts synergistically with Ldr3 to inhibit replication of an indirectly resistant mutant. Collectively, these results indicate that mutation of the HIV promoter can render the virus resistant to one or more shRNAs targeting distant sites in the viral genome by overwhelming of the RNAi pathway, but strategies to enhance RNAi may represent a means to address this problem.
Figure 6.

Enoxacin acts synergistically with RNA interference (RNAi) to inhibit a mutant indirectly resistant to Ldr3 alone. (a) Unprotected or Ldr3-protected cells were infected with wild-type (WT) or mutant HIV at a multiplicity of infection (MOI) of 0.005 in the presence of increasing amounts of enoxacin or carrier (dimethyl sulfoxide (DMSO)). Total viral burst sizes were calculated as previously described over 10 days and normalized to the burst size of WT virus in unprotected cells. Experiments were conducted in biological triplicate, and error bars represent 1 SD. *A significantly different burst size compared to the same virus and cell type without enoxacin (0 µmol/l) (P < 0.05). (b) Expected burst sizes for mutant LK-7 in the presence of enoxacin and the Ldr3 short hairpin RNA (shRNA) combined were calculated using a neutral model, in which the two inhibitors are assumed to act independently. Thus, the expected relative viral burst size in the presence of enoxacin and Ldr3 combined is equal to the relative burst size in the presence of enoxacin multiplied by the relative burst size in the presence of Ldr3, measured independently.22,23 If the observed relative burst size is less than the expected value, the inhibitors act synergistically. A synergism score was calculated by taking the ratio of expected to measured relative burst size. If enoxacin and Ldr3 act purely independently, the score =1, while a score >1 indicates synergism. Values reported as mean ± SD. *Synergism score significantly >1 (P < 0.05).
Discussion
Viral resistance generally involves direct mutation of a drug target, resulting for example in an enzyme active site that no longer binds the inhibitory molecule,24 or a RNA that is no longer degraded by RNAi.5 In these cases, cross-resistance can occur within specific drug classes. For instance, HIV resistance to one Protease inhibitor can confer HIV with cross-resistance to another Protease inhibitor,25,26 but not to a reverse transcriptase inhibitor. With sequence-specific therapies such as RNAi, it has widely been assumed that resistance to one short interfering RNA would not result in cross-resistance to another short interfering RNA targeting a distinct 21 nt sequence. Thus, combinatorial RNAi, in which multiple distinct 21 nt sequences are targeted simultaneously, has been suggested as a strategy to contend with resistance.7,27 Here, we show that evolution of transcriptional regulation can confer HIV with a generalized cross-resistance to RNAi. Thus, mutations in the viral promoter could alarmingly render the virus resistant to combinatorial RNAi and even to shRNAs to which the virus had never previously been exposed.
We identified mutations in U3 that may play a role in resistance by modulating the balance of activator and repressor recruitment to the HIV promoter.18 For example, deletion of the upstream NFAT site28 has been shown to increase LTR transcriptional activity and viral replication,29,30 and the fixed NFAT mutations in variant L-K7 and other mutants (Supplementary Table S1) could play similar roles in modulating transcription and replication. In addition, mutations in nuclear factor-κB sites—such as in L-K7, T-C19, C-CK1, and other mutants—could alter the differential binding affinities of various nuclear factor-κB dimers and thereby regulate transcriptional activity.31,32 While T-C19 did not show enhanced transcriptional elongation, the transcriptional upregulation may be subtler in this mutant and thus not detected with the specific assay we employed, or differences may present at other time points. Finally, several variants replicated with dynamics similar to WT virus (Supplementary Figure S2) and were likely propagated by replication in unprotected cells.
These results encourage thinking outside the target and designing combinations that take indirect mechanisms of resistance into account. For example, our results indicate that biochemical approaches to enhance RNAi19 (Figure 6) may aid in circumventing resistance involving general overwhelming of this pathway. Modulating expression of RNAi pathway components may also enhance RNAi activity;33 however, direct engineering of the RNAi pathway must be approached cautiously, as increasing expression of the shRNA itself may saturate or competitively inhibit endogenous miRNA activity.34,35 Alternatively, a combination of RNAi and non-RNAi therapies such as highly active antiretroviral therapies may also prevent resistance.8,36 In addition, the prescient inclusion of a CCR5 ribozyme and TAR decoy in addition to a shRNA targeting tat/rev in the recent phase I gene therapy trial9 results in a multilayered defense.
It is conceivable that indirect evasion could arise via elements other than the promoter, since for example sequence variation in Tat also impacts transcription,37 and it is possible that indirect evasion occurs for other antiviral therapies as well.38 Furthermore, whether analogous mechanisms of resistance exist for viruses with unique lifecycles remains an open question. For example, while poliovirus, respiratory syncytial virus, and hepatitis C virus do not have promoters that drive transcription, mutations that improve the initiation of positive strand RNA synthesis by the RNA-dependent RNA polymerase39,40 could result in indirect resistance to RNAi. Thus, broad sequencing approaches may aid in studying resistance mechanisms. Finally, fine-tuning of gene regulation by modulating the relative rates of mRNA synthesis and degradation may be a more general evolutionary mechanism, as recently investigated in yeast.41
In summary, we have shown that mutation of the HIV promoter can result in HIV cross-resistance to two distinct and spatially distant shRNAs targeting protein-coding sequences. This indirect and target region-independent mechanism of resistance occurred in the presence of combinatorial RNAi and could render such combinations ineffective. Such indirect mechanisms of resistance should arguably be considered when designing mono- or combinatorial anti-HIV therapies.
Materials and Methods
Cell culture, vector preparation, and cell line generation. Complimentary oligonucleotides (Invitrogen, Carlsbad, CA) including the appropriate loop and specific sequence for the targeted HIV regions (Supplementary Table S2) were annealed, phosphorylated, and ligated into the pSLIK vector (ATCC, Manassas, VA) as described.11 HEK 293Ts, SupT1s, and CEM green fluorescent protein cells were cultured, and lentiviral vectors were packaged as previously described.8 shRNA-expressing SupT1s were generated via transduction at multiplicity of infections of <0.1, and transduced populations were selected using 500 µg/ml of G418 sulfate (Sigma-Aldrich, St. Louis, MO) or cell sorting based on 1,000 ng/ml doxycycline-induced (Sigma-Aldrich) Venus expression.
HIV propagation experiments. Stocks of WT and mutant HIV NL4-3 were prepared using a single-LTR vector and titered as described.8 To initiate the short-term and long-term propagation experiments, 4 × 105 cells were infected as described,8 with 250 ng/ml of doxycycline to induce shRNA expression. Enoxacin (Sigma-Aldrich) dissolved in dimethyl sulfoxide (Sigma-Aldrich) at final concentrations of 25 and 50 µmol/l, or carrier (dimethyl sulfoxide) alone, was added as indicated. Infectious titers were measured using CEM green fluorescent protein indicator cells.8 For long-term cultures, supernatant was removed 8 days postinfection and stored at −80 °C, and 250 µl was used to initiate infections of fresh cultures of identical composition. Cultures that showed infectious titers greater than 1,000 IU/ml using the CEM green fluorescent protein indicator cells were considered to have escaped the RNAi.
Sequence analysis. Viral supernatant from putative escape cultures were used to infect a total of 1 × 105 naive SupT1s at a multiplicity of infection of 0.025 in the presence of 1 µmol/l saquinivir (NIH AIDS Research and Reference Reagent Program, Bethesda, MD) to limit infection to a single round. Genomic DNA was isolated 2 days postinfection using the Qiamp DNA Mini Kit (Qiagen, Venlo, the Netherlands). The LTR, psi region, tat, and vpu were PCR amplified using degenerate oligonucleotides binding to highly conserved regions (Supplementary Table S3) and TOPO cloned (Stratagene, La Jolla, CA) for sequencing. Python and the MUSCLE alignment algorithm were used for sequence analysis.
Transcriptional elongation assays. A total of 1 × 105 SupT1s were infected at a multiplicity of infection of 0.025 with WT or mutant virus in the presence of 1 µmol/l saquinivir to limit infection to a single round. Total RNA was extracted 3 days postinfection using Trizol (Invitrogen). Elongated transcripts were quantified using the single-step Quanititect SYBR Green Reverse Transcriptase-PCR kit (Qiagen) with primers to detect Tat and β-actin transcripts42,43 (Supplementary Table S4). Reverse transcriptase-quantitative PCR measurements were performed in technical triplicate.
Synergy analysis. Interaction between enoxacin and Ldr3 shRNA was calculated using a neutral model as described.22,23 Briefly, if two inhibitors of viral replication—A and B—act completely independently, the expected relative viral burst size in the presence of A and B combined is equal to the relative burst size in the presence of A multiplied by the relative burst size in the presence of B. A synergism score was calculated by taking the ratio of expected to measured burst sizes. A score >1 indicates synergism, a score = 1 indicates purely independent activity, and a score <1 indicates negative interactions.
Statistical analysis. Titers and fully elongated transcripts were compared using the Student's t-test (P < 0.05). Sequence diversity of virus exposed to different selective pressures was compared using the Kruskal–Wallis Rank sum test and Mann–Whitney U-test with the Holm–Bonferroni correction (P < 0.05).
SUPPLEMENTARY MATERIAL Figure S1. Regions sequenced. Figure S2. Burst size analysis of 12 mutants. Figure S3. Replication dynamics of four indirectly resistant mutants. Table S1. Fixed and low frequency mutations in functionally relevant regions of the HIV LTR. Table S2. Oligonucleotide sequences for pSLIK cloning. Table S3. Degenerate oligonucleotide sequences for PCR amplification of HIV from genomic DNA. Table S4. Oligonucleotide sequences for RT-QPCR.
Acknowledgments
We thank Kwang-Il Lim and Jonathan E. Foley for critical reading of the manuscript, Magdalena Niewiadomska-Bugaj for assistance with statistical analysis, and Siddharth S. Dey and Morgan N. Price for assistance with data analysis and representation. This work was funded by 2R01-GM073058, and a NSF graduate fellowship and SWE graduate scholarship to PSS. The authors declare no conflicts of interest for this work.
Supplementary Material
Regions sequenced.
Burst size analysis of 12 mutants.
Replication dynamics of four indirectly resistant mutants.
Fixed and low frequency mutations in functionally relevant regions of the HIV LTR.
Oligonucleotide sequences for pSLIK cloning.
Degenerate oligonucleotide sequences for PCR amplification of HIV from genomic DNA.
Oligonucleotide sequences for RT-QPCR.
REFERENCES
- UNAIDS (2010. AIDS Epidemic Update. Geneva.
- Smith RJ, Okano JT, Kahn JS, Bodine EN., and, Blower S. Evolutionary dynamics of complex networks of HIV drug-resistant strains: the case of San Francisco. Science. 2010;327:697–701. doi: 10.1126/science.1180556. [DOI] [PubMed] [Google Scholar]
- Haasnoot J, Westerhout EM., and, Berkhout B. RNA interference against viruses: strike and counterstrike. Nat Biotechnol. 2007;25:1435–1443. doi: 10.1038/nbt1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden D, Pusch O, Lee F, Tucker L., and, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Ooms M, Vink M, Das AT., and, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YP, von Eije KJ, Schopman NC, Westerink JT, ter Brake O, Haasnoot J.et al. (2009Combinatorial RNAi against HIV-1 using extended short hairpin RNAs Mol Ther 171712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Brake O, Konstantinova P, Ceylan M., and, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther. 2006;14:883–892. doi: 10.1016/j.ymthe.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Leonard JN, Shah PS, Burnett JC., and, Schaffer DV. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe. 2008;4:484–494. doi: 10.1016/j.chom.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A.et al. (2010RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma Sci Transl Med 236ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B. A new Houdini act: multiple routes for HIV-1 escape from RNAi-mediated inhibition. Future Microbiol. 2009;4:151–154. doi: 10.2217/17460913.4.2.151. [DOI] [PubMed] [Google Scholar]
- Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI.et al. (2006A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression Proc Natl Acad Sci USA 10313759–13764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Dykxhoorn DM, Kumar P, Ranjbar S, Song E, Maliszewski LE.et al. (2005Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV Blood 106818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J.et al. (2005Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs Nature 433769–773. [DOI] [PubMed] [Google Scholar]
- Applegate TL, Birkett DJ, Mcintyre GJ, Jaramillo AB, Symonds G., and, Murray JM. In silico modeling indicates the development of HIV-1 resistance to multiple shRNA gene therapy differs to standard antiretroviral therapy. Retrovirology. 2010;7:83. doi: 10.1186/1742-4690-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JN., and, Schaffer DV. Computational design of antiviral RNA interference strategies that resist human immunodeficiency virus escape. J Virol. 2005;79:1645–1654. doi: 10.1128/JVI.79.3.1645-1654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviran S, Shah PS, Schaffer DV., and, Arkin AP. Computational models of HIV-1 resistance to gene therapy elucidate therapy design principles. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD., and, Wray GA. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat Genet. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- Pereira LA, Bentley K, Peeters A, Churchill MJ., and, Deacon NJ. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000;28:663–668. doi: 10.1093/nar/28.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R.et al. (2008A small molecule enhances RNA interference and promotes microRNA processing Nat Biotechnol 26933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwase H, Momota K, Ohmine T, Komai T, Kimura T, Katsube T.et al. (1999A new fluoroquinolone derivative exhibits inhibitory activity against human immunodeficiency virus type 1 replication Chemotherapy 4548–55. [DOI] [PubMed] [Google Scholar]
- Nozaki-Renard J, Iino T, Sato Y, Marumoto Y, Ohta G., and, Furusawa M. Fluoroquinolones protect the human lymphocyte CEM cell line from HIV-1-mediated cytotoxicity. Cell Struct Funct. 1990;15:295–299. doi: 10.1247/csf.15.295. [DOI] [PubMed] [Google Scholar]
- Kapitzky L, Beltrao P, Berens TJ, Gassner N, Zhou C, Wüster A.et al. (2010Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action Mol Syst Biol 6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Cagney G., and, Krogan NJ. Quantitative genetic interactions reveal biological modularity. Cell. 2010;141:739–745. doi: 10.1016/j.cell.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F., and, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Hsieh SM, Chang SY, Hung CC, Sheng WH, Chen MY., and, Chang SC. Impact of first-line protease inhibitors on predicted resistance to tipranavir in HIV-1-infected patients with virological failure. BMC Infect Dis. 2009;9:154. doi: 10.1186/1471-2334-9-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Taylor J, Fessel WJ, Kaufman D, Towner W, Troia P.et al. (2010HIV-1 protease mutations and protease inhibitor cross-resistance Antimicrob Agents Chemother 544253–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre GJ, Groneman JL, Yu YH, Tran A., and, Applegate TL. Multiple shRNA combinations for near-complete coverage of all HIV-1 strains. AIDS Res Ther. 2011;8:1. doi: 10.1186/1742-6405-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA., and, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241:202–205. doi: 10.1126/science.3260404. [DOI] [PubMed] [Google Scholar]
- Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ., and, Lewis DB. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin Immunol. 2000;94:179–191. doi: 10.1006/clim.1999.4831. [DOI] [PubMed] [Google Scholar]
- Lu YC, Touzjian N, Stenzel M, Dorfman T, Sodroski JG., and, Haseltine WA. The NF kappa B independent cis-acting sequences in HIV-1 LTR responsive to T-cell activation. J Acquir Immune Defic Syndr. 1991;4:173–177. [PubMed] [Google Scholar]
- Leung TH, Hoffmann A., and, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Wang JK, Li TX, Bai YF., and, Lu ZH. Evaluating the binding affinities of NF-kappaB p50 homodimer to the wild-type and single-nucleotide mutant Ig-kappaB sites by the unimolecular dsDNA microarray. Anal Biochem. 2003;316:192–201. doi: 10.1016/s0003-2697(03)00049-6. [DOI] [PubMed] [Google Scholar]
- Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K.et al. (2010Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver J Clin Invest 1203106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L.et al. (2007Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC Nucleic Acids Res 355154–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR.et al. (2006Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways Nature 441537–541. [DOI] [PubMed] [Google Scholar]
- Huelsmann PM, Rauch P, Allers K, John MJ., and, Metzner KJ. Inhibition of drug-resistant HIV-1 by RNA interference. Antiviral Res. 2006;69:1–8. doi: 10.1016/j.antiviral.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Roof P, Ricci M, Genin P, Montano MA, Essex M, Wainberg MA.et al. (2002Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-kappaB and the Tat transactivator Virology 29677–83. [DOI] [PubMed] [Google Scholar]
- Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R.et al. (2011Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy Nature 47795–98. [DOI] [PubMed] [Google Scholar]
- Vogt DA., and, Andino R. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010;6:e1000936. doi: 10.1371/journal.ppat.1000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahias K, Ahmed-El-Sayed N, Masante C, Bitard J, Staedel C, Darfeuille F.et al. (2010Identification of a structural element of the hepatitis C virus minus strand RNA involved in the initiation of RNA synthesis Nucleic Acids Res 384079–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dori-Bachash M, Shema E., and, Tirosh I. Coupled evolution of transcription and mRNA degradation. PLoS Biol. 2011;9:e1001106. doi: 10.1371/journal.pbio.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JC, Miller-Jensen K, Shan PS, Arkin AP., and, Schaffer DV. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 2009;5:e1000260. doi: 10.1371/journal.ppat.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E., and, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. The EMBO Journal. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Regions sequenced.
Burst size analysis of 12 mutants.
Replication dynamics of four indirectly resistant mutants.
Fixed and low frequency mutations in functionally relevant regions of the HIV LTR.
Oligonucleotide sequences for pSLIK cloning.
Degenerate oligonucleotide sequences for PCR amplification of HIV from genomic DNA.
Oligonucleotide sequences for RT-QPCR.