

Abstract
To investigate the effects of the transient receptor potential vanilloid type 1 (TRPV1) channel on renal extracellular matrix (ECM) protein expression including collagen deposition and the transforming growth factor β (TGF-β)/Smad signaling pathway during salt-dependent hypertension, wild-type (WT) and TRPV1-null (TRPV1−/−) mutant mice were uninephrectomized and given deoxycorticosterone acetate (DOCA)-salt for 4 wks. TRPV1 gene ablation exaggerated DOCA-salt-induced impairment of renal function as evidenced by increased albumin excretion (μg/24 h) compared with WT mice (83.7 ± 7.1 versus 28.3 ± 4.8, P < 0.05), but had no apparent effect on mean arterial pressure (mmHg) as determined by radiotelemetry (141 ± 4 versus 138 ± 3, P > 0.05). Morphological analysis showed that DOCA-salt-induced glomerulosclerosis, tubular injury and macrophage infiltration (cells/mm2) were increased in TRPV1−/− compared with WT mice (0.74 ± 0.08 versus 0.34 ± 0.04; 3.14 ± 0.26 versus 2.00 ± 0.31; 68 ± 5 versus 40 ± 4, P < 0.05). Immunostaining studies showed that DOCA-salt treatment decreased nephrin but increased collagen type I and IV as well as phosphorylated Smad2/3 staining in kidneys of TRPV1−/− compared with WT mice. Hydroxyproline assay and Western blot showed that DOCA-salt treatment increased collagen content (μg/mg dry tissue) and fibronectin protein expression (%β-actin arbitrary units) in the kidney of TRPV1−/− compared with WT mice (26.7 ± 2.7 versus 17.4 ± 1.8; 0.93 ± 0.07 versus 0.65 ± 0.08, P < 0.05). Acceleration of renal ECM protein deposition in DOCA-salt-treated TRPV1−/− mice was accompanied by increased TGF-β1, as well as phosphorylation of Smad2/3 protein expression (%β-actin arbitrary units) compared with DOCA-salt-treated WT mice (0.61 ± 0.07 versus 0.32 ± 0.05; 0.57 ± 0.07 versus 0.25 ± 0.05; 0.71 ± 0.08 versus 0.40 ± 0.06, P < 0.05). These results show that exaggerated renal functional and structural injuries are accompanied by increased production of ECM protein and activation of the TGF-β/Smad2/3 signaling pathway. These data suggest that activation of TRPV1 attenuates the progression of renal fibrosis possibly via suppression of the TGF-β and its downstream regulatory signaling pathway.
INTRODUCTION
Hypertension is a leading risk factor contributing to the development and progression of chronic renal disease (1). Hypertension-induced renal injury displays several characteristics including proteinuria, inflammatory cell recruitment and accumulation of extracellular matrix (ECM) proteins in the glomerular and tubulointerstitial spaces (2,3). The resulting glomerulosclerosis and tubulointerstitial fibrosis are believed to lead to progressive renal insufficiency. Although accumulating evidence shows that hypertension-induced renal damage is associated with renal fibrotic responses, the exact molecular mechanisms responsible for renal fibrosis remain to be defined.
It has been demonstrated that increased ECM protein synthesis and/or decreased ECM degradation contributes to the development of renal glomerulosclerosis and tubulointerstitial fibrosis (3,4). To a large extent, the ECM accumulation is mediated by the profibrotic cytokine, transforming growth factor-β (TGF-β) (5). TGF-β plays a major role in the tissue response to injury by regulating both cellular proliferation and ECM turnover through the Smad signaling pathway (6,7). TGF-β is highly expressed in injured tissues, and TGF-β-dependent effects play a role in the pathogenesis of atherosclerosis, coronary artery disease and hypertension (8–11).
The transient receptor potential vanilloid type 1 (TRPV1) is a polymodal nociceptive transducer containing six trans-membrane domains that form a non-selective, calcium-preferring cation channel (12,13). Immunohistochemical labeling studies have shown that, in addition to the central nervous system, TRPV1 is expressed predominantly in unmyelinated C-fibers and thinly myelinated Aδ-afferent nerve fibers innervating various organs/systems, including the kidney and cardiovascular tissues (13). In addition to noxious heat, low pH and the “hot” pepper–derived vanilloid compound, capsaicin, several endogenous mediators such as anandamide and lipoxygenase metabolites of arachidonic acid may activate TRPV1 (12,14,15). Activation of TRPV1 expressed in sensory nerves causes release of a number of sensory neurotransmitters, commonly calcitonin gene-related peptide (CGRP) and substance P (13). Previously, we have found a compelling association between high salt intake and TRPV1 activation/upregulation in the face of salt load (15,16). Our recent studies have further shown that genetic deficiency of TRPV1 accelerates deoxycorticosterone acetate (DOCA)-salt-induced renal morphological injury accompanied with impaired renal function (17). The results suggest that activation of TRPV1 protects against renal injury during salt-dependent hypertension, albeit the underlying molecular mechanisms remain to be defined.
It is well established that the fibrotic response is a hallmark of renal injury resulting from hypertension. Thus, this study was to determine whether TRPV1 plays a role in attenuating the progression of renal functional and structural changes, specifically in ECM expansion, during DOCA-salt hypertension. Given that the TGF-β/Smad signaling pathway plays a vital role in the fibrogenic processes (6,7), we also examined the effects of TRPV1 on the regulation and expression of TGF-β1 and its downstream signaling proteins involving Smads in DOCA-salt hypertension.
MATERIALS AND METHODS
Animals
TRPV1-null (TRPV1−/−) mutant mice were kind gifts of David Julius (University of California, San Francisco, CA, USA) and were generated as described by Caterina et al. (12). After backcrossing of TRPV1−/− mice for six generations with C57BL/6 mice, TRPV1−/− mice were mated with each other to generate null mutant offspring to be used in the present study. Experiments were performed on 10- to 12-week-old male wild-type (WT, C57BL/6, Charles River Laboratories, Wilmington, MA, USA) and TRPV1−/− mice weighing 25 to 30 g. The mice were housed under controlled environmental conditions and maintained with standard food and water. All animal procedures were approved by the institutional animal care and use committee.
Experimental Protocol
DOCA-salt hypertension was performed as we described previously (17). Briefly, the mice were anesthetized with a single subcutaneous injection of ketamine (80 mg/kg) and xylazine (4 mg/kg). The left kidney of the animals was removed through a dorsal flank incision, a DOCA pellet (24 mg/10 g of body weight, Innovative Research of America, Sarasota, FL, USA) was implanted subcutaneously between the scapular blades. The mice receiving DOCA were fed with a standard chow and given 1% NaCl plus 0.2% KCl drinking solution throughout the 4 wk protocol. Control mice, including WT and TRPV1−/− mice, underwent uninephrectomy and consumed the standard chow and distilled water without receiving DOCA.
A day before euthanizing, the mice were placed in metabolic cages with free access to food and water for 24 h urine collection. At the end of the study, the mice were euthanized using pentobarbital sodium for tissue collection. Kidneys were removed, weighed, cleaned in ice-cold phosphate buffer, and snap frozen in liquid nitrogen for protein extraction or fixed in a 10% formaldehyde solution for histological studies.
Blood Pressure and Heart Rate (HR) Assay
Continuous mean arterial pressure (MAP) and HR data acquisition was performed with the use of the telemetry system (Data Sciences International, St. Paul, MN, USA) according to the manufacturer’s instruction. Briefly, the mice were anesthetized as described above. The catheter linked to the transmitter was inserted into the left carotid artery and the transmitter body placed subcutaneously in the lower right side of the abdomen. The skin incisions were then closed. The mice were returned to their individual cages and allowed recovery for 3 d before radiotelemetric recording started. MAP and HR were recorded continuously in DOCA-salt-treated WT and TRPV1−/− mice 1 wk before DOCA implantation and 4 wks after DOCA implantation.
Urinary Albumin Excretion Assay
Urinary albumin was measured with an enzyme linked immunosorbent assay (ELISA) kit (Exocell Inc, Philadelphia, PA, USA) following the manufacturer’s protocol. The rate of urinary albumin excretion was calculated based on the measurement of urinary albumin concentration and output. The values of urinary albumin were expressed as μg/24 h.
Morphological Assessment
The kidneys fixed with formaldehyde solution were embedded in paraffin and cut into 4-μm sections. The sections were stained with periodic acid–Schiff stain (PAS) and Masson trichrome stain. The glomerulosclerosis index was assessed in PAS-stained sections in 50 randomly selected glomeruli per section under 400× magnification. The glomerulosclerosis is defined as accumulation of ECM deposits and mesangial expansion, and the degree of sclerosis was graded using a semiquantitative scoring method as reported previously (18). Tubulointerstitial injury is defined as tubular atrophy, dilation, deposition of ECM and presence of inflammatory cells. The tubulointerstitial injury index was assessed in Masson trichrome-stained sections in 20 fields selected randomly from each kidney representing nearly the whole piece of cortex at 200× magnification, and the degree of tubulointerstitial injury was graded using a semiquantitative scoring method as described by Shih et al. (19). All scoring was performed in a blinded manner on coded slides.
Immunohistochemistry and Immunofluorescence
The kidney paraffin sections (3 μm thick) or cryosections (8 μm thick) were used for immunohistochemical or immunofluorescence staining using the following primary antibodies: monoclonal rat anti–mouse macrophage marker F4/80 (1:200, Serotec, Oxford, UK); polyclonal goat anti–human nephrin (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA); polyclonal rabbit anti–mouse collagen type I (1:80, Millipore, Billerica, MA, USA); polyclonal rabbit anti–mouse collagen type IV (1:100, Millipore); and polyclonal goat anti–human phosphorylated Smad2/3 (1:200, Santa Cruz Biotechnology). The paraffin sections were treated with 3% hydrogen peroxide for 5 min. To block nonspecific immuno-labeling, the paraffin sections or cryosections were incubated with 3% normal horse or bovine serum in phosphate buffer solution for 1 h at room temperature. The sections then were incubated with primary antibodies as described above at 4°C overnight. After being washed, the paraffin sections were incubated with horse anti–rat, anti–rabbit or anti–goat IgG–horseradish peroxidase (1:200 or 1:400, Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature, and visualized by incubating the sections with the substrate vector fast red or 3,3′-diaminobenzidine (Vector Laboratories). For immunofluorescence staining, the cryosections were incubated with a Cy3–conjugated bovine anti–rabbit IgG (1:800, Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature. Sections incubated with phosphate buffer, instead of the primary antibodies, were used as negative control. All sections were analyzed using light or fluorescence microscope and digitized using a high-resolution camera (Olympus HC-2000, Olympus, Tokyo, Japan). Macrophages positive for F4/80 were counted by examining 15 randomly selected cortical fields per section in a blind fashion under 400× magnification. The number of positive cells was expressed as cells per square millimeter. The degree of nephrin expansion was assessed using an in-house image analysis software, which determined the total area of a glomerulus occupied by nephrin. The degree of the nephrin expansion is expressed as the percentage of the total glomerular area.
Hydroxyproline Assay
The collagen content of renal tissue was determined by hydroxyproline assay. Kidney samples were processed as described previously (17). Hydroxyproline content was determined with a colorimetric assay and a standard curve of 0 to 5 μg hydroxyproline (Sigma-Aldrich Co., St. Louis, MO, USA). Data were expressed as mg collagen/μg dry weight, assuming that collagen contains an average of 13.5% hydroxyproline.
Western Blot Analysis
Kidney tissues were homogenized in lysis buffer containing protease inhibitors, separated on a 6 ~ 15% sodium dodecyl sulfate-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane as described previously (20). Blots were blocked 1 h at room temperature in 5% milk washing solution (50 mmol/L Tris-HCl, 100 mmol/L NaCl and 0.1% Tween-20 at pH 7.5). Subsequently, blots were incubated overnight at 4°C with polyclonal rabbit anti–rat fibronectin (1:800, Millipore); polyclonal rabbit anti–human TGF-β1 (1:500, Santa Cruz Biotechnology); monoclonal rabbit anti–human phospho-Smad2 (1:1000, Millipore); and polyclonal rabbit anti–human phospho-Smad3 (1:800, Invitrogen, Camarillo, CA, USA) in blocking solution, followed by incubation for 1 h with horseradish peroxidase–conjugated bovine anti–rabbit IgG (1:800 ~ 1000, Santa Cruz Biotechnology). Proteins were visualized with an enhanced chemiluminescence detection system (ECL) (Amersham Biosciences, Piscataway, NJ, USA). The densities of specific bands were determined using Scion Image Beta (version 4.02) software, and normalized to the total amount of β-actin loaded in each well.
Statistical Analysis
All values are expressed as mean ± SE. The significance of differences between groups for blood pressure data was evaluated with an analysis of variance (ANOVA) for repeated measures followed by a Bonferroni test. The differences among groups were analyzed using one-way ANOVA followed by a Bonferroni adjustment for multiple comparisons. Differences were considered statistically significant at P < 0.05.
RESULTS
As demonstrated in Table 1, there was no significant difference in body weight between groups studied at the beginning or end of the experimental period. There was no significant difference in the ratio of kidney to body weight between control WT and TRPV1−/− mice. DOCA-salt treatment significantly increased the ratio of kidney to body weight in WT and TRPV1−/− mice compared with their respective control mice (P < 0.05), but no difference was found between DOCA-salt-treated WT and TRPV1−/− mice (P > 0.05). As expected, DOCA-salt treatment resulted in a significant increase in urinary albumin excretion in WT and TRPV1−/− mice, with the excretion in TRPV1−/− mice being significantly greater than WT mice (P < 0.05).
Table 1.
Changes in metabolic and renal parameters in WT and TRPV1−/− mice after 4 weeks of DOCA-salt treatment.
| Parameters | WT | TRPV1−/− | DOCA-WTa | DOCA-TRPV1−/−b |
|---|---|---|---|---|
| BWc (g) | 28.8 ± 0.4d | 28.2 ± 0.3 | 29.1 ± 0.5 | 29.7 ± 0.3 |
| Kidney weight (mg/10 g of BW) | 77.1 ± 1.5 | 76.5 ± 1.2 | 109.6 ± 3.6e | 113.6 ± 3.1e |
| Urinary albumin (mg/24 h) | 6.9 ± 0.8 | 6.2 ± 0.7 | 28.3 ± 4.8e | 83.7 ± 7.1e,f |
| GSIg | 0.13 ± 0.03 | 0.09 ± 0.02 | 0.34 ± 0.04e | 0.74 ± 0.08e,f |
| TISh | 0.29 ± 0.10 | 0.21 ± 0.11 | 2.00 ± 0.31e | 3.14 ± 0.26e,f |
DOCA-WT, WT mice treated with DOCA-salt.
DOCA-TRPV1−/−, TRPV1−/− mice treated with DOCA-salt.
BW, body weight.
Values are mean ± SE (n = 6 to 8).
P < 0.05 compared to control WT or TRPV1−/− mice.
P < 0.05 compared to DOCA-WT mice.
GSI, glomerulosclerosis index.
TIS, tubulointerstitial injury score.
As shown in Figure 1, basal average 24 h MAP levels, as determined by radiotelemetry, were similar between WT and TRPV1−/− mice at the beginning of the DOCA-salt protocol. After initiation of the DOCA-salt protocol, MAP increased and reached the plateau in about 1 wk and remained at this level for 3 wks in both WT and TRPV1−/− mice with no difference between the two strains (P > 0.05). In addition, there was no difference in HR between WT and TRPV1−/− mice during the baseline and DOCA-salt treatment period.
Figure 1.

Time course responses of mean arterial pressure (A) and heart rate (B) to deoxycorticosterone acetate (DOCA)-salt treatment for 4 wks in WT and TRPV1−/− mice. Mean arterial pressure and heart rate were determined by radiotelemetry and averaged in 24 h. Values are mean ± SE (n = 6).
To examine whether the loss of TRPV1 affects renal morphological lesions during DOCA-salt hypertension, glomerulosclerosis and tubulointerstitial injury were studied. As illustrated in Figure 2, DOCA-salt treatment caused a more severe glomerulosclerosis in the kidneys of TRPV1−/− than in WT mice shown by PAS-staining. Likewise, DOCA-salt treatment induced tubular dilation, atrophy, interstitial cell infiltration and ECM accumulation in WT and TRPV1−/− mice, with more severe injury in the latter strain shown by Masson trichrome staining (Figure 2). Quantitative analysis showed that DOCA-salt treatment significantly increased glomerulosclerosis index and tubulointerstitial injury score in WT and TRPV1−/− mice, and the magnitude of the increases was greater in the latter group (P < 0.05) (Table 1). At the baseline, no specific renal histological lesions, in particular glomerular or tubulointerstitial lesion, were observed in control WT and TRPV1−/− mice.
Figure 2.
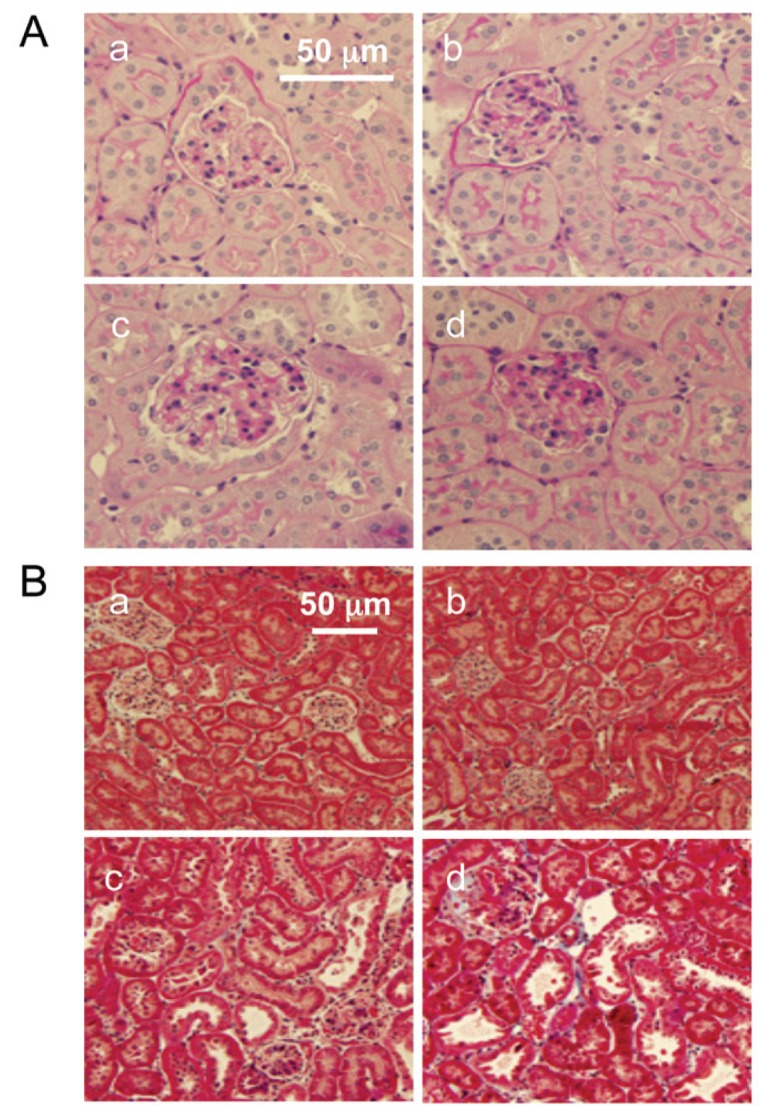
Changes in renal morphology in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. Representative PAS-stained (A) or Masson trichrome–stained (B) kidney sections showing glomerulosclerosis or tubulointerstitial injury in control WT and TRPV1−/− mice (a and b), and WT and TRPV1−/− mice (c and d) treated with DOCA-salt. Magnification, 400× (A) or 200× (B).
Given that hypertension-induced inflammatory responses exacerbate the disease state and organ damage, we assessed the changes in monocyte/macrophage infiltration in the kidney of WT and TRPV1−/− mice during DOCA-salt hypertension. As demonstrated in Figure 3, analysis of a marker of monocyte/macrophages (F4/80) showed significantly increased staining of F4/80 in the kidney of DOCA-salt-treated WT and TRPV1−/− mice, relative to the control WT and TRPV1−/− mice. Quantification of the F4/80 staining revealed a significant increase in the number of monocytes/macrophages infiltrating into the tubulointerstitial space in DOCA-salt-treated WT mice, and the inflammatory cell infiltration was accelerated in DOCA-salt-treated TRPV1−/− mice.
Figure 3.
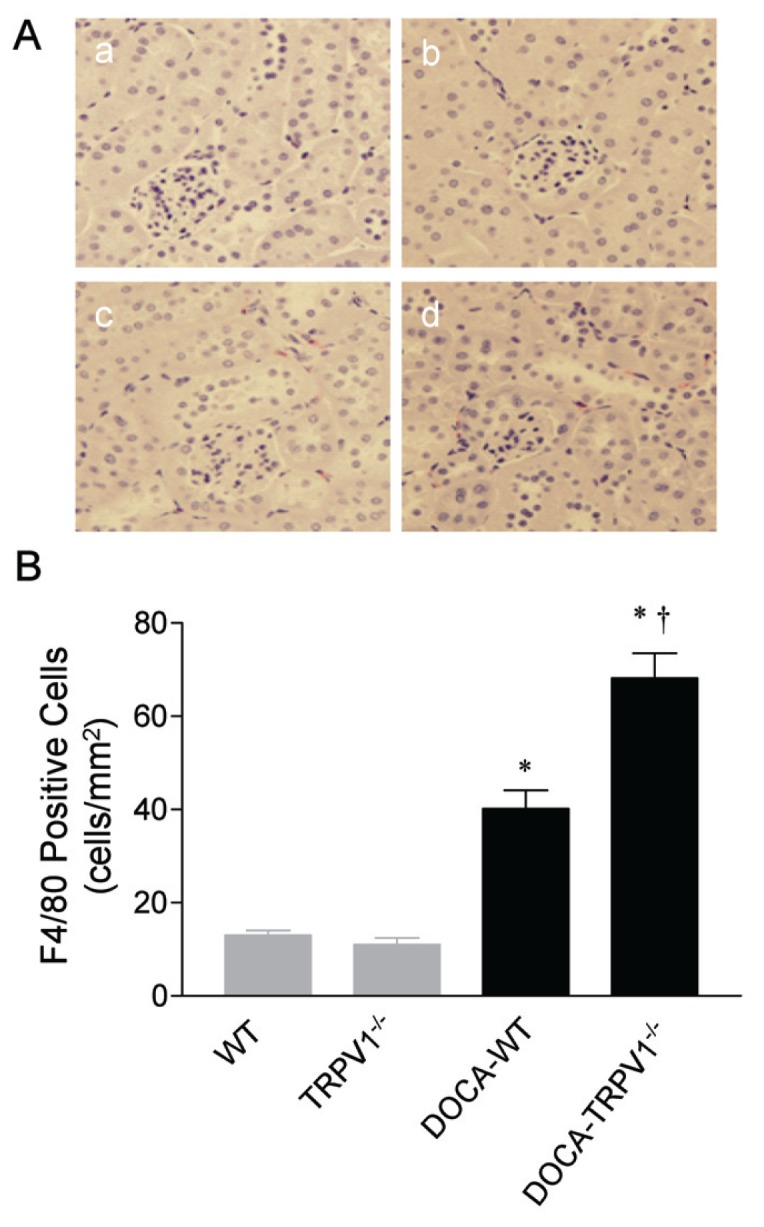
Changes in renal cortical immunostaining of F4/80-positive cells (monocytes/macrophages) in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A) Representative immunostained kidney sections showing the immunostaining of F4/80-positive cells (monocytes/macrophages in red) in control WT and TRPV1−/− mice (a and b), and WT and TRPV1−/− mice (c and d) treated with DOCA-salt. Magnification, 400×. (B) Bar graph shows the changes in renal cortical F4/80-positive cells (monocytes/macrophages) in WT and TRPV1−/− mice with or without DOCA-salt treatment. Values are mean ± SE (n = 7 to 8). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
Given that podocyte may play a role in urinary albumin excretion, we determined the effects of TRPV1 ablation on nephrin expression in podocytes. As illustrated in Figure 4, the area of nephrin staining decreased significantly in DOCA-salt-treated WT and TRPV1−/− mice compared with the control groups, and the magnitude of decreases was greater in DOCA-salt-treated TRPV1−/− than in WT mice (P < 0.05).
Figure 4.
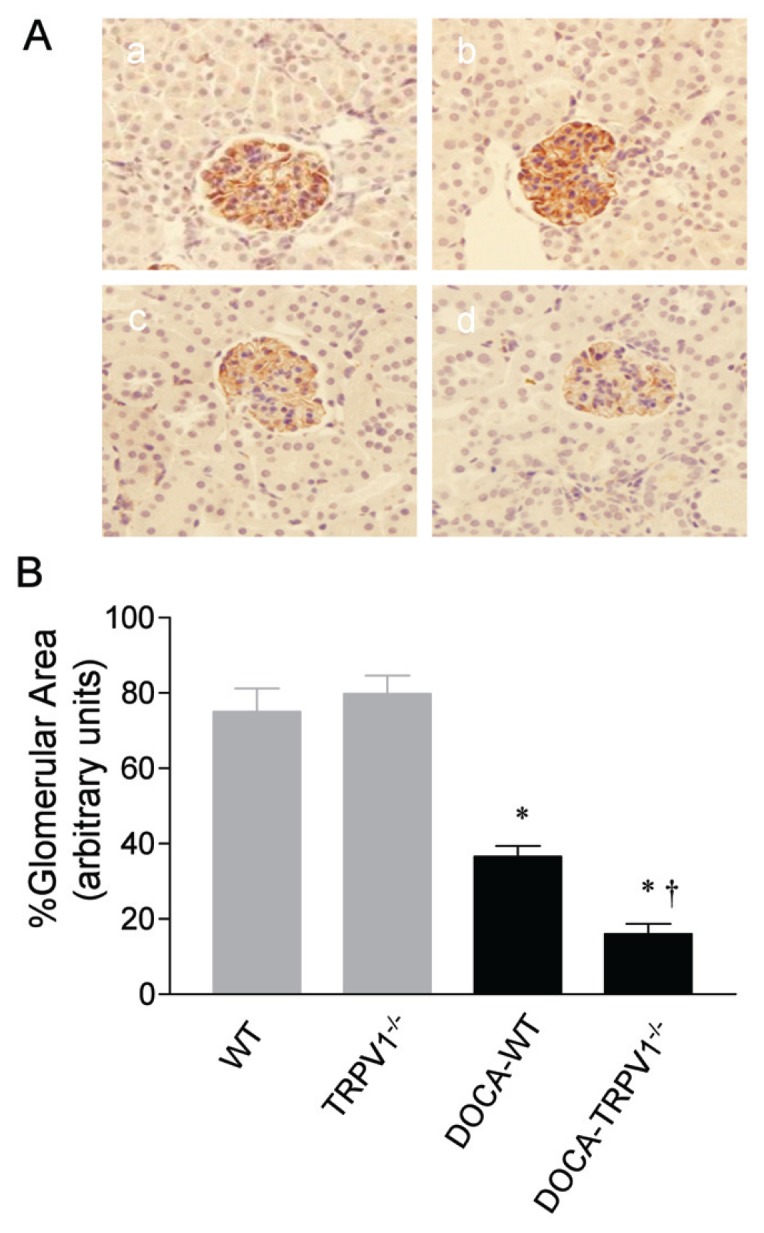
Changes in renal glomerular immunostaining of nephrin in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A) Representative immunostained kidney sections showing the immunostaining of nephrin in brown in control WT and TRPV1−/− mice (a and b), and WT and TRPV1−/− mice (c and d) treated with DOCA-salt. Magnification, 400×. (B) Bar graph shows the changes in renal glomerular immunostaining of nephrin in WT and TRPV1−/− mice with or without DOCA-salt treatment. Values are mean ± SE (n = 7 to 8). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
Immunofluorescent staining to assess DOCA-salt-induced renal collagen deposition, specifically collagen types I and IV, in WT and TRPV1−/− mice is shown in Figure 5. DOCA-salt treatment caused an overall increase in the intensity of immunostaining for collagen types I and IV in WT mice, whereas these changes were accelerated in TRPV1−/− mice. No difference in the intensity of immunostaining for collagen types I and IV was observed between control WT and TRPV1−/− mice. Quantification of changes in the collagen content by hydroxyproline assay confirmed the immunohistochemical observations. Accordingly, the total collagen content determined by hydroxyproline assay was greater in DOCA-salt-treated TRPV1−/− than in control TRPV1−/− or DOCA-salt-treated WT mice (P < 0.05). The collagen content in the kidney tended to be higher in DOCA-salt-treated WT but was not statistically significant when compared with control WT mice (P > 0.05).
Figure 5.
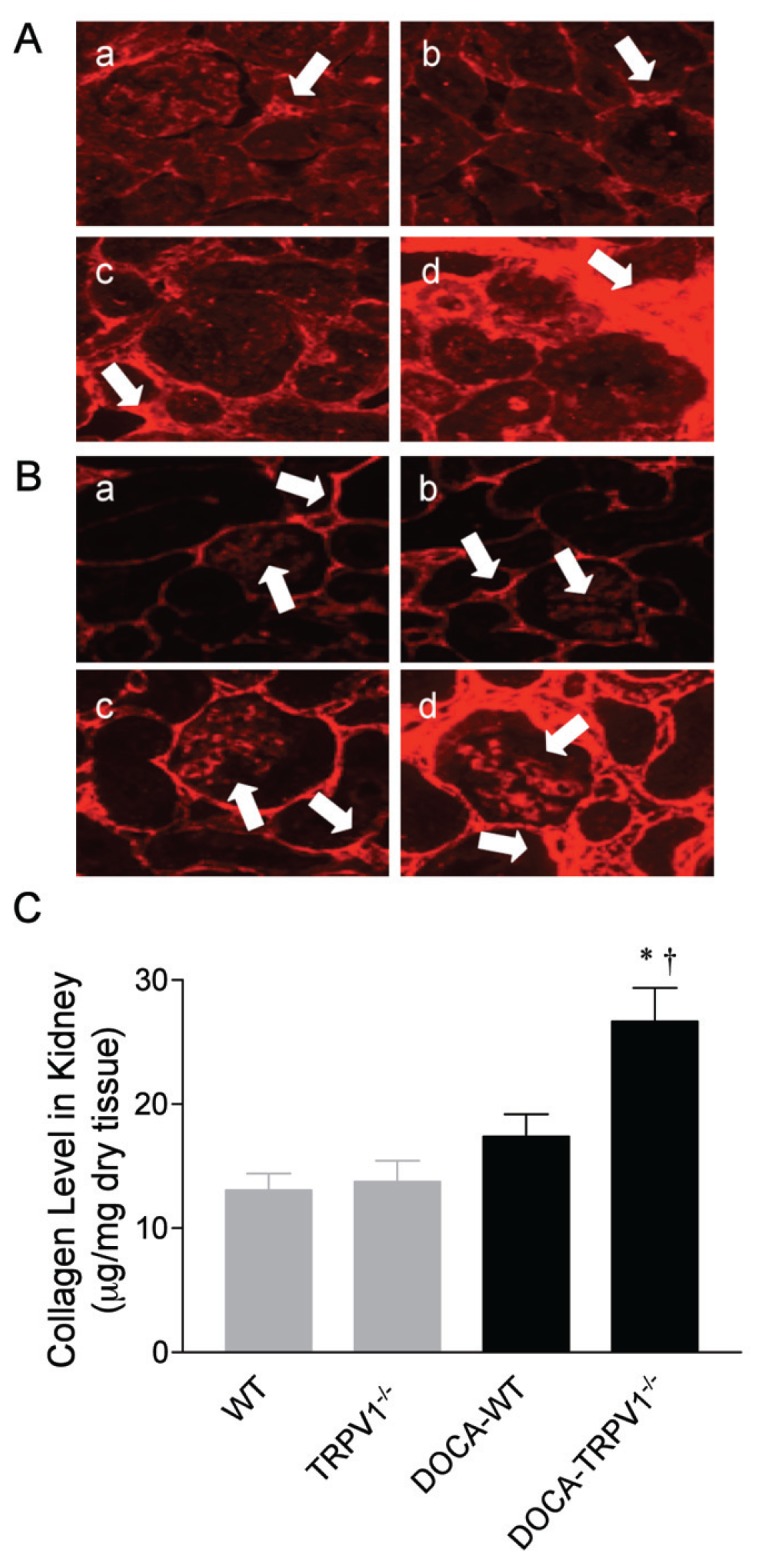
Changes in renal cortical immunolocalization of collagen type I and type IV and collagen levels in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A, B) Representative immunostained kidney sections showing the immunostaining of collagen type I and type IV (in red) in control WT and TRPV1−/− mice (a and b), and WT and TRPV1−/− mice (c and d) treated with DOCA-salt. Arrows point to collagen type I-positive staining (A) or collagen type IV-positive staining (B). Magnification, 400×. (C) Bar graph shows the changes in renal collagen levels in WT and TRPV1−/− mice with or without DOCA-salt treatment. Values are mean ± SE (n = 7 to 8). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
Similar to the changes in collagen protein, quantitative analysis of fibronectin protein expression by Western blot showed that DOCA-salt-treated WT exhibited an increase in renal fibronectin protein expression (P < 0.05), and the changes were accelerated by deletion of the TRPV1 receptor (Figure 6).
Figure 6.
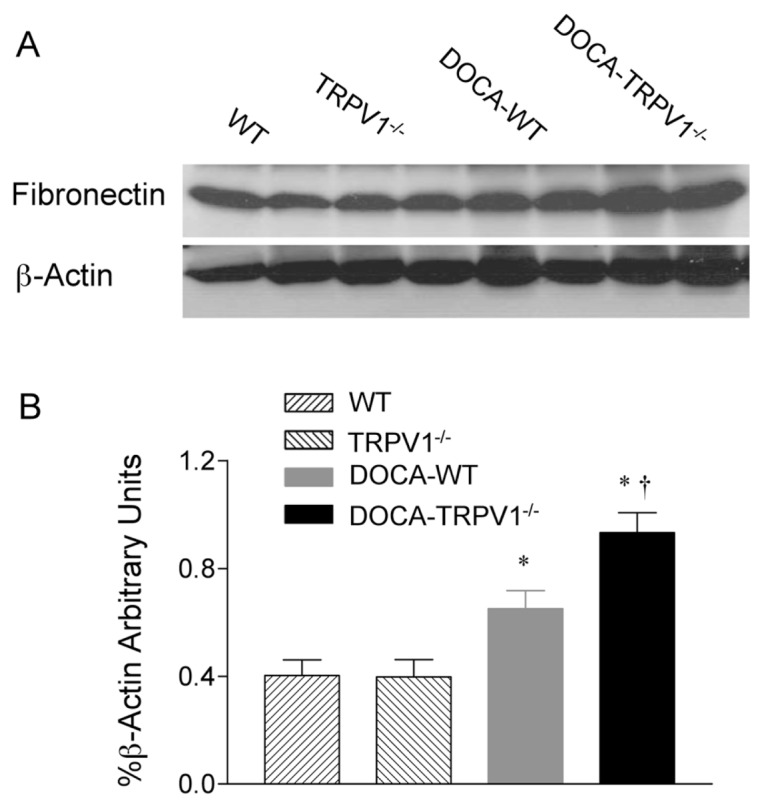
Changes in renal fibronectin protein expression in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A) Representative Western blot of renal fibronectin in WT and TRPV1−/− mice with or without DOCA-salt treatment. (B) Bar graph shows the relative optical density values for renal fibronectin in WT and TRPV1−/− mice with or without DOCA-salt treatment. Results were expressed as ratio of fibronectin to corresponding β-actin. Values are mean ± SE (n = 4 to 5). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
To understand the mechanisms contributing to renal fibrosis during DOCA-salt hypertension, we focused on the effects of TRPV1 ablation on the TGF-β1/Smad signaling pathway. As illustrated in Figure 7, a significant increase in protein expression of TGF-β1 determined by Western blot was observed in kidneys of DOCA-salt hypertensive WT and TRPV1−/− mice compared with their respective controls, with protein expression of TGF-β1 in TRPV1−/− mice being significantly greater than in WT mice (P < 0.05). No significant difference was seen in renal TGF-β1 protein expression between control WT and TRPV1−/− mice. These results support the notion that the loss of TRPV1 increases TGF-β1 protein expression in the kidney in DOCA-salt hypertension.
Figure 7.
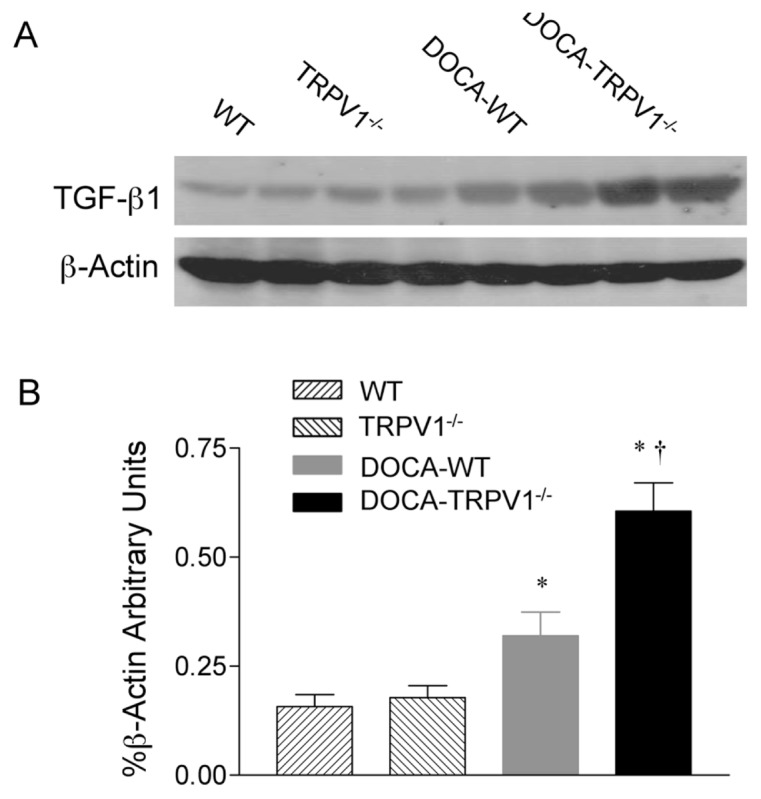
Changes in renal TGF-β1 protein expression in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A) Representative Western blot of renal TGF-β1 in WT and TRPV1−/− mice with or without DOCA-salt treatment. (B) Bar graph shows the relative optical density values for renal TGF-β1 in WT and TRPV1−/− mice with or without DOCA-salt treatment. Results were expressed as ratio of TGF-β1 to corresponding β-actin. Values are mean ± SE (n = 4 to 5). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
Since Smad2/3 is known to play a crucial role in TGF-β1-induced ECM deposition, we investigated the effects of TRPV1 ablation on the regulation of phosphorylated Smad2/3 during DOCA-salt hypertension. As shown in Figure 8, immunohistochemical staining demonstrated that phosphorylated Smad2/3-positive cells were located in glomeruli, tubules, and cortical interstitial area. The phosphorylated Smad2/3-positive cells increased in DOCA-salt-treated WT and TRPV1−/− mice, with a greater intensity in the latter group. Consistently, Western blot analysis showed that a significant increase in renal protein expression of phosphorylated Smad2 or Smad3 was found in DOCA-salt-treated WT and TRPV1−/− mice relative to their respective controls (P < 0.05) (Figure 9). DOCA-salt-induced increases in protein expression of phosphorylated Smad2 or Smad3 were accelerated in TRPV1−/− compared with WT mice (P < 0.05). There was no significant difference in phosphorylated Smad2 and Smad3 between control WT and TRPV1−/− mice. These data indicate that TRPV1 is likely to govern the regulation of the TGF-β1/Smad2/3 signaling pathway during DOCA-salt hypertension.
Figure 8.
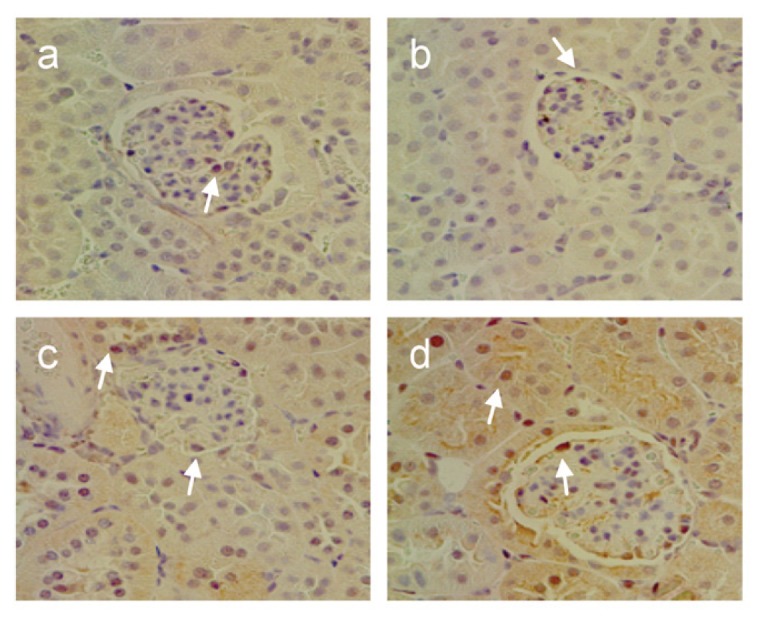
Changes in renal cortical immunostaining of phosphorylated Smad2/3-positive cells in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. Representative immunostained kidney sections showing the immunostaining of phosphorylated Smad2/3 in brown in control WT and TRPV1−/− mice (a and b), and WT and TRPV1−/− mice (c and d) treated with DOCA-salt. Arrows point to phosphorylated Smad2/3-positive cells. Magnification, 400×.
Figure 9.
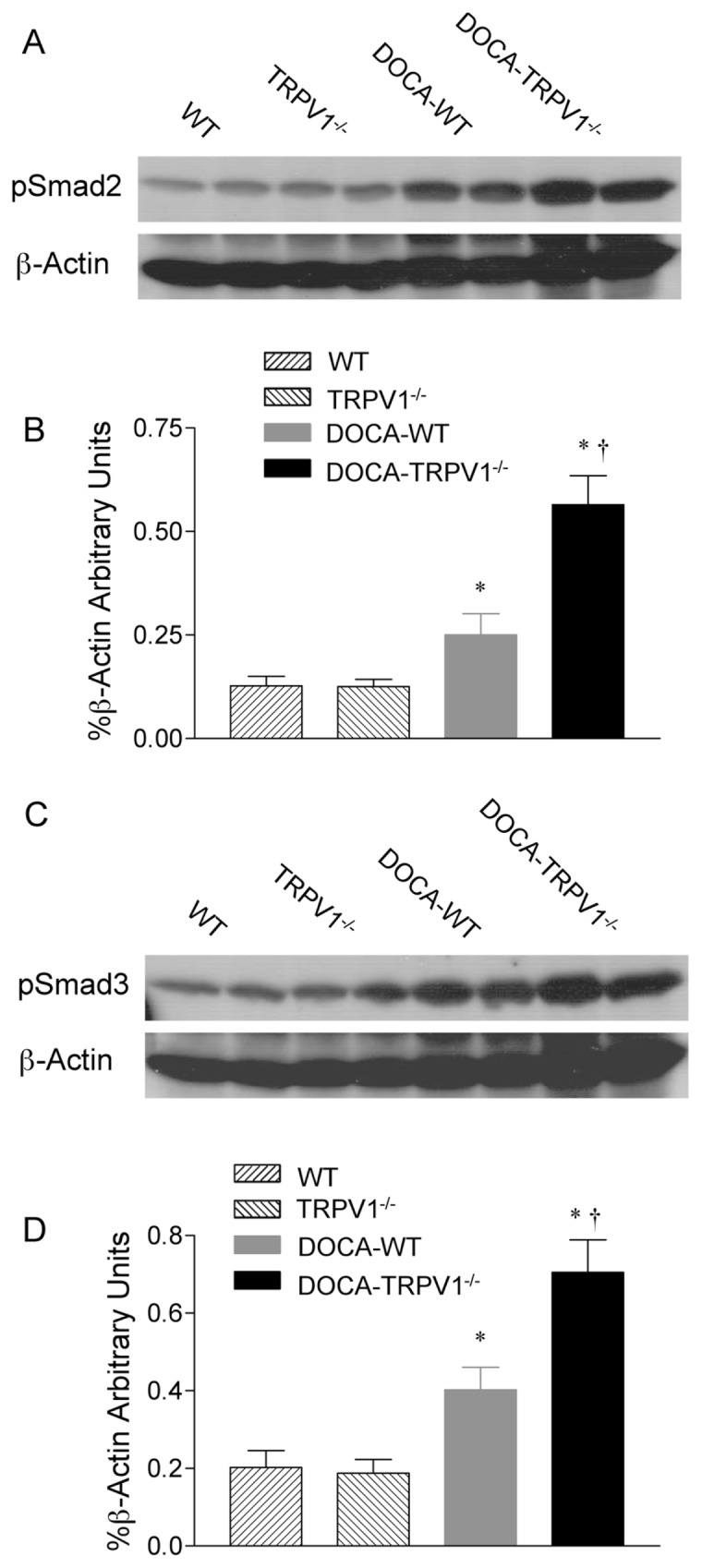
Changes in renal phosphorylated Smad2 (pSmad2) and phosphorylated Smad3 (pSmad3) protein expression in WT and TRPV1−/− mice after 4 wks of deoxycorticosterone acetate (DOCA)-salt treatment. (A, C) Representative Western blot of renal pSmad2 (A) and pSmad3 (C) in WT and TRPV1−/− mice with or without DOCA-salt treatment. (B, D) Bar graphs show the relative optical density values for renal pSmad2 (B) and pSmad3 (D) in WT and TRPV1−/− mice with or without DOCA-salt treatment. Results were expressed as ratio of pSmad2 and pSmad3 to corresponding β-actin. Values are mean ± SE (n = 4 to 5). *P < 0.05 compared to control WT or TRPV1−/− mice; †P < 0.05 compared to DOCA-WT mice.
DISCUSSION
The results of the present study show that TRPV1 ablation leads to exaggerated albuminuria, glomerulosclerosis and tubulointerstitial injury in response to DOCA-salt treatment. These functional and structural impairments are accompanied by enhancements in ECM protein production and activation of the TGF-β/Smad2/3 signaling pathway. These findings indicate that TRPV1 exerts an antifibrotic effect to protect the kidney against injury induced by DOCA-salt hypertension, and that the protective effect of TRPV1 involves the regulation of TGF-β and its downstream signaling pathway, including members of the Smad family, including Smad2/3.
We found that urinary albumin excretion, an important predictor for end-stage renal disease, was higher in DOCA-salt hypertensive mice than in control mice. In addition, urinary albumin excretion during hypertension was exaggerated by TRPV1 ablation. Previous studies indicated that podocyte injury may result in proteinuria (21). Podocytes are susceptible to injury as seen in many forms of glomerular diseases, including focal segmental glomerulosclerosis (22). Emerging evidence suggests that podocyte damage is characterized by losing nephrin and P-cadherin. Consistently, our data show that DOCA-salt treatment led to decreases in nephrin, and the decreases were aggravated in TRPV1−/− mice.
The progression of renal disease displays several characteristics, including inflammatory cell recruitment and accumulation of ECM proteins in the interstitium (3). These processes lead to renal fibrosis that ultimately contributes to kidney failure. To explore the antifibrotic role of TRPV1 in DOCA-salt-induced renal injury, we set to examine the premise that TRPV1 activation suppresses ECM protein formation through inhibiting the TGF-β/Smad signaling pathway. We observed that, compared to WT mice, TRPV1−/− mice displayed a further increase in ECM protein accumulation in the kidney in response to DOCA-salt treatment, evidenced by further increased expression of fibronectin, collagen type I and collagen type IV protein. These results are consistent with the notion that TRPV1 activation impedes ECM protein deposition.
It has been established that TGF-β is an important mediator in the fibrotic process. TGF-β1, the major isoform of the TGF-β superfamily, is produced by fibroblasts under various pathophysiological conditions, and activation of TGF-β1 stimulates cellular differentiation, proliferation, migration and ECM protein expression (6,7). Indeed, the dynamics of ECM protein synthesis and degradation is tightly regulated by TGF-β-mediated actions (5). The importance of TGF-β1-mediated actions in the onset and development of renal disease, including diabetic nephropathy and hypertension-induced nephrosclerosis, has been well established (5,23,24). Hypertension-induced kidney damage is characterized by overexpression of TGF-β1 mRNA and protein in experimental animal models as well as in humans (23–25). Furthermore, specific inhibition of TGF-β1 activity by a neutralizing antibody, or other interventions, successfully prevents mesangial glomerulosclerosis, ECM over-expression, and renal insufficiency in various animal models with or without hypertension (26–28). Consistent with these studies, our data show that TGF-β1 protein is overexpressed in the kidneys of WT mice four wks after DOCA-salt treatment. Moreover, deletion of TRPV1 further increases TGF-β1 protein expression in response to DOCA-salt treatment, indicating that TRPV1 may act as an endogenous renoprotective means to limit renal fibrosis via possibly interfering with TGF-β1 expression and action.
While TGF-β action may involve multiple downstream signaling pathways and cross-talks, the intracellular Smad pathway may play a crucial role, contributing to TGF-β-dependent structural manifestation of renal fibrotic responses. Recently, a more complex TGF-β/Smads signaling pathway has been reported. Activated TGF-β binds to a heteromeric complex of type I (TGF-βRI) and type II (TGF-βRII) receptors, which induces intracellular signals via phosphorylation of TGF-βRI-associated Smads (29,30). TGF-β signals through two distinct type I receptors, ALK1 and ALK5, and their respective signal transducers: Smad2 and Smad3 for ALK5 and Smad1 and Smad5 for ALK1 (30). Although it has been suggested that the TGF-β/Smad2/3 signaling pathway mediates organ fibrosis and that TGF-β/Smad1 mediates bone morphogenetic protein (BMP) signals for formation of bone and cartilage, elevated expression of Smad1 protein has been found in human and animal diabetic kidneys as well as in vitro models of fibrogenic responses (31,32,33).
Our data show that DOCA-salt hypertension increases phosphorylated Smad2 and Smad3 in the kidneys of WT mice, indicating that these Smads are activated in response to DOCA-salt treatment and may contribute to glomerulosclerosis and renal interstitial fibrosis during DOCA-salt hypertension. Furthermore, deletion of TRPV1 enhances DOCA-salt-induced increases in phosphorylated Smad2/3 protein expression. These findings support the premise that, in addition to regulating TGF-β1 protein expression, TRPV1 is capable of suppressing TGF-β1 activity, possibly via downregulating its profibrotic regulatory protein Smad2/3. Our observations demonstrate a novel aspect of TRPV1-dependent regulation of the TGF-β/Smad2/3 signaling pathway in response to DOCA-salt treatment. Although the interrelationship of TRPV1 and the TGF-β/Smad1 signaling pathway during DOCA-salt treatment is not the focus of the present study, it deserves further investigation given its potential role in tissue fibrosis.
It is known that the inflammatory response is a major driving force for profibrotic activities and that a number of inflammatory mediators may activate the TGF-β/Smad signaling pathway (34–36), leading to renal injury during hypertension (37–39). The inflammatory response is especially a key step bringing about renal injury during salt-dependent hypertension. Our recent study showed that an increased inflammatory response characterized by enhanced infiltration of monocytes/macrophages occurred in DOCA-salt-treated TRPV1−/− mice (40). The results from the present study further demonstrate that the increased monocyte/macrophage recruitment is associated with enhanced activation of the TGF-β/Smad signaling pathway in DOCA-salt-treated TRPV1−/− mice. Taken together, these data indicate that the protective effects of TRPV1 against DOCA-salt-induced renal fibrosis may be mediated, at least in part, by inhibiting inflammatory activation of the TGF-β/Smad signaling pathway.
Additionally, the TGF-β/Smad signaling pathway may be activated by a number of cellular stress signals, including oxidative stress, angiotensin II and mechanical stress (5,29,36). A possible link between TRPV1 and renal oxidative stress during salt load or salt-dependent hypertension has been established (17,41). Evidence shows that high salt intake increases renal superoxide production and expression of the NAD(P)H oxidase subunits in sensory denervated rats treated with capsaicin (41), indicating that TRPV1-positive sensory nerves play a counterregulatory role in preventing renal oxidative stress induced by salt load. Moreover, urinary 8-isoprostane, an oxidative stress marker, is increased significantly in DOCA-salt-treated TRPV1−/− mice, suggesting that TRPV1 activation leads to a diminution in renal oxidative stress during DOCA-salt hypertension (17). Thus, TRPV1 activation may inhibit the TGF-β/Smad signaling pathway via interfering with oxidative stress generation, and future elucidation on this possibility may provide further insight into TRPV1-dependent regulation of renal fibrosis.
In summary, we have demonstrated that renal ECM deposition resulting from DOCA-salt hypertension is exaggerated in TRPV1−/− mice that have an equivalent level of blood pressure as those of WT mice. Moreover, TRPV1 ablation leads to significant increases in the TGF-β1/Smad2/3 protein expression during DOCA-salt hypertension, indicating that activation of TRPV1 in response to DOCA-salt treatment may play a counterregulatory protective role against renal matrix remodeling through inhibiting the TGF-β/Smad (for example, TGF-β/Smad2/3) signaling pathway.
Hypertension-induced renal damage is characterized with increased ECM deposition. Our results provide convincing evidence for a beneficial role of TRPV1 activation in limiting renal fibrosis induced by DOCA-salt hypertension. These findings would expand our knowledge about the molecular mechanisms by which renal fibrosis develops in salt-dependent hypertension. In addition, these findings suggest that activation of TRPV1 may be a novel and important intervention to combat renal fibrosis during salt-dependent hypertension.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grants HL-57853, HL-73287 and DK67620 and a grant from the Michigan Economic Development Corporation. The authors thank Beihua Zhong for her excellent technical assistance.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Atkins RC. The epidemiology of chronic kidney disease. Kidney Int. 2005;94:S14–18. doi: 10.1111/j.1523-1755.2005.09403.x. [DOI] [PubMed] [Google Scholar]
- 2.Hartner A, Veelken R, Wittmann M, Cordasic N, Hilgers KF. Effects of diabetes and hypertension on macrophage infiltration and matrix expansion in the rat kidney. BMC Nephrol. 2005;6:6. doi: 10.1186/1471-2369-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- 4.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–73. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Koka V, Lan HY. Transforming growth factor-β and Smad signaling in kidney disease. Nephrology (Carlton) 2005;10:48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 6.Chabrier PE. Growth factors and vascular wall. Int Angiol. 1996;15:100–3. [PubMed] [Google Scholar]
- 7.Kretzschmar M, Massague J. SMADs: mediators and regulators of TGF-beta signaling. Curr Opin Genet Dev. 1998;8:103–11. doi: 10.1016/s0959-437x(98)80069-5. [DOI] [PubMed] [Google Scholar]
- 8.Spriewald BM, Ensminger SM, Billing JS, Morris PJ, Wood KJ. Increased expression of transforming growth factor-beta and eosinophil infiltration is associated with the development of transplant arteriosclerosis in long-term surviving cardiac allografts. Transplantation. 2003;76:1105–11. doi: 10.1097/01.TP.0000076467.83192.42. [DOI] [PubMed] [Google Scholar]
- 9.Ryan ST, Koteliansky VE, Gotwals PJ, Lindner V. Transforming growth factor-beta-dependent events in vascular remodeling following arterial injury. J Vasc Res. 2003;40:37–46. doi: 10.1159/000068937. [DOI] [PubMed] [Google Scholar]
- 10.Mata-Greenwood E, Meyrick B, Steinhorn RH, Fineman JR, Black SM. Alterations in TGF-beta1 expression in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol. 2003;285:L209–21. doi: 10.1152/ajplung.00171.2002. [DOI] [PubMed] [Google Scholar]
- 11.Porreca E, et al. Increased transforming growth factor-beta production and gene expression by peripheral blood monocytes of hypertensive patients. Hypertension. 1997;30:134–9. doi: 10.1161/01.hyp.30.1.134. [DOI] [PubMed] [Google Scholar]
- 12.Caterina MJ, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–13. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 13.Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- 14.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Kaminski NE, Wang DH. VR1–mediated depressor effects during high-salt intake: role of anandamide. Hypertension. 2005;46:986–91. doi: 10.1161/01.HYP.0000174596.95607.fd. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Kaminski NE, Wang DH. Endocannabinoid regulates blood pressure via activation of the TRPV1 in Wistar rats fed a high-salt diet. J Pharmacol Exp Ther. 2007;321:763–9. doi: 10.1124/jpet.106.112904. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Babánková D, Huang J, Swain GM, Wang DH. Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension. 2008;52:264–70. doi: 10.1161/HYPERTENSIONAHA.108.110197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito T, Sumithran E, Glasgow EF, Atkins RC. The enhancement of aminonucleoside nephrosis by the co-administration of protamine. Kidney Int. 1987;32:691–9. doi: 10.1038/ki.1987.262. [DOI] [PubMed] [Google Scholar]
- 19.Shih W, Hines WH, Neilson EG. Effects of cyclosporin A on the development of immune-mediated interstitial nephritis. Kidney Int. 1988;33:1113–8. doi: 10.1038/ki.1988.119. [DOI] [PubMed] [Google Scholar]
- 20.Wang YP, et al. Lipopolysaccharide triggers late preconditioning against myocardial infarction via inducible nitric oxide synthase. Cardiovasc Res. 2002;56:33–42. doi: 10.1016/s0008-6363(02)00506-0. [DOI] [PubMed] [Google Scholar]
- 21.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005–15. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 22.Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest. 2001;108:1583–7. doi: 10.1172/JCI14629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ying WZ, Sanders PW. The interrelationship between TGF-beta1 and nitric oxide is altered in salt-sensitive hypertension. Am J Physiol. 2003;285:F902–8. doi: 10.1152/ajprenal.00177.2003. [DOI] [PubMed] [Google Scholar]
- 24.Socha MJ, Manhiani M, Said N, Imig JD, Motamed K. Secreted protein acidic and rich in cysteine deficiency ameliorates renal inflammation and fibrosis in angiotensin hypertension. Am J Pathol. 2007;171:1104–12. doi: 10.2353/ajpath.2007.061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu S, Liu Y, Wang L, Meng QH. Transforming growth factor-beta1 is associated with kidney damage in patients with essential hypertension: renoprotective effect of ACE inhibitor and/or angiotensin II receptor blocker. Nephrol Dial Transplant. 2008;23:2841–6. doi: 10.1093/ndt/gfn159. [DOI] [PubMed] [Google Scholar]
- 26.Ziyadeh FN, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitrans-forming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA. 2000;97:8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuda H, et al. Development of gene silencing pyrrole-imidazole polyamide targeting the TGF-beta1 promoter for treatment of progressive renal diseases. J Am Soc Nephrol. 2006;17:422–32. doi: 10.1681/ASN.2005060650. [DOI] [PubMed] [Google Scholar]
- 28.Ellmers LJ, et al. Transforming growth factor-beta blockade down-regulates the renin-angiotensin system and modifies cardiac remodeling after myocardial infarction. Endocrinology. 2008;149:5828–34. doi: 10.1210/en.2008-0165. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 30.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest. 2005;128:585S–90S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- 31.Abe H, et al. Type IV collagen is transcriptionally regulated by Smad1 under advanced glycation end product (AGE) stimulation. J Biol Chem. 2004;279:14201–6. doi: 10.1074/jbc.M310427200. [DOI] [PubMed] [Google Scholar]
- 32.Matsubara T, et al. Expression of Smad1 is directly associated with mesangial matrix expansion in rat diabetic nephropathy. Lab Invest. 2006;86:357–68. doi: 10.1038/labinvest.3700400. [DOI] [PubMed] [Google Scholar]
- 33.Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforming growth factor-β receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 2007;282:10405–13. doi: 10.1074/jbc.M611742200. [DOI] [PubMed] [Google Scholar]
- 34.Jones LK, et al. IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis. Nephrol Dial Transplant. 2009;24:3024–32. doi: 10.1093/ndt/gfp214. [DOI] [PubMed] [Google Scholar]
- 35.Keophiphath M, et al. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol. 2009;23:11–24. doi: 10.1210/me.2008-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol. 2004;286:F606–16. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- 38.Muller DN, et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–93. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elmarakby AA, et al. Chemokine receptor 2b inhibition provides renal protection in angiotensin II-salt hypertension. Hypertension. 2007;50:1069–76. doi: 10.1161/HYPERTENSIONAHA.107.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Wang DH. Aggravated renal inflammatory responses in TRPV1 gene knockout mice subjected to DOCA-salt hypertension. Am J Physiol. 2009;297:F1550–9. doi: 10.1152/ajprenal.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Chen AF, Wang DH. Enhanced oxidative stress in kidneys of salt-sensitive hypertension: role of sensory nerves. Am J Physiol. 2006;291:H3136–43. doi: 10.1152/ajpheart.00529.2006. [DOI] [PubMed] [Google Scholar]