

Abstract
Oncolytic viruses are generally designed to be cancer selective on the basis of a single genetic mutation. JX-594 is a thymidine kinase (TK) gene-inactivated oncolytic vaccinia virus expressing granulocyte-macrophage colony-stimulating factor (GM-CSF) and lac-Z transgenes that is designed to destroy cancer cells through replication-dependent cell lysis and stimulation of antitumoral immunity. JX-594 has demonstrated a favorable safety profile and reproducible tumor necrosis in a variety of solid cancer types in clinical trials. However, the mechanism(s) responsible for its cancer-selectivity have not yet been well described. We analyzed the replication of JX-594 in three model systems: primary normal and cancer cells, surgical explants, and murine tumor models. JX-594 replication, transgene expression, and cytopathic effects were highly cancer-selective, and broad spectrum activity was demonstrated. JX-594 cancer-selectivity was multi-mechanistic; replication was activated by epidermal growth factor receptor (EGFR)/Ras pathway signaling, cellular TK levels, and cancer cell resistance to type-I interferons (IFNs). These findings confirm a large therapeutic index for JX-594 that is driven by common genetic abnormalities in human solid tumors. This appears to be the first description of multiple selectivity mechanisms, both inherent and engineered, for an oncolytic virus. These findings have implications for oncolytic viruses in general, and suggest that their cancer targeting is a complex and multifactorial process.
Introduction
Oncolytic viruses represent an emerging therapeutic platform for the treatment of human cancer with unique attributes compared with conventional therapeutic modalities.1,2,3,4 One especially favorable feature of these therapeutic agents is their proposed selectivity for replication in cancer cells, while sparing normal cells and tissues, thus limiting off-target cell killing and toxicities. Oncolytic viruses can also be engineered to express therapeutic and monitoring transgenes. The expression of these proteins can also be highly cancer-selective if controlled by late promoters that are turned on after replication is initiated, for example. Since these agents can amplify themselves within cancer tissues, and simultaneously undergo clearance from normal tissues, they have the potential for a very large therapeutic index between cancer and normal tissues. This large therapeutic index is in marked contrast to the narrow therapeutic index for typical chemotherapeutics. The safety of oncolytic viruses in humans has been excellent to date, with no reported treatment-related deaths following treatment of over 500 patients.5 Understanding the therapeutic index for oncolytic viruses in relevant and representative human tissues, and elucidating the selectivity mechanisms involved, is critical for the effective design and development of these agents.
The mechanisms responsible for oncolytic virus cancer-selectivity include both inherent tumor tropism (e.g., vesicular stomatitis virus, refs. 6,7 and reovirus, refs. 8,9), and engineered selectivity (e.g., adenovirus, refs. 10,11 measles virus, refs. 12,13 and herpes simplex virus, refs. 14,15) deletion mutants. Poxviruses have biological characteristics that make them particularly promising as oncolytic therapeutics: (i) intravenous stability and efficient delivery to metastatic tumors through the blood, (ii) rapid and motile spread within tumors, (iii) large transgene-encoding capacity, and (iv) a large safety experience in humans as a vaccine.16 The tissue and/or cancer tropism of poxviruses, however, is poorly understood. Vaccinia virus binding to cells is mediated by the heparan sulfate, a ubiquitous cell surface-associated carbohydrate moiety of glycoproteins.17 Cellular uptake is mediated through a ubiquitous macropinocytosis mechanism in which the virus mimics an apoptotic bleb.18 Therefore, the exquisite selectivity of specific poxviruses for certain species or tissues is mediated within the cytoplasm of infected cells.
JX-594, the lead product within the oncolytic poxvirus class, is an engineered, targeted, and transgene-“armed” oncolytic poxvirus engineered from the Wyeth vaccine strain (Dryvax; Wyeth laboratories, Madison, NJ). JX-594 has an engineered disruption of the thymidine kinase (TK) gene for attenuation.19,20 In addition, the human CSF2 (granulocyte-macrophage colony-stimulating factor; GM-CSF) complementary DNA is expressed under the control of a synthetic (i.e., engineered) early-late promoter, and the lac-Z marker transgene is under control of the p7.5 promoter.19,20 The use of this attenuated vaccinia virus in cancer therapy was initially proposed purely as a vehicle to express the immunostimulatory cytokine GM-CSF, in solid tumors after intratumoral injection as a means to induce an anticancer immune response;19 GM-CSF had previously been identified as a potent activator of antigen-presenting cells in preclinical models.21 Kirn subsequently hypothesized that this agent could be used as a replication-selective, systemically active, and intravenously administered targeted oncolytic therapeutic for cancer.16,22
JX-594 has been well-tolerated in phase 1 and 2 clinical trials to date, both by intratumoral injection19,22 and by intravenous administration.23 Virus replication, GM-CSF expression and biological activity, and tumor responses have been reported in a diverse spectrum of cancer types; of note, infection and necrosis of distant, noninjected tumors have been shown reproducibly. While transient flu-like symptoms are common, no significant normal tissue toxicity has been reported to date.
Other oncolytic vaccinia viruses in preclinical and clinical development include vvDD (a TK- and vaccinia growth factor-deleted mutant),24 JX-963 (vvDD expressing GM-CSF; Jennerex, San Francisco, CA),25 JX-795 (TK- and B18R-deletion mutant expressing IFN-β Jennerex, San Francisco, CA),26 VV-FCU1 (Transgene SA, Illkirch-Graffenstaden, France),27 and GL-ONC (a TK-, hemagglutinin-, F14.5L- mutant expressing imaging transgenes; Genelux, San Diego, CA).28 This oncolytic virus class is therefore growing rapidly, and elucidating the selectivity mechanisms that these agents exploit will be critical to maximize patient benefit and the design of future products in this class.
Despite the clinical progress with JX-594, the mechanisms responsible for JX-594 cancer-selectivity remain poorly understood. Kim et al. demonstrated that a TK-deleted vaccinia virus (Western Reserve strain) replicated to higher titers in MCF-7 and Panc-1 cancer cell lines compared to immortalized serum starved MRC-5 fibroblasts.20 This study did not assess JX-594 specifically, and it did not evaluate TK-deleted vaccinia in truly normal and non-immortalized cells. Likewise, the infected cancer cells were tumor cell lines that had been serially passaged in vitro for over a decade and therefore are unlikely to bear resemblance to cancer cells in patients. No selectivity mechanisms were evaluated. We therefore elected to determine whether JX-594 was truly cancer-selective at a cellular level by studying diverse cancer and normal cell populations immediately after being obtained from patients. Once cancer-selectivity was demonstrated convincingly, we worked to elucidate cellular factors that were driving the cancer-selective replication and cytolysis of JX-594. We hypothesized that cellular TK levels would correlate with JX-594 activity since JX-594 has an inactivated TK gene; TK levels in cancer cells are reportedly constitutively higher than in normal cells.29 We also hypothesized that JX-594 cancer-selectivity was driven by epiderma growth factor (EGF) receptor pathway activation; this pathway is known to be required for efficient vaccinia replication and carcinogenesis (activated in over 90% of all human solid tumors). Finally, we explored whether the disruption of the type-I IFN clearance mechanism in cancer cells was exploited by JX-594.
Results
JX-594 efficiently kills tumor cells, including tumor initiating cells of various origins
The ED50 of JX-594 was assessed on a panel of cancer cell lines of different tissue origins (NCI60 cell line panel). Low concentrations of JX-594 (<1 pfu/cell) were effective at reducing cell viability of cell lines of various tissue origins, including colon (both Ras mutant and wild-type), prostate, breast, ovarian, lung, kidney, skin, and brain cancer cells; leukemia cells were relatively resistant to JX-594 killing compared with other tumor types (Figure 1a).
Figure 1.

JX-594 replicates and induces cytolysis in a broad spectrum of cancer cell types, including cell lines, tumor initiating cells, and primary human tumor tissue. (a) JX-594 ED50 (pfu/cell) on human cell lines in the NCI cell line panel upon JX-594 infection at multiple multiplicities of infection (MOI) 72 hours postinfection (n = 3). Ras mutational status for colorectal cancer cell lines is indicated. Error bars = SD. (b) Percent viability (compared to untreated control) over time of three tumor initiating cell lines (lung or colon) following infection with JX-594 or UV-inactivated JX-594 control at an MOI of 3 (n =3). Error bars = SD. (c) Human tumor biopsy material was sliced into 5 × 5 × 5 mm segments and infected with 5 × 106 pfu JX-594 suspended in 100 µl α-MEM serum-free medium ex vivo. Samples were homogenized and infectious virus quantitated 48 hours postinfection. MEM, minimum essential medium; NCI, National Cancer Institute; NSCLC, non-small-cell lung cancer; UV, ultraviolet.
Tumor initiating cells (TICs) of two different origins (lung and colon; representing large solid tumor populations clinically) were incubated with JX-594 or ultraviolet-inactivated JX-594 control. JX-594 showed a strong cytotoxic effect on all three TIC cell lines, with less than 10% residual cell viability after 5 days of infection (Figure 1b). Ultraviolet-inactivated JX-594 had no effect on cell killing.
JX-594 selectively replicates in primary human tumor biopsy tissue
In order to best model JX-594 infection of tumors in patients, surgically resected human primary tumor tissue material, and when possible, companion normal tissue from the same patient, was assayed for ex vivo sensitivity to JX-594 infection by monitoring expression of JX-594-driven β-galactosidase (β-gal) or green fluorescent protein (GFP), and for viral replication in a subset of samples. A total of 29 patients had tumor tissue samples obtained from surgical resection specimens acutely. In addition, 8 of these patients also had adjacent normal tissue obtained and assayed from the same surgical specimen. Cancer types included colorectal (n = 4), ovarian (n = 6), endometrial (n = 7), glioblastoma (n = 5), and non-small cell lung (n = 4). Nine tumor samples had viral replication assayed (viral burst assay) 48 hours postinfection (Figure 1c). In total, 29 tumor samples, and 8 normal tissue samples were similarly assayed, and 17 out of 29 tumors (57%) showed JX-594-driven β-gal activity or GFP fluorescence following ex vivo infection (relative to uninfected tumor tissue) (Table 1). Ovarian tumor explants (n = 6) exhibited particular sensitivity to JX-594 ex vivo (83%). The eight normal tissues tested displayed either no apparent susceptibility to JX-594 (n = 7), or less sensitivity to JX-594 infection (n = 1) than companion tumor tissue (Note: invasion of tumor cells into normal tissue cannot be ruled out in the single normal tissue sample which exhibited GFP positivity) (Table 1). A significant difference between infectivity of tumor and normal tissue was observed (P = 0.04; Fisher's exact test). As a more quantitative indication of the differential sensitivity of primary human tumor and normal tissues to JX-594 infection, and to confirm that reporter transgene expression correlated with replication as predicted, a subset of paired tumor and normal samples from four individuals were assayed for ex vivo viral amplification. These samples were selected on the basis that (i) the tumor specimen provided was scored as susceptible to JX-594–driven gene expression, and (ii) the responsive tumor specimens were accompanied by a normal organ tissue biopsy sample. Virus amplification in the tumor tissue ~100- to 3,000-fold higher than in the companion normal tissue (Figure 2a).
Table 1. JX-594 preferentially infects and expressed its transgene in primary human tumor biopsy tissue ex vivo.
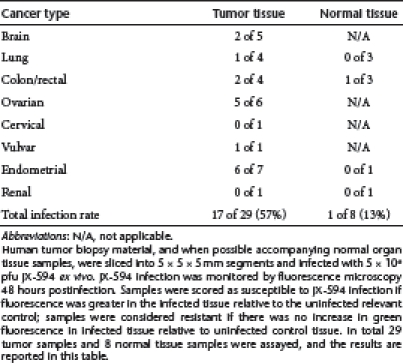
Figure 2.

JX-594 preferentially infects tumor tissue, while sparing normal tissue. (a) Human tumor biopsy material, and when possible accompanying adjacent normal organ tissue samples, were sliced into 5 × 5 × 5 mm segments and infected with 5 × 106 pfu JX-594 or JX-594-GFP+/β-gal− ex vivo. When possible, paired normal and tumor tissue samples wherein the tumor displayed virus-driven fluorescent reporter expression, were homogenized and titered to quantify infectious virus recovered from the specimen 48 hours postinfection. The ratio of JX-594-GFP+/β-gal− in tumor tissue versus the corresponding normal tissue is plotted. (b) Donor PBMC from a healthy volunteer were cultured for 24 hours in the presence or absence of PHA (5 µg/ml) before infection with JX-594. PBMC or U2OS cell cultures were then inoculated with JX-594 at an MOI = 1 pfu/cell and after 24, 48, or 72 hours. β-gal activity was detected by flow cytometry using ImaGene Green LacZ detection kit (n =1). (c) JX-594 output from duplicate culture/infection conditions as measured by plaque assay (n = 1). β-gal, β-galactosidase; GFP, green fluorescent protein; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cells; PHA, phytohemagglutinin.
Peripheral blood mononuclear cells are resistant to JX-594 replication
In its clinical application as an oncolytic viral therapy for cancer, JX-594 can be found in circulation of patients shortly after intravenous or intratumoral administration, and at later times post-treatment as in vivo replication occurs.22 As blood cells will come into contact with JX-594, either through direct intravenous delivery or during secondary spread following in vivo replication, it is important to determine whether blood cells become infected by JX-594. Peripheral blood mononuclear cells (PBMC) were isolated from naive whole blood, and inoculated with JX-594, and virus-driven β-gal activity was quantified by flow cytometry. As a comparison, U2OS human osteosarcoma cells were similarly infected and assayed. PBMC displayed between 0.02 and 0.73% β-gal expressing cells over a 24–72 hours period following infection with JX-594 at multiplicity of infection (MOI) of 1 (Figure 2b). To address any concern that activation of PBMC may enhance their susceptibility to viral infection, donor PBMC (collected as described) were activated with phytohemagglutinin (PHA) before infection with JX-594. JX-594-driven β-gal expression in PBMC stimulated with PHA (5 µg/ml) occurred in only 0.05–9.63% over the same timeframe (Figure 2b). However, background β-gal expression in uninfected PHA-stimulated PBMC ranged from 0.12 to 3.23% in this same timeframe. In contrast, U2OS human osteosarcoma cells infected with JX-594 at MOI of 1 showed 28.11% of cells expressing virus-driven β-gal activity at 24 hours postinfection, increasing to 43.35% at 48 hours, and peaking at 98.15% by 72 h postinfection (Figure 2b). PBMC were therefore highly refractory to JX-594 viral gene expression, relative to the human cancer cell line U2OS, at a high MOI over 72 hours of infection.
In duplicate samples, PBMC and U2OS infected with JX-594 underwent three freeze-thaw cycles before titering on U2OS cells to assess virus production (Figure 2c). JX-594 did not replicate significantly in JX-594-infected PBMC, even after PHA stimulation. In contrast, U2OS cells produced ~100-fold higher pfu when compared to PBMC at 24 hours postinfection, and ~1,000-fold more by 48–72 hours.
Selective replication of JX-594-luc+/β-gal− in tumors in vivo
As an in vivo demonstration of the tumor-selectivity of JX-594 replication, CD1 nude mice bearing subcutaneous SW620 human colon carcinoma xenograft tumors were treated with JX-594-firefly luciferase (luc)+/β-gal− via the intravenous route followed by bioluminescence imaging by in vivo imaging system (IVIS; Xenogen, Hopkinton, MA). Bioluminescence imaging has previously been shown to correlate with vaccinia titer.30 As shown in Figure 3a, luminescence is highly tumor-selective (days 3 and 6). As a more quantitative measure of longer term replication of intravenously delivered virus in vivo, mice bearing subcutaneous SW620 tumors were euthanized and multiple tissues collected 7 days postinfusion for viral titering (pfu/g tissue). Infectious JX-594-luc+/β-gal− pfu were ~5 or more logs higher in tumor tissues compared with any other organ tissue tested, including brain, heart, kidneys, liver, lungs, ovaries, and spleen (Figure 3b). Concomitant treatment of mice with universal IFN-α resulted in reduced JX-594-luc+/ β-gal− output in normal tissues while not affecting JX-594-luc+/β-gal− titers recovered from tumors (Figure 3b).
Figure 3.

In vivo biodistribution of JX-594-luc+/β-gal− infection and replication following intravenous administration. (a) Athymic nude mice with established subcutaneous SW620 human colon carcinoma tumors were treated intravenously via the tail vein with 107 pfu JX-594-luc+/β-gal−. Three and 6 days postinfection, luminescence was assessed emanating from the tumor and normal tissue with IVIS system. (b) Athymic nude mice bearing subcutaneous SW620 tumors treated with intravenous JX-594-luc+/β-gal− at a dose of 1 × 107 pfu in a, as well as mice treated with JX-594-luc+/β-gal− and universal IFN-α were sacrificed 7 days following JX-594-luc+/β-gal− treatment. Organs were collected and the presence of virus was quantified by plaque assay in U2OS cells (n = 6 per group). Error bars = SD. (c) Female transgenic FVB/N mice with autochthonous ovarian tumors were treated with 1 × 108 pfu JX-594-luc+/β-gal− intraperitoneally, and 5 days later luminescence was assessed with a Xenogen 200 Series IVIS system. β-gal, β-galactosidase; IFN, interferon; IVIS, in vivo imaging system; luc, firefly luciferase; MOI, multiplicity of infection; shRNA, short hairpin RNA; TK, thymidine kinase; TKKD, thymidine kinase knockdown.
JX-594-luc+/β-gal− selectivity was also assessed in a transgenic mouse model of ovarian cancer; in this model the transforming region of SV40 is driven by the Mullerian inhibitory substance type II receptor gene promoter resulting in bilateral ovarian carcinomas.31 Mice were treated with 108 pfu JX-594-luc+/β-gal− via the intraperitoneal route and assessed by IVIS for bioluminescence imaging on day 5 (Figure 3c); luciferase expression was only observed at the site of the cancerous tissue in the ovaries.
JX-594 replication correlates with cellular TK levels
JX-594 is a Wyeth strain vaccinia virus with a disruption and inactivation of the viral TK gene; thus infection should be dependent on the presence of sufficient cellular TK. Cancer cells are known to have high constitutive TK concentrations compared to normal cells. To assess the dependence of JX-594 replication on cellular TK levels, HeLa cells with high levels of endogenous TK were infected with a lentivirus expressing a TK-directed short hairpin RNA (shRNA). Reduction in endogenous TK levels was confirmed (Figure 4a). Both JX-594 and wild-type Wyeth control replication was subsequently assayed in the HeLa cell line as well as the TK-knockdown clone. JX-594 burst size was attenuated in cells with reduced TK levels when compared to control cells; no effect on wild-type Wyeth virus was observed in the TK-knockdown clone (Figure 4b). Decreased JX-594 output upon TK knockdown was confirmed using two additional TK-knockdown clones (Supplementary Figure S1).
Figure 4.

JX-594 replication is attenuated in cancer cells depleted of cellular thymidine kinase (TK). (a) Western blotting confirming knockdown of endogenous TK in HeLa cells performed by retroviral transduction using shRNA against TK, or a control shRNA. (b) JX-594 and wild-type Wyeth control replication in HeLa cells or TK-knockdown clones, following infection at MOI 0.1 for 48 hours before performing plaque assays on infected material (n =3). Error bars = SD. MOI, multiplicity of infection; shRNA, short hairpin RNA.
JX-594-GFP+/β-gal− replication is dependent on EGFR/Ras/MAPK pathway signaling
Since wild-type vaccinia virus has been shown to be dependent on EGFR signaling,32 we sought to investigate whether JX-594 replication is dependent on EGFR/Ras/MAPK (mitogen-activated protein kinase) signaling. An inhibitor specific for extracellular signal-regulated kinase (U0126) was used to interrogate JX-594 dependence on this pathway at concentrations that did not affect cell viability (data not shown). Incubation of two human colorectal cancer cell lines (SW620 and HCT116) with U0126 inhibited JX-594-GFP+/β-gal− replication (Figure 5).
Figure 5.

JX-594-GFP+/β-gal− replication is attenuated in the presence of an Erk inhibitor. Two human colon carcinoma cell lines (SW620 and HCT116) were infected with JX-594-GFP+/β-gal− at an MOI of 0.1 for 50 hours in the presence or absence of the Erk inhibitor U0126. JX-594-GFP+/β-gal− output from triplicate culture/infection conditions was measured by plaque assay (n = 3). Error bars = SD. β-gal, β-galactosidase; DMSO, dimethyl sulfoxide; Erk, extracellular signal-regulated kinase; GFP, green fluorescent protein.
Selective inhibition of in vitro JX-594-GFP+/β-gal− replication in normal human fibroblasts but not in cancer cells
Previous reports have demonstrated that TK-deleted vaccinia virus preferentially infects cancer cells as compared to serum-starved MRC-5 cells (secondary human lung fibroblasts).20 In addition, it is known that cancers often have a defective IFN response.6 To demonstrate cancer-selective replication of JX-594 with greater stringency, infection experiments on GM38 cells (human fibroblasts) and SW620 cells were conducted in the presence and absence of exogenously supplied IFN-α, a downstream mediator of IFN-β.7 Cells were infected with JX-594-GFP+/β-gal− in vitro, in the presence and absence of exogenous IFN-α, and the progression of JX-594-GFP+/β-gal− infection was monitored over 48 hours by fluorescence microscopy. JX-594-GFP+/β-gal− transgene expression in the presence of IFN-α was markedly higher in SW620 cells versus normal human fibroblasts (Figure 6a). These results are in accordance with decreased JX-594-luc+/β-gal− output detected in normal tissues following treatment of tumor-bearing mice with JX-594-luc+/β-gal− and universal IFN-α (Figure 3b).
Figure 6.

JX-594-GFP+/β-gal− replication is attenuated in the presence of interferon on normal cells, but not on cancer cells. (a) SW620 human colon carcinoma cells (interferon-resistant) and GM38 normal human fibroblasts (interferon-sensitive) were inoculated with JX-594-GFP+/β-gal− (MOI 0.001–1) in the presence or absence of 200 IU/ml exogenous IFN-α and transgene expression (GFP) was monitored 48 hours postinfection using a Zeiss Axiovision microscope and Axiovision acquisition and image storage software. (b) GM38 normal human fibroblasts and UACC257 human melanoma cells (interferon-resistant) were seeded (3 × 104 cells per well) in the presence or absence of varying concentrations of IFN-α and infected with JX-594-GFP+/β-gal− at various MOIs, and 72 hours later cell viability was assessed by CellTiter Blue assay (n = 4). β-gal, β-galactosidase; GFP, green fluorescent protein; IFN, interferon; MOI, multiplicity of infection.
UACC257 melanoma cells were infected with JX-594-GFP+/β-gal− in the presence of increasing concentrations of IFN-α and evaluated for viability 72 hours later. In the absence of IFN-α UACC257 cells were efficiently killed with an EC50 of less than 5 × 102 pfu when compared to the EC50 on GM38 normal fibroblasts (2.7 × 103 pfu). However, in the presence of low concentrations of IFN-α (250 IU), the EC50 of JX-594-GFP+/β-gal− on GM38 cells shifted to 1.7 × 104 pfu while UACC257 cells were not protected with IFN-α treatment (even at a high dose of 1,000 IU) (Figure 6b).
Discussion
We describe here multiple distinct cancer-selectivity mechanisms, both inherent and engineered, for the oncolytic virus JX-594. To our knowledge, this is the first such description in the literature. To date, oncolytic viruses have been proposed to be cancer-selective on either an inherent or an engineered basis, with a single mechanism being proposed for the cancer-selectivity of each virus. In this study we demonstrate three mechanisms contributing to the tumor-selectivity of JX-594, a TK gene-inactivated Wyeth vaccinia virus expressing GM-CSF and lac-Z transgenes. Two mechanisms that drive JX-594 replication are inherent to cancer cell biology: (i) activation of the EGFR/Ras/MAPK signal transduction pathway and (ii) inactivation of the type-I IFN response pathway. We predicted that these pathways would be critical since vaccinia virus is known to manipulate these cellular pathways for (i) its replication and spread and (ii) to avoid IFN-mediated clearance; this was confirmed experimentally as published here. In fact, vaccinia secretes viral proteins from the infected cell that activate the EGFR/Ras/MAPK pathway (vaccinia growth factor)33 or partially inactivate type-I IFNs (e.g., B18R),34 and inhibitors of EGFR were demonstrated to block vaccinia replication.32,35 A third mechanism that drives selective replication relates to the engineered inactivation of the TK gene in JX-594 and the resulting reliance on high cellular TK concentrations for replication; TK activity is constitutively high in many cancer cells.29 Given prior data that a TK-inactivated vaccinia virus is attenuated in normal cells,36 we theorized that cellular TK levels would correlate with JX-594 replication. Since TK is an E2F-responsive gene, TK levels in cancer cells are generally higher and constitutive compared to normal cells (including cycling normal cells). Therefore, three distinct mechanisms for the cancer-selectivity of JX-594 are reported herein, and both inherent and engineered mechanisms contribute. Nevertheless, it is highly likely that additional cellular mediators are involved and will be discovered.
JX-594 demonstrated broad spectrum efficacy against diverse solid tumor types, both in cell lines and in ex vivo assays with primary human tissue explants. Of note, lung and colorectal TICs were also sensitive to JX-594-mediated destruction. This is of significance as TICs are known to be generally resistant to currently approved chemotherapies.37 Since JX-594 is able to target cancer cells with common genetic signatures, and because vaccinia uptake into cells is via a mechanism that is ubiquitous (apoptotic bleb mimicry),18 broad spectrum activity was predicted. The EGFR/Ras pathway, for example, is activated in the vast majority of human carcinomas (>90%), either by activation of the receptor or downstream signaling molecules (e.g., Ras, Raf).38,39,40,41 It remains to be determined which downstream mediators in the EGFR/Ras pathway drive JX-594 replication, spread, and cytolysis most potently. Since JX-594 is activated for replication and spread by EGFR pathway signaling, regardless of which component in the pathway is activated, JX-594 can potentially be more broadly effective than EGFR inhibitors (e.g., cetuximab, erlotinib). Tumors can develop resistance to these inhibitors by activating downstream signaling molecules (e.g., Ras) or parallel, redundant pathways.42,43 JX-594 was active against EGFR inhibitor-resistant tumors both in vitro and in patients. The exact prevalence of IFN resistance mechanisms and elevated TK levels, two other predictors of JX-594 sensitivity, for specific cancer patient populations remains to be established. The cellular uptake of vaccinia by receptor- independent mechanisms may result in broader spectrum activity than is reported with other oncolytic viruses. For example, Ad5 uptake is coxsackievirus-adenovirus receptor (CAR) -dependent, thus resulting in poor uptake and efficacy against tumors with low CAR expression.44
Significant questions still remain with regard to the cancer-targeting selectivity of JX-594. In clinical trials of intratumoral and/or intravenous administration of JX-594, cancer-selectivity and safety have been established. Transient flu-like symptoms have been the predominant toxicity, while organ tissue toxicities have not been reproducibly demonstrated to date.19,22,23 Nevertheless, it is theoretically possible that certain normal cell populations within the body could be susceptible to toxicity. Highly proliferative cells in the bone marrow do not appear to be susceptible since intravenous therapy in patients does not result in reproducible durable cytopenias23; interestingly, leukemia cells and proliferating normal PBMC were also highly resistant. Although gut crypt epithelial cells are highly proliferative, gut toxicity was not described in either GLP toxicology studies nor in >100 patients treated to date.20,22,23 It is possible that these normal proliferating cells remain resistant due to the fact that the IFN resistance mechanism is intact in these tissues, in contrast to tumors. The toxicity of JX-594 and related viruses at sites of wound healing remains to be determined. To date, the agent has been used without toxicity in patients receiving tumor biopsies or percutaneous needle insertions.22,23 Finally, the roles of vaccinia proteins that interact with the EGFR (e.g., VGF), nucleotide synthesis (e.g. ribonucleotide reductase) or type-I IFNs (e.g. B18R) remain to be elucidated in human tumors. Safety may be enhanced by the multiple cancer-selectivity mechanisms involved, rather than reliance on a single mechanism for cancer targeting.
In this publication we describe the cancer-selectivity of an oncolytic virus in a number of highly relevant in vitro and in vivo models. These methods have major implications for the field since they are likely more relevant to human cancer patient safety with oncolytic viruses than previously published methods. To date, most reported selectivity data has been derived from “normal” cell lines such as fibroblasts that do not reflect normal tissues in patients. First, these cells are not epithelial, and second they are immortalized and therefore not normal. In addition, the contribution of endogenous IFN on virus inhibition is often ignored in these assessments. Our goal was to assess potential selectivity and safety in a variety of normal tissues, including those most likely to be exposed after intravenous administration: vascular endothelial cells and blood cells. Instead of using immortalized cell lines, we tested primary cells directly obtained from humans without extensive serial passaging. This assay could form the basis for a predictive test that which is more likely to be more clinically useful than assays that assess a single genetic feature of a target tumor. Although these methods represent a major advance for the field, our methods do still have some limitations. First, it is extremely difficult to assess any immune-mediated toxicities that could occur in vivo. Second, the assessment of JX-594 selectivity in mice suffers from the fact that viral replication is markedly higher in human versus murine tissues; therefore, the tumor-selectivity shown after intravenous infusions may be due not only to differences in cancer versus normal cells, but also due to species differences. Orthotopic tumor implantation may also be more reflective of the clinical situation. Rabbit tumor models may be superior since normal rabbit tissues appear to be more susceptible to wild-type Wyeth vaccinia (parental strain for JX-594) than mouse tissues.20 Primate models may be more relevant for safety assessments, but tumor-targeting cannot be simultaneously assessed.
These findings have significant implications for future development in the field. Given the multiple levels of safety and selectivity we describe with JX-594, it is likely that many of these same mechanisms are important for other oncolytic viruses. These findings may help to explain controversies in the literature surrounding the predictive utility of single genetic markers for infection and replication. Given the complexity of cancer genetics and virus replication, it is highly unlikely that a single genetic feature, or even activation of an entire signal transduction pathway, will be completely predictive on its own for cell sensitivity to an oncolytic virus. The excellent safety profile described for JX-594 and other oncolytic viruses entering the clinic to date5 may be a result of this multi-mechanistic selectivity. Due to selective amplification in tumors (e.g., several logs increase over a 1 week period) with simultaneous clearance from normal tissues, targeted oncolytic viruses have the potential for a much larger therapeutic index than other cancer therapeutics such as chemotherapy. These findings also have implications for combination therapy with JX-594; inhibitors of the EGFR signaling pathway, for example, should be avoided for simultaneous treatment and should be used sequentially. Finally, the high degree of cancer-selectivity demonstrated with JX-594 suggests that safe and systemically delivered multi-mechanistic poxvirus products are possible. By engineering these agents with multiple transgenes encoding for therapeutic proteins, high-level cancer specific expression of multiple therapeutic proteins could be achieved in solid tumors. Multiple mechanisms-of-action can therefore be designed and engineered into these products. This may represent a breakthrough for the in vivo systemic delivery and efficacy of cancer biologics in general.
Materials and Methods
Viruses. Vaccinia virus JX-594 was previously described.19,20 Serial derivatives of JX-594 were used in the laboratory in order to easily assess JX-594 replication. These include JX-594-GFP+/β-gal and JX-594-luc+/β-gal which express GFP and luc, respectively, instead of β-gal under the synthetic early/late promoter pSE/L in the vaccinia TK gene locus. The vaccinia virus Wyeth strain was obtained from American Type Culture Collection (distributed by Cedarlane Laboratories, Burlington, Ontario, Canada). JX-594 was propagated in HeLa cells; all other viruses were propagated in U2OS cells.
Cell lines. U2OS (human osteosarcoma), SW620, HCT116 (human colorectal cancer), and UACC257 cells (human melanoma) were obtained from the NCI/NIH (Bethesda, MD) and maintained in Dulbecco's modified Eagle medium (DMEM) + 10% fetal bovine serum (FBS) (HyClone, Hudson, NH). GM38 (human skin fibroblasts) were obtained from Dr Bruce McKay (Ottawa Hospital Research Institute, Ottawa, Ontario, Canada) and were propagated in DMEM + 10% FBS. Susceptibility of the NCI tumor cell line panel to JX-594 was screened by the In Vitro Cell Line Screening Program (DTP; SAIC/NCI-Frederick, Frederick, MD). Sixty different human tumor cell lines representing leukemia, melanoma, and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney were incubated with serially diluted JX-594 virus inocula, in the range of 2–2 × 105 pfu/200 µl/well. Cell death was determined after 72 hours by 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay as described.45 ED50 values are reported and represent the initial virus MOI to achieve 50% cell killing compared to untreated control cells at 72 hours postinoculation.
Infection of TICs with JX-594. TIC cultures (TriStar Technology Group, Rockville, MD) were established and propagated under spherogenic conditions as described.46 Cells were cultured for 20–30 days in serum-free medium containing EGF (tebu-bio, Le Perray en Yvelines, France) and basic fibroblast growth factor (tebu-bio) before plating for the cytotoxicity assay. The cytotoxicity of the JX-594 virus was measured by the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) in a 96-well microplate format. TIC cultures, two from each of colorectal cancer47 and non-small-cell lung cancer,46 were supplemented with a single dose of oncolytic virus or control virus (MOI = 3) and kept in culture for 24, 48, 72, 96, and 120 hours, respectively. A single cell density, determined to be in the linear range of the assay at all timepoints, was chosen and three replicates for each experimental point were included. At the end of the incubation, cell viability was determined by adding the CellTiter-Glo Reagent (Promega), and measuring the luminescent signal. JX-594 cytotoxicity was evaluated in comparison to its inactivated control. Suitable cell aliquots were withdrawn from master TIC cultures, collected by low-speed centrifugation (900 rpm for 5 minutes at 20 °C), enzymatically dissociated in 250 µl of Accumax Reagent (Sigma-Aldrich, Oakville, Ontario, Canada) for 5–10 minutes at 37 °C, and counted. By low-speed centrifugation (900 rpm for 5 minutes at 20 °C), 2.0 × 105 cells of each master cell culture were collected and enzymatically dissociated as above, then diluted in 5 ml of TIC basal medium for washing. Cell pellets were collected by low-speed centrifugation (900 rpm for 5 minutes at 20 °C), and resuspended again in 5 ml of TIC basal medium plus growth factors at a final concentration of 40,000 cells/ml. Fifty microliters of cell suspension were added to each well of five 96-well black microplates with clear, flat bottom. A single cell density of 2,000 cells was chosen to be within the linear range of the assay at all timepoints for all TIC lines. Cells were seeded in a sufficient number of wells for performing two single-dose infections, with JX-594 virus and ultraviolet-inactivated JX-594 virus, at five time points (24, 48, 72, 96, and 120 hours), including three replicates for each experimental point. In addition, 1:2 serial cell dilutions from 2,000 to 125 cells/well were also plated in triplicate for each timepoint to obtain internal reference cell titration curves. Control wells containing medium without cells were used to obtain background assay values. After plating, cells were incubated for 2 hours at 37 °C, before the addition of 50 µl/well of a viral suspension containing 6,000 pfu (to infect cells at an MOI of 3). Initially, 50 µl of viral stocks (108 pfu/ml) were diluted 1:10 in 500 µl to obtain a 107 pfu/ml concentration; a second dilution 1:10 was prepared to obtain 500 µl of 106 pfu/ml concentration. The whole volume (500 µl) was further diluted by addition of 3.65 ml of culture medium to reach the final concentration of 1.2 × 105 pfu/ml. All dilutions were made in TIC basal medium plus GFs. Then cell cultures were incubated at 37 °C in the presence of the viral agents for 24, 48, 72, 96, or 120 hours. At the end of each incubation time the number of viable cells was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega), essentially by following the manufacturer's recommendations. Luminescence was recorded using a Perkin Elmer Wallac 1450 MicroBeta TriLux detector (PerkinElmer, Waltham, MA).
Human tumor biopsy infection with JX-594 ex vivo. Surgical biopsy material was obtained from patients at the Ottawa Hospital General Campus undergoing surgery to biopsy or remove solid organ malignancies under Protocol 2004266-01H. A total of 29 tumor samples, of which 8 were accompanied by normal tissue from the affected organ, were assayed for their sensitivity to JX-594 or JX-594-GFP+/β-gal−. Informed consent was obtained from all patients before surgery. Samples were received in cell culture medium at room temperature from the Ottawa Hospital General Campus and were processed within 2–16 hours. Small slices (~5 × 5 × 5 mm) of specimens were prepared in a sterile biosafety cabinet using a sterile surgical blade and forceps. A single tissue portion was placed into one well of a 24-well sterile tissue culture plate for infection. Samples were inoculated with 5 × 106 pfu JX-594 or JX-594-GFP+/β-gal− in 100 µl of α-minimum essential medium (α-MEM) serum-free medium. Virus was allowed to adsorb for 2 hours at 37 °C, at which time 1.9 ml of α-MEM supplemented with 10% FBS was added. Infected tissue specimens were incubated in a humidified incubator at 37 °C for 24 or 48 hours before imaging JX-594-driven β-gal activity or GFP. Briefly, endogenous β-gal activity in explant tissues was blocked by incubation with 300 µmol/l chloroquine for 2 hours at 37 °C. Following this blocking step, chloroquine-containing medium was aspirated and tissues were incubated with 500 µl of a 33 µmol/l working stock of C12FDG substrate for 30 minutes at 37 °C before adding phenyl-ethyl β-D-thiogalactopyranoside (PETG) to a final concentration of 1 mmol/l to stop the reaction. Explants were photographed under a Leica MZ FLIII fluorescent dissecting microscope (at ×2–4 magnification; Leica, Wetzlar, Germany) and images were stored using Adobe Photoshop version 7.0.
The determination of positive or negative responses in terms of tissue sensitivity to infection with JX-594 or JX-594-GFP+/β-gal− is qualitative. Tissue samples were considered to be susceptible to JX-594 infection if green fluorescence following C12FDG staining was greater in the infected tissue compared to the background fluorescence in the uninfected relevant control. Tumor tissue samples were scored as resistant if there was no increase in green fluorescence in infected tissue relative to uninfected control tissue. Normal tissue samples were scored as resistant if there was no increase in green fluorescence in infected tissue relative to uninfected tissue or if the normal tissue was less susceptible than companion tumor tissue.
When possible, paired normal and tumor tissue samples were homogenized and titered by plaque assay to quantify infectious virus recovered from the specimen 48 hours postinfection.
Plaque assay for JX-594 quantification in cells infected in vitro. Cells infected with JX-594, JX-594-GFP+/β-gal− or JX-594-luc+/β-gal− in vitro underwent three freeze-thaw cycles before plating. Tissue samples infected with virus were thawed once, and homogenized before plating. Samples were serially diluted in serum-free α-MEM tissue culture medium and 1 ml of each dilution was plated onto a monolayer of U2OS cells, established by plating 106 cells/well of a 6-well plate the day before initiating the plaque assay. After 2 hours of incubation at 37 °C, a semi-solid overlay of carboxymethylcellulose in α-MEM supplemented with 10% FBS was applied. Plates were incubated for an additional 48 hours, at which time plaques were fixed and stained with 0.1% Crystal violet in 80% methanol.
Ex vivo PBMC infection. Blood was drawn from healthy volunteers into heparinized vacutainer tubes and diluted 1:3 in phosphate-buffered saline (PBS). Diluted blood was overlayed onto 5 ml Ficoll-Paque Plus (Amersham-Pharmacia, Uppsala, Sweden) and the buffy coat layer containing PBMC was isolated following centrifugation at 400g for 25 minutes at room temperature. PBMC were washed twice in PBS and resuspended to 106 cells in 1 ml of DMEM + 10% FBS. Cells were cultured for 24 hours in the presence or absence of PHA (5 µg/ml) before infection with JX-594. Cultures of resting or stimulated PBMC were inoculated with JX-594 at MOI of 1 pfu/cell for 2 hours at 37 °C. Cultures were supplemented with DMEM containing 10% FBS and cultured for an additional 24–72 hours before flow cytometric analysis to detect β-gal activity, using the ImaGene Green lacZ Gene Expression Kit (Molecular Probes, Eugene, OR) according to manufacturers' instructions. Briefly, cells were incubated in 300 µmol/l chloroquine reagent for 2 hours at 37 °C and washed in PBS. Cells were resuspended in 1 ml of 33 µmol/l C12FDG substrate reagent and incubated at 37 °C for 30 minutes before stopping the reaction with 1 mmol/l PETG. Infection of cells and cell viability were quantitated by flow cytometric analysis using a Beckman-Coulter Epics XL flow cytometer (Beckman Coulter, Mississauga, Ontario, Canada) and Expo32 acquisition software. Detectors for C12FDG and propidium iodide were FL1 and FL2, respectively. U2OS cells were similarly infected in parallel, albeit without additional PHA stimulation. Duplicates of all culture/infection conditions were prepared and were frozen at −80 °C until thawing to assay JX-594 viral output by plaque assay.
Murine tumor models. Athymic nude mice (Charles River, Wilmington, MA) were implanted with subcutaneous human SW620 colon cancer xenografts by injecting 5 × 106 SW620 cells in 200 µl PBS, which were allowed to grow for 14 days. At this time, all mice were treated with 107 pfu of JX-594-luc+/β-gal−, by the intravenous route (tail vein). Mice receiving universal IFN-α (PBL Interferon Source, Piscataway, NJ) were given an intraperitoneal injection of 5 × 104 IU at the time of JX-594 infusion.
Thirteen-week-old female FVB/N mice expressing the transforming region of SV40 driven by the Mullerian inhibitory substance type II receptor gene promoter31 were treated with a single intraperitoneal dose of JX-594-luc+/β-gal− (1 × 108 pfu).
All experiments were conducted with the approval of the University of Ottawa Animal Care and Veterinary Service.
Bioluminescent imaging of JX-594 replication. Mice were injected with d-luciferin (Molecular Imaging Products Company, Ann Arbor, MI; 200 µl intraperitoneally at 10 mg/ml in PBS) to image JX-594-driven luc expression. Mice were anesthetized under 3% isofluorane and imaged with the 200 Series in vivo Imaging System (Xenogen). Data acquisition and analysis was performed using Living Image v2.5 software (Xenogen). In vivo imaging was performed at the indicated timepoints postinfection.
In vivo biodistribution. Nude mice bearing SW620 human colon cancer xenograft subcutaneous tumors were injected with 107 pfu JX-594 IV. Seven days post-injection, mice were euthanized and tumors, liver, lungs, brain, spleen, heart, ovaries, and kidneys were collected and frozen at −80 °C. Subsequently, tissues were thawed, homogenized, and infectious virus titer was quantified by plaque assay as previously described.
TK knockdown. The Tk1 knockdown in HeLa cells was achieved through delivery of shRNA via lentiviral transduction. Short hairpins against human Tk1 were designed and cloned into a bicistronic lentivector pLVTHM which contains a GFP marker downstream of the shRNA sequence; transduced cells stably expressing shRNA targeting Tk1 were selected based on expression of GFP. The stability of these Tk1 knockdown cells was monitored using a Leica MZ FLIII fluorescent microscope and showed no signs of loss of the lentiviral vector. Parental or TK-knockdown HeLa cell clones were infected with JX-594 or Wyeth wild-type virus at MOI of 0.1 for a total of 48 hours before assessing virus replication.
Western blotting. Three different hairpins were found to successfully knockdown Tk1 levels in HeLa cells using western blotting. Mouse anti-human Tk1 monoclonal antibody (clone 3B3.E11) from Novus Biologicals (Littleton, CO) was used at a 1:500 dilution, and the goat anti-mouse IgG-HRP conjugated secondary antibody (Bio-Rad, Mississauga, Ontario, Canada) was used at a 1:1,000 dilution. The HRP-tagged secondary was detected by the enhanced chemiluminescence method using the SuperSignal West Pico Chemiluminescent substrate kit according to manufacturer's instructions (Pierce, Rockford, IL). The Tk1 level in the original HeLa (control) and the knockdown clones (A4, A5, D14) was quantified using the BioRad GS-800 Calibrated Densitometer (Bio-Rad, Hercules, CA) and QuantityOne software.
In vitro infection with extracellular signal-regulated kinase inhibitor. Human colorectal cancer cell lines SW620 (provided by the NCI) and HCT116 (NCI) (106 cells/well) were pretreated for 2 hours with various concentrations of U0126 (Sigma, Oakville, Ontario, Canada). Cells were then inoculated with recombinant JX-594-GFP+/β-gal− at MOI of 0.1 pfu/cell for 2 hours in serum-free medium. Cells were cultured in the presence of serum for an additional 48 hours, were harvested and virus replication was quantified by plaque assay and in duplicate cultures.
JX-594-GFP+/β-gal− imaging and cell viability assay in presence of IFN. SW620 human colon carcinoma cells (provided by the NCI), and GM38 normal human fibroblasts (provided by Bruce McKay; OHRI, Ottawa, Ontario, Canada) were plated at 105 cells/well and cultured overnight in the presence or absence of 200 IU/ml human IFN-α (Schering Canada, Pointe-Claire, Quebec, Canada). Cells were then inoculated with JX-594-GFP+/β-gal− at MOIs of 1.0, 0.1, 0.01, and 0.001 pfu/cell for 2 hours in serum-free medium. Cells were cultured in the presence of serum for an additional 48 hours, over which time cells were monitored for susceptibility to viral gene expression (GFP) by fluorescence microscopy.
GM38 normal human fibroblasts and UACC257 human melanoma cells were seeded (3 × 104 cells/well) in the presence or absence of varying concentrations of IFN-α (Schering Canada, Pointe-Claire, Quebec, Canada) and infected with JX-594 at a various MOIs. Seventy-two hours later, cell viability was assessed by CellTiter-Blue assay (Promega) according to manufacturer's instructions. Fluorescence was read on a Fluoroskan (Thermo Scientific, Hudson, NH) at an excitation of 530 nm and emission of 590 nm.
Statistical analyses. All statistical analyses were performed using the GraphPad Prism 5.0 software (GraphPad Software, La Jolla, CA). The unpaired t-test was used to assess differences in virus output in HeLa versus TK-knockdown cells. The Fisher's exact test was used to assess differences in infectivity of tumor and normal tissue explants. GraphPad Prism software was also used to calculate the IC50 of JX-594 in normal and tumor cells in the presence and absence of IFN.
SUPPLEMENTARY MATERIAL Figure S1. Reduced TK levels affect JX-594 output in multiple TK-knockdown clones.
Acknowledgments
This study was supported by a grant of the Terry Fox Foundation (J.C.B.) and the Ottawa Regional Cancer Foundation (B.C.V.). C.J.B., T.H., and D.H.K. are employees of JENNEREX, Inc., and J.C.B., is a consultant and shareholder in Jennerex. Jennerex, Inc. holds the license for JX-594. The other authors declared no conflict of interest.
Supplementary Material
Reduced TK levels affect JX-594 output in multiple TK-knockdown clones.
REFERENCES
- Parato KA, Lichty BD., and, Bell JC. Diplomatic immunity: turning a foe into an ally. Curr Opin Mol Ther. 2009;11:13–21. [PubMed] [Google Scholar]
- Cattaneo R, Miest T, Shashkova EV., and, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JC, Lichty B., and, Stojdl D. Getting oncolytic virus therapies off the ground. Cancer Cell. 2003;4:7–11. doi: 10.1016/s1535-6108(03)00170-3. [DOI] [PubMed] [Google Scholar]
- Liu TC, Hwang TH, Bell JC., and, Kirn DH. Development of targeted oncolytic virotherapeutics through translational research. Expert Opin Biol Ther. 2008;8:1381–1391. doi: 10.1517/14712598.8.9.1381. [DOI] [PubMed] [Google Scholar]
- Liu TC, Galanis E., and, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N.et al. (2000Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus Nat Med 6821–825. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S.et al. (2003VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents Cancer Cell 4263–275. [DOI] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA., and, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Norman KL., and, Lee PW. Reovirus as a novel oncolytic agent. J Clin Invest. 2000;105:1035–1038. doi: 10.1172/JCI9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M.et al. (1996An adenovirus mutant that replicates selectively in p53-deficient human tumor cells Science 274373–376. [DOI] [PubMed] [Google Scholar]
- Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A.et al. (2000An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy Nat Med 61134–1139. [DOI] [PubMed] [Google Scholar]
- Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC., and, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA.et al. (2001Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice Blood 973746–3754. [DOI] [PubMed] [Google Scholar]
- Mineta T, Rabkin SD, Yazaki T, Hunter WD., and, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- Mineta T, Rabkin SD., and, Martuza RL. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 1994;54:3963–3966. [PubMed] [Google Scholar]
- Kirn DH., and, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Chung CS, Hsiao JC, Chang YS., and, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol. 1998;72:1577–1585. doi: 10.1128/jvi.72.2.1577-1585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laliberte JP., and, Moss B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc Natl Acad Sci USA. 2009;106:17517–17521. doi: 10.1073/pnas.0909376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA.et al. (1999Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma Cancer Gene Ther 6409–422. [DOI] [PubMed] [Google Scholar]
- Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE.et al. (2006Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF Mol Ther 14361–370. [DOI] [PubMed] [Google Scholar]
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K.et al. (1993Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity Proc Natl Acad Sci USA 903539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC.et al. (2008Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial Lancet Oncol 9533–542. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ.et al. (2011Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans Nature 47799–102. [DOI] [PubMed] [Google Scholar]
- McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK.et al. (2001Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes Cancer Res 618751–8757. [PubMed] [Google Scholar]
- Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F.et al. (2007Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963 J Clin Invest 1173350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn DH, Wang Y, Le Boeuf F, Bell J., and, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foloppe J, Kintz J, Futin N, Findeli A, Cordier P, Schlesinger Y.et al. (2008Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus Gene Ther 151361–1371. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Liang C, Yu YA, Chen N, Dandekar T., and, Szalay AA. The highly attenuated oncolytic recombinant vaccinia virus GLV-1h68: comparative genomic features and the contribution of F14.5L inactivation. Mol Genet Genomics. 2009;282:417–435. doi: 10.1007/s00438-009-0475-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengstschläger M, Pfeilstöcker M., and, Wawra E. Thymidine kinase expression. A marker for malignant cells. Adv Exp Med Biol. 1998;431:455–460. [PubMed] [Google Scholar]
- Luker KE, Hutchens M, Schultz T, Pekosz A., and, Luker GD. Bioluminescence imaging of vaccinia virus: effects of interferon on viral replication and spread. Virology. 2005;341:284–300. doi: 10.1016/j.virol.2005.06.049. [DOI] [PubMed] [Google Scholar]
- Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X.et al. (2003Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer Cancer Res 631389–1397. [PubMed] [Google Scholar]
- Yang H, Kim SK, Kim M, Reche PA, Morehead TJ, Damon IK.et al. (2005Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction J Clin Invest 115379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Twardzik DR, Marquardt H., and, Todaro GJ. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature. 1985;313:491–492. doi: 10.1038/313491a0. [DOI] [PubMed] [Google Scholar]
- Alcami A., and, Efstathiou S. Soluble chemokine binding proteins are also encoded by herpesviruses. Immunol Today. 2000;21:526–527. doi: 10.1016/s0167-5699(00)01632-7. [DOI] [PubMed] [Google Scholar]
- Langhammer S, Koban R, Yue C., and, Ellerbrok H. Inhibition of poxvirus spreading by the anti-tumor drug Gefitinib (Iressa) Antiviral Res. 2011;89:64–70. doi: 10.1016/j.antiviral.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Buller RM, Smith GL, Cremer K, Notkins AL., and, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A.et al. (2008Identification and expansion of the tumorigenic lung cancer stem cell population Cell Death Differ 15504–514. [DOI] [PubMed] [Google Scholar]
- Li D, Xie K, Wolff R., and, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M.et al. (2005KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib PLoS Med 2e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi AS, Palmedo G, Flaig MJ, Puchta U, Reckwerth A, Rütten A.et al. (2003Mutations of the BRAF gene in benign and malignant melanocytic lesions J Invest Dermatol 1211160–1162. [DOI] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I., and, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Giaccone G., and, Wang Y ( Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat Rev. 2011;37:456–464. doi: 10.1016/j.ctrv.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ.et al. (2008Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer J Clin Oncol 261626–1634. [DOI] [PubMed] [Google Scholar]
- Douglas JT, Kim M, Sumerel LA, Carey DE., and, Curiel DT. Efficient oncolysis by a replicating adenovirus (ad) in vivo is critically dependent on tumor expression of primary ad receptors. Cancer Res. 2001;61:813–817. [PubMed] [Google Scholar]
- Weislow OS, Kiser R, Fine DL, Bader J, Shoemaker RH., and, Boyd MR. New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst. 1989;81:577–586. doi: 10.1093/jnci/81.8.577. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A.et al. (2008Identification and expansion of the tumorigenic lung cancer stem cell population Cell Death Differ 15504–514. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C.et al. (2007Identification and expansion of human colon-cancer-initiating cells Nature 445111–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reduced TK levels affect JX-594 output in multiple TK-knockdown clones.