

Abstract
Histone methylation is an important regulator of gene expression; its coordinated activity is critical in complex developmental processes such as hematopoiesis. Disruptor of telomere silencing 1-like (DOT1L) is a unique histone methyltransferase that specifically methylates histone H3 at lysine 79. We analyzed Dot1L-mutant mice to determine influence of this enzyme on embryonic hematopoiesis. Mutant mice developed more slowly than wild-type embryos and died between embryonic days 10.5 and 13.5, displaying a striking anemia, especially apparent in small vessels of the yolk sac. Further, a severe, selective defect in erythroid, but not myeloid, differentiation was observed. Erythroid progenitors failed to develop normally, showing retarded progression through the cell cycle, accumulation during G0/G1 stage, and marked increase in apoptosis in response to erythroid growth factors. GATA2, a factor essential for early erythropoiesis, was significantly reduced in Dot1L-deficient cells, whereas expression of PU.1, a transcription factor that inhibits erythropoiesis and promotes myelopoiesis, was increased. These data suggest a model whereby DOT1L-dependent lysine 79 of histone H3 methylation serves as a critical regulator of a differentiation switch during early hematopoiesis, regulating steady-state levels of GATA2 and PU.1 transcription, thus controlling numbers of circulating erythroid and myeloid cells.
Introduction
Among the first differentiated cell types to emerge in the developing mammalian embryo are the blood cells. In the mouse, the process of blood development, hematopoiesis, begins at approximately embryonic day 7.0-7.5 (E7.0-E7.5), when cells originating in the primitive streak migrate to the site of yolk sac formation.1 By E7.5, the cells coalesce into blood islands, where they mature, proliferate, and differentiate.2 These early hematopoietic progenitors, termed primitive erythroid colony-forming cells, are nucleated red cells, which express primitive globins and can carry oxygen to nourish the developing embryo on the initiation of blood flow after E8.5.1,3–5 The presence of these primitive progenitors is transient, peaking in numbers at E8.0 and disappearing by E9.0,2 whereas the progeny erythrocytes persist throughout gestation.6 After E8.5, a second wave of hematopoietic progenitors emerges from a variety of sites, including the vasculature about the aorta-gonad-mesonephros and the yolk sac. These cells enter the circulation and migrate to the developing fetal liver. There, they proliferate and undergo “definitive” maturation, giving rise to multiple adult hematopoietic lineages, including mature, enucleated erythrocytes.7 This multistep process of hematopoiesis and the fate decisions of the developing cells are regulated by the precisely controlled, sequential induction and silencing of gene expression in response to a variety of growth and differentiation factors.8 The identity of the cell-type specific genes that direct differentiation, the factors controlling their expression, and the mechanisms by which their expression is regulated are currently active areas of investigation.
Although many mechanisms probably contribute to the coordination of gene expression during hematopoiesis, it is becoming increasingly clear that epigenetic change is an important strategy used by eukaryotes to accomplish such complex regulation.9–12 One of the most widely studied regulators of epigenetic change is the enzymatic modification of histones. In eukaryotic cells, histones and histone-associated proteins along with DNA are packaged into nucleosomes, which are the fundamental repeating structural units of chromatin.13 The histones can be posttranslationally modified in a variety of ways, including acetylation, phosphorylation, ubiquitination, and methylation. These modifications influence chromatin structure, facilitate interactions between nucleosomes, recruit nonhistone proteins to the DNA, and can potentially regulate transcription.13,14
Histone methylation has been shown to play an important role in regulating both chromatin function and gene expression.15 Most histone lysine methyltransferase (HLMT) family members contain a conserved SET domain, which is required for enzymatic activity and is believed to be a signature motif for this class of enzymes.13,14,16 One member of this family that is an exception to this rule is disruptor of telomere silencing 1 (Dot1), an HLMT first identified in Saccharomyces cerevisiae.17 Unlike other HLMT, Dot1, as well as its mammalian homologue DOT1-like (DOT1L), lacks a SET domain.13,17 Further, the substrate residue for Dot1/DOT1L is lysine 79 of histone H3 (H3K79), which is not located on the N-terminal tail of H3, but rather is located within a loop in the globular domain of the histone that is exposed on the nucleosome surface.15 These unique aspects of DOT1L may underlie the distinctive biologic roles that have been identified for this enzyme relative to other HLMTs.13,14,18,19 The consequences of H3K79 methylation are incompletely understood, but this histone modification has been shown to be strongly associated with actively transcribed chromatin in several mammalian cell lines.20
In a recent study, mice were generated in which DOT1L activity was disrupted by targeted mutation. This study focused on the embryonic development of Dot1L knockout (KO) mice, DOT1L function in embryonic stem (ES) cell growth, and aberrant telomere elongation in Dot1L KO ES cells.21 Defects in yolk sac angiogenesis were reported, but no alterations in hematopoiesis were described. In other studies, however, H3K79 methylation was observed in the β-globin locus, suggesting a possible role for this histone modification in erythropoiesis.20,22,23 To date, no detailed molecular level studies of the role of DOT1L in hematopoiesis have been carried out.
Here, we report the analysis of a mouse line in which a gene trap has been used to abrogate DOT1L activity. Our results confirm that DOT1L function is essential for embryogenesis, with embryonic lethality occurring at mid-gestation in Dot1L-deficient mice. Notably, we observed a defect in early erythropoiesis as a primary contributor to this embryonic lethality. By examining gene expression in hematopoietic progenitor cells of these embryos, we found that 2 genes critical for early hematopoietic fate decision, GATA2 and PU.1, were differentially affected by the loss of DOT1L function. This work highlights the importance of H3K79 methylation in embryogenesis and has begun to shed light on the molecular basis of DOT1L function in embryonic hematopoiesis.
Methods
Generation of Dot1L KO mice
A mouse ES cell line (Sequence Tag, RRR032) that contains a gene trap integration within exon 13 of the Dot1L gene and a β-geo selection cassette was obtained from Bay Genomics. Dot1L-mutant mice were generated per standard protocols according to blastocyst injection and mouse breeding protocols from Bay Genomics (http://baygenomics.ucsf.edu/protocols/comp1/blastocyst.html). Dot1L heterozygous (Dot1L+/−) F1 mice were generated by breeding chimeras with C57Bl/6 females (The Jackson Laboratory). Stocks of heterozygous mice were maintained by continuous backcrossing to C57Bl/6 stocks.
Preparation of single yolk sac cell suspension
E8.5 or E10.5 embryos were obtained from timed mating between heterozygous Dot1L-mutant mice. Yolk sacs were incubated in 0.1% collagenase (StemCell Technologies Inc)/phosphate-buffered saline (PBS)/20% fetal bovine serum at 37°C for 30 minutes. The digested yolk sacs were aspirated through 25- and 30-gauge needles sequentially and then filtered through a 70-μM nylon mesh (BD Biosciences). Genotyping was performed on DNA isolated from corresponding embryo tissues.
Culture of mouse embryonic fibroblasts
E10.5 embryos were dissociated by aspiration through a 25-gauge needle. Cells were incubated in 12-well plates at 37°C in complete Dulbecco modified Eagle medium (DMEM)/10% fetal calf serum (FCS) until confluent. Mouse embryonic fibroblasts (MEFs) were then incubated in DMEM/0.1% FCS for 72 hours, restimulated with DMEM/20% FCS for 24 hours, and harvested for analysis.
Extraction of histones from both starved and restimulated MEFs
MEFs were washed twice with ice-cold PBS plus 5mM sodium butyrate then trypsinized and resuspended (107 cells/mL) in Extraction Buffer (TEB: 0.5% Triton X-100 [vol/vol], 2mM phenylmethylsulfonyl fluoride, 0.02% [wt/vol] NaN3 in PBS). The MEF/TEB suspensions were incubated on ice for 10 minutes and centrifuged at 800g for 10 minutes at 4°C. The supernatant was discarded, and the pelleted nuclei were washed in one-half the volume of TEB and centrifuged as before. The nuclear pellets were resuspended in 0.2N HCl at a density of 4 × 107/mL at 4°C overnight. The samples were centrifuged (800g), and supernatant was analyzed. Protein content was determined by the Bradford assay (Bio-Rad), and aliquots were stored at −80°C.
Western blot analyses
Western blot analyses of the extracted histones were performed with enhanced chemiluminescence following standard protocols. Antibodies against mono-methyl (ab2886), di-methyl (ab3594), and tri-methyl H3K79 (ab2621) were obtained from Abcam Inc. Antibodies against unmodified histone H3 and methylated H3K4, H3K9, H3K27, and H3K36 were all obtained from Cell Signaling Technologies.
Whole-mount immunolabeling for fluorescence
Embryonic membranes were isolated from timed matings at E10.5. Yolk sacs were fixed with 3% paraformaldehyde for 5 minutes and then transferred to fresh 3% paraformaldehyde for 10 minutes. Yolk sacs were permeabilized in PBS containing 0.2% Triton X-100 for 30 minutes. Nonspecific binding was blocked by incubation with 3% BSA plus 5% donkey serum in PBS (1 hour at 20°C × 2). Tissues were then incubated with anti–platelet endothelial cell adhesion molecule 1 (CD31; BD PharMingen) at 4°C overnight, then washed 5 times in 0.5 mL of blocking solution for 1 hour each. A fluorescein isothiocyanate–coupled donkey antirat secondary antibody (1:75 dilution in blocking solution, 4°C overnight) was used for detection (Jackson ImmunoResearch Laboratories Inc). The yolk sacs were then mounted onto glass slides with the use of an antiphotobleaching medium for immunofluorescence detection.
Analysis of primitive and definitive erythropoiesis
Primitive erythropoiesis was measured by performing colony-forming assays as described.2 Briefly, cells (3 × 104) from E8.5 embryo and yolk sac were plated in methylcellulose containing 10% plasma-derived serum (Antech Inc), 5% protein-free hybridoma medium (Gibco-BRL), and cytokines KL (100 ng/mL) and erythropoietin (2 U/mL; PeproTech Inc). Colonies were analyzed for identity, size, and number after 5-7 days of in vitro culture.
To assess definitive erythropoiesis, erythroid burst-forming unit (BFU-E) and granulocyte, macrophage colony-forming unit (CFU-GM) assays were performed. Dissociated cells (5 × 104) from each E10.5 yolk sac were plated in M3434 methylcellulose medium (StemCell Technologies) in 35-mm culture dishes. In some experiments, cells were plated on medium (M3334) that only sustained the growth of erythroid CFU and mature BFU-E colonies. These cells were cultured at 37°C and scored according to the manufacturer's recommendations. Single-colony area was measured with ImageJ software (National Institutes of Health).24
Cell sorting with c-kit and analysis of c-kit+ cells
Single-cell suspensions from yolk sacs were incubated for 30 minutes at 4°C with anti–c-Kit antibody conjugated to phycoerythrin cyanine 5.5 (eBioscience Inc). The c-kit+ cells were isolated by cell sorting with the use of a BD FACSAria cell sorter (BD Biosciences). Sorted cells (1.5 × 104) were cultured, and colony growth was analyzed as described in “Analysis of primitive and definitive erythropoiesis.”
RNA extraction and real-time polymerase chain reaction
Total RNA from sorted, c-kit+ yolk sac cells were extracted with the use of TRIzol reagent (Invitrogen) according to the manufacturer's instructions and used to make cDNA with the use of the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Sequences of real-time polymerase chain reaction (PCR) primers for candidate genes were obtained from Primer Bank (http://pga.mgh.harvard.edu/primerbank/). Quantitative, real-time PCR was performed and analyzed with the 7500 real-time PCR system (Applied Biosystems). Fold change was calculated with the Δ-Δ Ct method.25
Apoptosis analysis and cell-cycle analysis
Single-cell suspensions from E10.5 yolk sacs were cultured in M3334 medium at 37°C for 4 days for the apoptosis analysis and 3 days for cell-cycle studies. For apoptosis, 1 × 105 cells from each sample were stained with annexin V–phycoerythrin (BD Biosciences) and Topro 3 (Molecular Probes), following the instructions of the manufacturer. For cell-cycle studies, the cells were stained with propidium iodide. All analyses were performed by flow cytometry with the use of a FACSCalibur (BD Biosciences).
G1E cell culture and chromatin immunoprecipitation of the Gata2 and Sfpi1/Pu1 loci
G1E cells were provided by Dr Mitchell Weiss (University of Pennsylvania) and maintained according to published protocols.26 Chromatin immunoprecipitation (ChIP) assays were performed as described.27 ChIP antibodies included rabbit polyclonal anti–histone H3 mono-methyl K79 (H3K79me), di-methyl K79 (H3K79me2), and tri-methyl K79 (H3K79me3; Abcam Inc). Normal rabbit serum (BioSource International) was used as negative control. After immunoprecipitation, real-time PCR was performed to quantify precipitated DNA with the use of the 7500 real-time PCR system. Primer pairs for analyzing the Gata2 locus were designed with MacVector software (MacVector Inc). Pu1/Sfpi1 primer pairs were previously described.28 Sequences can be found in supplemental Figure 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Results
Generation of mouse lines containing mutant allele of Dot1L
To generate an in vivo model of DOT1L function in the mouse, an ES cell line was obtained from Bay Genomics with an insertion mutation at the Dot1L locus. Four primers were designed to determine the exact position of the gene trap insertion. Three forward primers were within intron 11 of the Dot1L gene (F1, F2, F3), whereas the reverse primer was within the inserted gene trap (R1) (Figure 1A). Sequence analysis identified the gene trap cassette within exon 13 after the 26th nucleotide. This position is 5′ to the region encoding the critical nucleosome binding region of DOTL1 (amino acids 390-407), which is essential for DOT1L enzymatic activity.20 Thus, insertion in this area would be predicted to result in a substantial disruption of DOT1L function. Indeed, this gene trap has been used recently to analyze DOT1L loss of function in fibroblasts.20 Embryos were generated with the gene-trapped stem cells, and genotyping PCR primers were used to distinguish wild-type (WT), Dot1L heterozygous mutant, and KO mice and embryos (Figure 1B).
Figure 1.

Generation of mice with mutant Dot1L alleles. (A) Schematic representation of the gene trap construct used to generate the mutant Dot1L alleles (KO). The numbered boxes represent exons of the Dot1L gene. Arrows represent PCR primers used to amplify DNA from ES cells to identify the exact point of insertion of the gene trap. (B) PCR genotyping with the use of DNA from embryos generated by interbreeding heterozygous Dot1L mutant mice. PCR primers were generated that distinguished WT and KO alleles. As expected, both alleles were detected in heterozygous embryos (lane 3). (C) Absence of H3K79 methylation in MEFs from Dot1L−/− embryos. MEFs were derived from E10.5 embryos and either serum-starved for 24 hours (left bands) or treated overnight with 20% FCS (right bands). Histones were acid-extracted from cells, and proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Blots were probed with antibodies against histone H3 lysine 79 mono- (H3K79me), di- (H3K79me2), and tri-methylation (H3K79me3). Other antibodies against histone H3 modifications were used to probe the blot: unmodified histone H3 (H3), dimethylated lysine 4 (H3K4me2), dimethylated lysine 9 (H3K9me2), dimethylated lysine 27 (H3K27me2), and lysine 36 (H3K36me2).
DOT1L deficiency causes lethal, embryonic anemia
To assess the functional consequences of Dot1L loss, MEFs were derived from WT, heterozygous, and homozygous mutant (KO) E10.5 embryos. In MEFs from Dot1L-KO embryos, there was no significant expression of Dot1L mRNA (supplemental Figure 2). Because antibodies were not available to measure protein expression, DOT1L activity was assessed. Histones were prepared from each genotype, and immunoblot analysis showed that mono-, di-, and tri-methylation of H3K79, the reported substrate for DOT1L,15 were completely abrogated in the KO MEFs, whereas in both WT and heterozygous MEF, H3K79 methylation was unaffected (Figure 1C). The patterns of H3K79 methylation were not influenced by either the presence or absence of serum (Figure 1C compare left with right panels), conditions known to influence histone methylation.29 Other H3 lysine methylation marks, including dimethylation of K4, K9, K27, and K36, were unaffected in the mutant MEFs (Figure 1C). These results confirm that DOT1L is specific for H3K79 and is the only detectible H3K79 methyltransferase during embryogenesis, consistent with previous reports.15,21
The Dot1L+/− mice appear phenotypically normal and fertile. However, on examination of > 150 pups, no viable Dot1L KO offspring were produced after breeding heterozygotes (Table 1), indicating that Dot1L deficiency resulted in embryonic lethality. To identify the timing and the cause of the Dot1L KO-dependent lethality, WT, heterozygous, and KO embryos were examined at several time points during development. The genotype distribution at E9.5 occurred at expected Mendelian ratios, and there were no distinguishable phenotypic differences among WT, heterozygous, and KO embryos harvested from the litters at this time point. At E10.5, the distribution of viable embryos was close to the expected Mendelian ratios, but most KO embryos were reduced in size compared with WT littermates (Figure 2A-B). In addition, Dot1L KO embryos exhibited a striking near absence of red blood cells, which resulted in an overall paler appearance than the WT embryos (Figure 2A-B).
Table 1.
Survival
| E10.5 | E11.5 | E13.5 | P21 | |
|---|---|---|---|---|
| Total embryos | 227 | 40 | 18 | 151 |
| Dot1L+/+ (WT) | 61 (27%) | 14 (35%) | 4 (22%) | 53 (35%) |
| Dot1L+/− (Het) | 114 (50%) | 19 (48%) | 12 (67%) | 98 (65%) |
| Dot1L−/− (KO) | 52 (23%) | 7 (18%) | 0 (2)* (11%) | 0 (0%) |
Embryos were dead but not resorbed.
Figure 2.
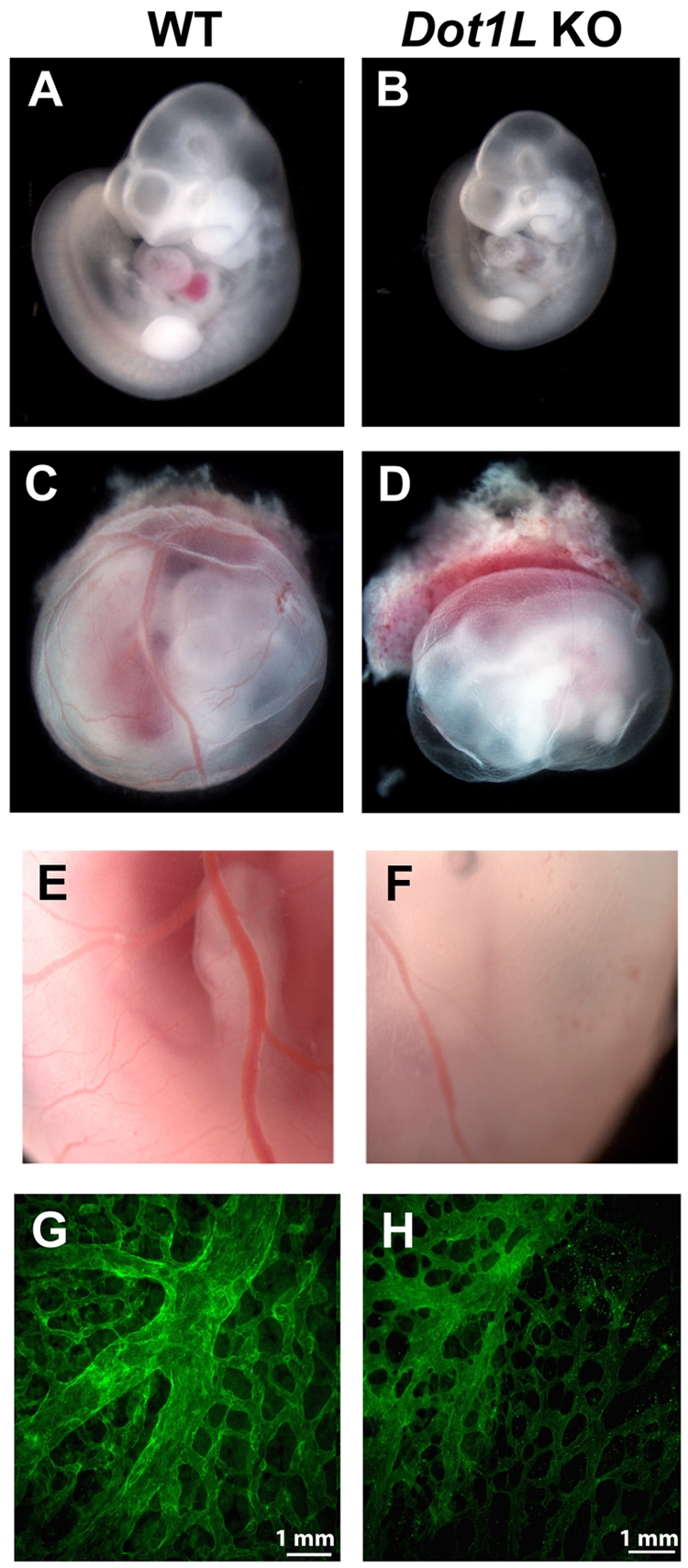
Defects in hematopoiesis and vascular development in Dot1L KO embryos. (A-B) Representative pictures of WT (A) and Dot1L KO (B) embryos at E10.5, showing reduced embryo size as well as significant anemia in the Dot1L KO embryos. Note the overall pale appearance of the KO embryo. (C-D) Representative pictures of whole-mount WT (C) and Dot1L KO (D) embryos with surrounding yolk sac show that yolk sacs from Dot1L KO contain less blood and exhibit a reduction in the number of vessels compared with those from WT. (A-D) Embryos were viewed with the use of a NIKON SMZ800 stereo microscope with Plan 1× objective lenses (total 10×), and images were taken with a LEICA DFC480 camera with the use of Leica Firecam software at room temperature. (E-F) Representative enlarged pictures (20×) show that blood vessels in KO yolk sacs exhibit abnormal caliber and contain less blood compared with WT. (G-H) Representative immunofluorescence confocal images of E10.5 yolk sacs stained with anti-CD31 show a mild defect in vascular remodeling in the Dot1L KO embryos (H). Pictures are representative of 7-10 individual embryos. Pictures were viewed with an Olympus 1 × 71 microscope with 10× UPLANF1 objective lens, and images were taken by Olympus DP71 camera with the use of Olympus DP71 controller software. The fluorochrome was fluorescein isothiocyanate.
Examination of the yolk sacs showed that those from KO embryos also contained less blood and exhibited altered vessels in comparison to the yolk sacs from WT embryos (Figure 2C-F). Immunofluorescence analysis of WT and KO yolk sacs with the use of anti–platelet endothelial cell adhesion molecule (CD31) showed that both contained abundant endothelial cells, which constitute the complex vascular plexus network present at this time. In WT yolk sacs at E10.5, progression of angiogenic remodeling was observed, as indicated by the large caliber vessels of the vitelline artery and vein. In Dot1L KO yolk sacs, the remodeling process was present; however, the vessels appeared disorganized and exhibited relatively deficient vascular branching, similar to the Dot1L-deficient yolk sacs described by Jones et al21 (Figure 2G-H).
At E11.5, the percentage of viable Dot1L KO embryos was much lower than the expected Mendelian ratio, and no Dot1L-mutant embryos survived at or beyond E13.5 (Table 1). Collectively, these data show that the Dot1L KO embryos do not survive past mid-gestation, a phenomenon probably due to the severe anemia and associated defects in vascular remodeling.
DOT1L controls yolk sac erythropoiesis
The severe anemia observed in the KO embryos (Figure 2) suggested that DOT1Ls is essential for hematopoiesis. Because it is thought that a primitive erythroid defect would contribute to anemia observed before E13,7,30,31 we first investigated whether the loss of DOT1L affects the formation of primitive erythroid progenitor cells. At E10.5, most circulating, differentiated erythroid cells should be derived from primitive erythropoiesis.7 A nonquantitative analysis of blood smears at this time point showed normal-appearing, nucleated erythroid cells in the Dot1L KO embryos of various stages of maturation (supplemental Figure 3). We carried out more quantitative CFU assays to determine the ability of primitive progenitors to proliferate and form colonies in response to growth factors. With the use of E8.5 embryos and yolk sacs as a source of primitive erythroid progenitors, we found that the Dot1L KO cells gave rise to significantly fewer colonies when we applied equal numbers of the cells onto each culture dish (Figure 3A). In addition, with the use of image analysis, we observed that Dot1L KO colonies were only approximately one-half the average size of WT or heterozygous colonies (Figure 3BG-H; P < .01).
Figure 3.

Loss of DOT1L results in the impairment of both primitive and definitive yolk sac erythropoiesis. (A-B) Identical numbers of dissociated E8.5 yolk sac cells were cultured for 7 days in media specifically supporting the growth of primitive erythroid colonies. Colonies were counted (A) and sizes were measured (B) with ImageJ software (National Institutes of Health). (C-D) Identical numbers of dissociated E10.5 yolk sac cells were cultured in methylcellulose growth medium, which supports both erythroid and granulocyte-macrophage progenitors. Colonies were scored and counted (C) after 10-12 days in culture. (D) The areas of multiple, single BFU-E and CFU-GM colonies were measured to estimate colony size with ImageJ software (National Institutes of Health). (E) Identical numbers of dissociated E10.5 yolk sac cells were plated in methylcellulose medium, which only supports the growth of erythroid progenitors (erythroid CFUs and mature BFU-Es). Colonies were identified and counted after 3 days of culture. (F) C-kit+ cells were isolated from individual yolk sacs by sorting. Identical numbers from each genotype were cultured in methylcellulose medium supporting both erythroid and myeloid lineages for 10 days. Total erythroid (BFU-E) and myeloid (CFU-GM) colonies were counted. (G-L) Representative images of primitive erythroid (G-H), BFU-E (I,J), and CFU-GM (K-L) colonies showing size and appearance differences between WT (G,I,K) and Dot1L KO (H,J,L). (G-H) Derived from experiments described in Figure 3A and B. (I-L) From the experiment described in Figure 3C and D. Cultures were viewed with a NIKON ECLIPSE TS100 microscope equipped with 10×-20× phase contrast, numerical aperture 1.2 objective lens. Images were taken with a LEICA DFC480 camera with the use of Leica Firecam software at room temperature. *Average number (A,C,E) and size (B,D,F) of KO colonies were significantly different from WT and heterozygotes (P < .01, t test). Error bars represent standard errors within each group.
We next analyzed definitive hematopoiesis at E10.5, when the anemic phenotype of Dot1L KO embryos is obvious, but embryonic viability remains unaffected. At this time, the fetal liver is not yet completely developed, and a substantial reservoir of embryonic hematopoiesis occurs in cells derived from the yolk sac.2,7,32 Yolk sacs from E10.5 embryos were dissociated and plated under conditions that support multipotent, erythroid and myeloid progenitor cell differentiation. The number of definitive erythroid progenitors was measured by counting BFU-E colonies, which have a characteristic structure under light microscopy (Figure 3I). The total number of BFU-E colonies obtained from KO yolk sac cells was much lower than the number obtained from WT or heterozygous embryos (Figure 3C). In contrast, the number of CFU-GMs was not significantly different among WT, heterozygote, and KO yolk sacs (Figure 3C). The few BFU-E colonies generated from Dot1L KO yolk sacs were approximately 3 times smaller than WT colonies, suggesting that Dot1L-deficient erythroid progenitors are defective in their ability to optimally proliferate in response to growth factors in the media (Figure 3D,I-J; P < .01). No significant difference in the size of the CFU-GM colonies was observed among the different genotypes (Figure 3D,K-L).
We next plated equal numbers of dissociated yolk sac cells on media that only supports the outgrowth of erythroid CFU colonies (Figure 3E). Under these conditions, significantly fewer KO erythroid colonies were derived than from either WT (P < .01) or heterozygous (P < .02) cells. The slight difference between WT and heterozygous colony numbers was not statistically significant (P = .38). Primitive and definitive WT and Dot1L KO erythroid colonies were all positive for benzidine staining, indicating normal globin production in the KO cells. However, the staining was less intense in KO colonies, because of relatively lower cell numbers in each colony (supplemental Figure 4). We also collected cells from primitive and definitive erythroid colonies and analyzed them by Giemsa staining. No gross structural differences between WT and Dot1L KO were observed at a single-cell level (supplemental Figure 5).
The observed defect in erythropoiesis could be from either a reduction in the relative number of erythroid progenitors or a reduction in the developmental potential of these cells that are present. Hematopoietic progenitors in the yolk sac are enriched within the CD41+/c-kit+ (CD117) population.33 Thus, we analyzed CD41+/c-kit+ frequency in E10.5 embryos. No significant differences were observed in the relative percentages of these populations between WT and Dot1L KO yolk sacs, although the absolute numbers of cells were lower (supplemental Figure 6). Likewise, we detected no differences in the percentages of total CD45+/c-kit+ cells in yolk sacs (supplemental Figure 7). Therefore, the definitive progenitors in Dot1L KO yolk sacs emerge relatively normally.
C-kit is expressed by most common hematopoietic progenitor cells,34 including those that give rise to erythrocytes. Identical numbers of sorted, c-kit+ yolk sac cells from each genotype were cultured under conditions that support both erythroid and myeloid differentiation. After 10 days of culture, no BFU-E colonies developed from KO, c-kit+ yolk sac cells, whereas myeloid colonies (CFU-GM) formed normally (Figure 3F). In contrast, c-kit+ progenitors from WT and heterozygous tissues formed BFU-E and CFU-GM colonies as expected.
Dot1L deficiency blocks cell-cycle progression and promotes apoptosis of hematopoietic progenitor cells
Defective erythropoiesis in the Dot1L KO could result from either an alteration in cell-cycle progression or increased apoptotic death of progenitor cells or both. To distinguish between these possibilities, we plated cells from individual E10.5 yolk sacs on erythroid growth media for development into erythroid colonies. After 4 days of growth, we harvested the cells and analyzed their cell-cycle profile by propidium iodide staining and flow cytometry. An analysis of 7 WT and 8 KO embryos showed a significant defect in the KO cells' progression through the cell cycle. We found that in WT colonies, an average of 66% of cells were in G0/G1 stages, whereas 20% and 14% of cells were in the S and G2/M stages, respectively. In sharp contrast, 82% of KO cells were found to be in the G0/G1 stages, whereas 10% and 8% of the cells had progressed to S and G2/M stages (Figure 4A).
Figure 4.

Defects in cell-cycle progression and increased apoptosis caused by Dot1L KO during erythropoiesis. (A) Representative histograms showing cell-cycle progression of cultured WT and KO yolk sac cells. Dissociated yolk sac cells of WT and Dot1L KO embryos were plated in methylcellulose growth medium, which supports erythroid lineage growth, and were cultured for 4 days. The cells were harvested, and cell cycle was analyzed by propidium iodide staining and flow cytometry. Differences in percentages of cells in all stages of the cell cycle were significantly different (P < .02, t test). (B) Analysis of apoptosis was examined in yolk sac cells cultured in erythropoietin-containing growth medium by annexin V staining and flow cytometry.
We also examined apoptosis by annexin V staining in E10.5 yolk sac cells. After 3 days of culture in erythroid growth media, Dot1L KO cells showed significantly more apoptosis than either WT or heterozygous cells (Figure 4B). These data indicate that DOT1L loss in yolk sac–derived, erythroid progenitors induces both an increased G0/G1 period and an increased susceptibility to apoptosis. We postulate that the requirement for DOT1L in timely cell-cycle progression and survival of the erythroid lineage may be partly responsible for the reduced number and size of erythroblast colonies generated by KO yolk sacs.
DOT1L controls GATA2 and PU.1/Sfpi1 transcription
To more precisely understand the mechanism by which DOT1L affects erythropoiesis, the expression levels of genes known to play an important role in erythroid development were examined in sorted, c-kit+ WT and KO E10.5 yolk sac cells. We observed no significant changes in the expression of globin genes in Dot1L-deficient cells (supplemental Figure 8), consistent with positive benzidine staining of KO erythroid colonies (supplemental Figure 4). Remarkably, among the genes profiled by reverse transcription–PCR, only 4 were significantly affected by DOT1L loss: GATA2, which was reduced; and PU.1, SCF, and TRAP220, which were all increased (Figure 5A-B). The c-kit+, hematopoietic progenitors showed a consistent elevation in PU.1 levels relative to WT (Figure 5A), whereas GATA2 expression was decreased by ∼ 40% (Figure 5A). These data suggest that the maintenance of a normal ratio of GATA2 to PU.1 is influenced by H3K79 methylation and are consistent with the reported reciprocal expression and functional relationship between GATA2 and PU.1 within hematopoietic progenitors.27,35–37
Figure 5.

Gene expression analysis of WT and Dot1L KO progenitor cells in yolk sac. (A-B) mRNA was extracted from c-kit+ yolk sac progenitor cells sorted by fluorescence-activated cell sorting. Reverse transcription was performed, and genes known to be important in erythropoiesis were assessed by quantitative reverse transcription–PCR. mRNA was obtained from c-kit+, sorted cells from 3 to 10 individual yolk sacs. (*P < .01, t test).
H3K79 methylation in the Gata2 and PU.1/Sfpi1 loci
Because DOT1L loss affected GATA2 and PU.1 expression, we sought to determine the H3K79 methylation status within each locus with the use of ChIP analysis. To accumulate chromatin sufficient for this analysis, we used G1E cells,26 a proerythroblast line that expresses high levels of GATA2 and low levels of PU.1 (supplemental Figure 9). We found that in the actively transcribed Gata2 locus, H3K79 mono-, di-, and tri-methylation are all enriched at the 1S promoter, the first exon after 1S, the 1G promoter, the first exon after 1G, and intron 4, which contains an important regulatory element38–40 (Figure 6A). In the silenced Pu1/Sfpi1 locus, we found that only mono-methylation was enriched in regulatory elements and in the promoter (Figure 6B). Although these results do not show that H3K79 methylation directly regulates transcriptional activity of either Gata2 or Pu1/Sfpi1, they do correlate with the expression of these genes, suggesting a role for the histone mark in the regulation of their transcription. These results are in accord with previous studies showing that H3K79 di- and tri-methylation are associated with regions of active gene expression, whereas mono-methylation is indicative of gene regulatory elements.20
Figure 6.
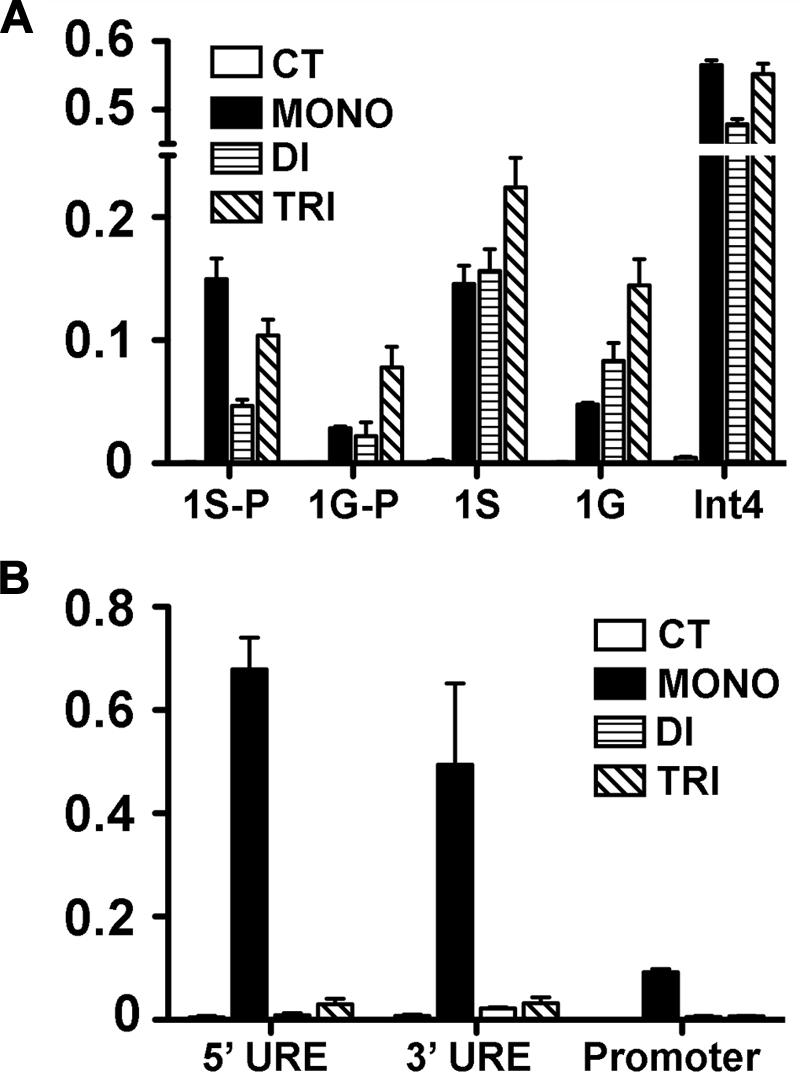
H3K79 methylation in the Gata2 and Pu1/Sfpi1 loci. ChIP was performed on chromatin from the G1E cell line to assess H3K79 mono-, di-, and tri-methylation of promoter and regulatory sites of Gata2 and Pu1/Sfi1 loci in G1E cells. At the Gata2 locus (A), which is actively transcribed, the 1S and 1G promoters (1S-P, 1G-P) and the first exon following each promoter (1S, 1G), as well as a regulatory element in intron4 (Int4), were examined. At the silenced Pu1/Sfi1 locus (B), we examined 2 upstream regulatory elements (UREs), 5′URE and 3′URE, as well as the promoter region. The data represent the mean of 3 independent experiments.
Discussion
Given the special position of lysine 79 on histone H3 and the unique structure of its methyltransferase-DOT1L, we sought to examine the effects of a germline KO of the Dot1L gene on embryonic growth and development. While these studies were ongoing, another group showed that DOT1L loss resulted in embryonic lethality.21 That group found that by E10.5, the viability of Dot1L KO embryos was below the expected Mendelian ratio and that no KO embryos survived beyond this time. The lethality in these embryos was mostly ascribed to defective yolk sac angiogenesis. We too observed similar defective yolk sac angiogenesis in Dot1L−/− embryos (Figure 2). However, because we also observed a selective and profound defect in erythropoiesis, we propose that the vessel-remodeling defects observed in the Dot1L-mutant extraembryonic tissues are largely a consequence of altered hematopoiesis and reduced blood flow in these embryos. Indeed, an essential role for blood flow and hemodynamic forces in driving angiogenic remodeling of the yolk sac has been shown previously.4
Although Dot1L deficiency also resulted in embryonic lethality in our hands, our embryos survived somewhat longer than was reported in the study mentioned earlier. In contrast to that study, we found that at E10.5 embryo viability appeared normal. However, most KO embryos were reduced in size compared with WT littermates, and no Dot1L-mutant embryos survived at or beyond E13.5. We believe this discrepancy may be due to the different methods used to generate the mutant embryos. The mutant described by Jones et al21 was generated by deletion of exons 5 and 6 of Dot1L, whereas the gene trap mutation used in our studies resulted in an interruption of the entire 3′ of the gene, after exon 13.
Despite the differences in the generation of the KO, both Jones et al21 and our findings show that Dot1L KO results in the absence of H3K79 mono-, di- and tri-methylation. Notably, the conclusion of Jones et al21 about the absence of mono-methyl H3K79 was based on mass spectrometry rather than immunoblot analysis, which was the basis of our conclusion. This was because the mono-methyl H3K79 antisera reportedly cross-reacted with histone H3 in their hands. In our studies, this antisera worked well for Western blotting, was specific, and had minimal cross-reactivity with histone H3 (Figure 1C). The reason for this difference is not clear; however, it may be related to batch-to-batch variability of this polyclonal rabbit antisera or variability in secondary antibodies or means of detection.
Another group also examined DOT1L loss of function with the use of the same gene trap used in our study.20 However, we have determined that the gene trap insertion is within exon 13, rather than in intron 12, as was previously suggested.20 In agreement with our results, that study also reported specificity of the mono-methyl H3K79 antisera without cross-reactivity with H3 by immunoblot. In contrast to our findings, that group concluded that mono-methyl H3K79 was increased in the Dot1L KO cells. This conclusion was based on ChIP analyses, which showed enriched mono-methyl H3K79 in certain loci. The reason for this difference is not certain, but it is tempting to speculate that this result could reflect some level of cross-reactivity of the antisera with formalin-fixed H3 in the immunoprecipitations.
Primitive versus definitive erythropoiesis
Our results clearly show that Dot1L deficiency results in a significant defect in erythrocyte development, manifested by the severe anemia observed in E10.5 Dot1L−/− embryos. At distinct times in development, the yolk sac is a source of progenitor cells for both primitive and definitive erythropoiesis.2,7 Our colony-forming studies on cells from E8.5 and E10.5 embryos showed that DOT1L loss caused a defect in erythroid progenitors derived from both the primitive and definitive lineages. Analysis of isolated c-kit+ progenitors at E10.5 indicates that this defect resides in this cell population (Figure 3F). Although the total number of erythroid progenitors was reduced in the KO embryos, the relative percentages of these cells appeared normal (supplemental Figures 6-7). This fact, coupled with direct visualization of blood at E10.5 (supplemental Figure 3), indicates that both primitive and definitive progenitors can be produced and can mature normally. However, their common impaired expansion and survival in colony-forming assays suggests a shared deficiency or inappropriate response to erythroid growth stimuli, resulting in failure to form large colonies in vitro and anemia in vivo.36,41–44 This putative-altered response to growth factors is probably the primary cause of lethality in these embryos after E10.5. Collectively, these data indicate that DOT1L plays an essential role in the growth and differentiation of the erythroid lineage during early hematopoiesis.
GATA2 and PU.1 expression regulates erythroid formation
Expression analysis of several genes considered to be important in erythropoiesis showed only a few significant differences in c-kit+ progenitors derived from E10.5, KO yolk sac cells compared with WT cells, including down-regulation of GATA2 (Figure 5A). Whereas a ∼ 40% decrease may appear modest at first glance, it is comparable with that of Gata2+/− mice, which exhibit decreased colony-forming hematopoietic progenitors.45 In addition to the decreased GATA2, there was concomitant up-regulation of PU.1. Notably, the relative expression levels of GATA2 and PU.1 are known to be critical for determining hematopoietic progenitor cell fate: high GATA2, low PU.1 in erythroid cells; and low GATA2, high PU.1 in myeloid cells.36,44 Selective defects in erythropoiesis caused by reduced GATA2 with corresponding increases in PU.1 transcription have been previously reported.27 Another GATA family transcription factor important in erythroid fate, GATA1, remained unchanged in KO samples, although the expression levels were quite low in c-kit+ cells, consistent with published data.27
Our gene expression results are consistent with a proposed model of erythroid differentiation37 and provide us with a possible explanation for the anemic phenotype observed in Dot1L KO embryos. We propose that in the absence of DOT1L in erythroid progenitors, Gata2 transcription is significantly reduced, and at the same time there is an increase in PU.1 levels. We speculate that the reduction in GATA2 levels results in de-repression of the Pu1/Sfpi1 locus, enabling increased expression of PU.1.27,37,46 As a consequence, PU.1 function becomes predominant, a situation known to block erythropoiesis while allowing myelopoiesis.37 We did not observe an accompanying, compensatory increase in granulocyte/monocyte development. This observation is consistent with the report that a selective loss of GATA2 does not result in increased myelopoiesis at the expense of erythropoiesis.47 This implies that the effects of Dot1L loss (eg, gene expression changes, altered cell growth, etc) affect only those cells that have already committed to the erythroid lineage.
Thus, our data are consistent with a model by which DOT1L-dependent H3K79 methylation is an important regulator of the differentiation switch during early hematopoiesis, resulting in appropriate steady-state levels of GATA2 and PU.1. By maintaining the proper ratio of these 2 transcription factors at this early stage of hematopoiesis, appropriate proportions of circulating erythroid and myeloid cells are ensured. In future studies, it will be important to examine the effect of DOT1L on other epigenetic modifications, such as acetylation and methylation changes in the Gata2 and Pu1/Sfpi1 loci, and whether any observed changes in epigenetic status might affect transcriptional activation of these genes.
Supplementary Material
Acknowledgments
We thank E. Chen for critical reading of the manuscript.
This work was supported by the National Institutes of Health (grants R21AI076863, K22AI057562) and the Department of Pathology and Laboratory Medicine at the University of Kansas Medical Center.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.F., Y.Y., M.M.O., M.Z., J.N.C., J.B.J., and J.L.V. performed the research; P.E.F. and J.L.V. designed the research; and P.E.F., Y.F., T.A.F., and J.L.V. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Patrick E. Fields, Department of Pathology and Laboratory Medicine, Division of Cancer and Developmental Biology, Institute for Reproductive Health and Regenerative Medicine, University of Kansas Medical Center, 3901 Rainbow Blvd, Kansas City, KS 66160; e-mail: pfields@kumc.edu.
References
- 1.McGrath KE, Koniski AD, Malik J, Palis J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood. 2003;101(5):1669–1676. doi: 10.1182/blood-2002-08-2531. [DOI] [PubMed] [Google Scholar]
- 2.Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. 1999;126(22):5073–5084. doi: 10.1242/dev.126.22.5073. [DOI] [PubMed] [Google Scholar]
- 3.Ji RP, Phoon CK, Aristizabal O, McGrath KE, Palis J, Turnbull DH. Onset of cardiac function during early mouse embryogenesis coincides with entry of primitive erythroblasts into the embryo proper. Circ Res. 2003;92(2):133–135. doi: 10.1161/01.res.0000056532.18710.c0. [DOI] [PubMed] [Google Scholar]
- 4.Lucitti JL, Jones EA, Huang C, Chen J, Fraser SE, Dickinson ME. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development. 2007;134(18):3317–3326. doi: 10.1242/dev.02883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trimborn T, Gribnau J, Grosveld F, Fraser P. Mechanisms of developmental control of transcription in the murine alpha- and beta-globin loci. Genes Dev. 1999;13(1):112–124. doi: 10.1101/gad.13.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baron MH, Fraser ST. The specification of early hematopoiesis in the mammal. Curr Opin Hematol. 2005;12(3):217–221. doi: 10.1097/01.moh.0000163217.14462.58. [DOI] [PubMed] [Google Scholar]
- 7.McGrath K, Palis J. Ontogeny of erythropoiesis in the mammalian embryo. Curr Top Dev Biol. 2008;82:1–22. doi: 10.1016/S0070-2153(07)00001-4. [DOI] [PubMed] [Google Scholar]
- 8.Kim SI, Bresnick EH. Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene. 2007;26(47):6777–6794. doi: 10.1038/sj.onc.1210761. [DOI] [PubMed] [Google Scholar]
- 9.Ginder GD, Gnanapragasam MN, Mian OY. The role of the epigenetic signal, DNA methylation, in gene regulation during erythroid development. Curr Top Dev Biol. 2008;82:85–116. doi: 10.1016/S0070-2153(07)00004-X. [DOI] [PubMed] [Google Scholar]
- 10.Wozniak RJ, Bresnick EH. Epigenetic control of complex loci during erythropoiesis. Curr Top Dev Biol. 2008;82:55–83. doi: 10.1016/S0070-2153(07)00003-8. [DOI] [PubMed] [Google Scholar]
- 11.Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26(47):6697–6714. doi: 10.1038/sj.onc.1210755. [DOI] [PubMed] [Google Scholar]
- 12.Hu X, Ybarra R, Qiu Y, Bungert J, Huang S. Transcriptional regulation by TAL1: a link between epigenetic modifications and erythropoiesis. Epigenetics. 2009;4(6):357–361. doi: 10.4161/epi.4.6.9711. [DOI] [PubMed] [Google Scholar]
- 13.Ng HH, Feng Q, Wang H, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16(12):1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109(6):745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 15.Feng Q, Wang H, Ng HH, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Hayashizaki Y, Kone BC. Structure and regulation of the mDot1 gene, a mouse histone H3 methyltransferase. Biochem J. 2004;377(Pt 3):641–651. doi: 10.1042/BJ20030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singer MS, Kahana A, Wolf AJ, et al. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150(2):613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conde F, Refolio E, Cordon-Preciado V, et al. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics. 2009;182(2):437–446. doi: 10.1534/genetics.109.101899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25(19):8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steger DJ, Lefterova MI, Ying L, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28(8):2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones B, Su H, Bhat A, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4(9):e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Im H, Park C, Feng Q, et al. Dynamic regulation of histone H3 methylated at lysine 79 within a tissue-specific chromatin domain. J Biol Chem. 2003;278(20):18346–18352. doi: 10.1074/jbc.M300890200. [DOI] [PubMed] [Google Scholar]
- 23.Sawado T, Halow J, Im H, et al. H3 K79 dimethylation marks developmental activation of the beta-globin gene but is reduced upon LCR-mediated high-level transcription. Blood. 2008;112(2):406–414. doi: 10.1182/blood-2007-12-128983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Weiss MJ, Yu C, Orkin SH. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol Cell Biol. 1997;17(3):1642–1651. doi: 10.1128/mcb.17.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou ST, Khandros E, Bailey LC, et al. Graded repression of PU. 1/Sfpi1 gene transcription by GATA factors regulates hematopoietic cell fate. Blood. 2009;114(5):983–994. doi: 10.1182/blood-2009-03-207944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoogenkamp M, Krysinska H, Ingram R, et al. The Pu. 1 locus is differentially regulated at the level of chromatin structure and noncoding transcription by alternate mechanisms at distinct developmental stages of hematopoiesis. Mol Cell Biol. 2007;27(21):7425–7438. doi: 10.1128/MCB.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kourmouli N, Jeppesen P, Mahadevhaiah S, et al. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J Cell Sci. 2004;117(Pt 12):2491–2501. doi: 10.1242/jcs.01238. [DOI] [PubMed] [Google Scholar]
- 30.Brotherton TW, Chui DH, McFarland EC, Russell ES. Fetal erythropoiesis and hemoglobin ontogeny in tail-short (Ts/+) mutant mice. Blood. 1979;54(3):673–683. [PubMed] [Google Scholar]
- 31.Kingsley PD, Malik J, Fantauzzo KA, Palis J. Yolk sac-derived primitive erythroblasts enucleate during mammalian embryogenesis. Blood. 2004;104(1):19–25. doi: 10.1182/blood-2003-12-4162. [DOI] [PubMed] [Google Scholar]
- 32.Lux CT, Yoshimoto M, McGrath K, Conway SJ, Palis J, Yoder MC. All primitive and definitive hematopoietic progenitor cells emerging before E10 in the mouse embryo are products of the yolk sac. Blood. 2008;111(7):3435–3438. doi: 10.1182/blood-2007-08-107086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikkola HK, Fujiwara Y, Schlaeger TM, Traver D, Orkin SH. Expression of CD41 marks the initiation of definitive hematopoiesis in the mouse embryo. Blood. 2003;101(2):508–516. doi: 10.1182/blood-2002-06-1699. [DOI] [PubMed] [Google Scholar]
- 34.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life. 2009;61(8):800–830. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 35.Ling KW, Ottersbach K, van Hamburg JP, et al. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J Exp Med. 2004;200(7):871–882. doi: 10.1084/jem.20031556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai FY, Keller G, Kuo FC, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371(6494):221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 37.Zhang P, Behre G, Pan J, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU. 1. Proc Natl Acad Sci U S A. 1999;96(15):8705–8710. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grass JA, Jing H, Kim SI, et al. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol. 2006;26(19):7056–7067. doi: 10.1128/MCB.01033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khandekar M, Brandt W, Zhou Y, et al. A Gata2 intronic enhancer confers its pan-endothelia-specific regulation. Development. 2007;134(9):1703–1712. doi: 10.1242/dev.001297. [DOI] [PubMed] [Google Scholar]
- 40.Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH. Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem. 2007;282(19):14665–14674. doi: 10.1074/jbc.M700792200. [DOI] [PubMed] [Google Scholar]
- 41.Koury MJ, Bondurant MC. Maintenance by erythropoietin of viability and maturation of murine erythroid precursor cells. J Cell Physiol. 1988;137(1):65–74. doi: 10.1002/jcp.1041370108. [DOI] [PubMed] [Google Scholar]
- 42.Koury MJ, Bondurant MC. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. 1990;248(4953):378–381. doi: 10.1126/science.2326648. [DOI] [PubMed] [Google Scholar]
- 43.Yu H, Bauer B, Lipke GK, Phillips RL, Van Zant G. Apoptosis and hematopoiesis in murine fetal liver. Blood. 1993;81(2):373–384. [PubMed] [Google Scholar]
- 44.Zhang P, Zhang X, Iwama A, et al. PU. 1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000;96(8):2641–2648. [PubMed] [Google Scholar]
- 45.Rodrigues NP, Janzen V, Forkert R, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106(2):477–484. doi: 10.1182/blood-2004-08-2989. [DOI] [PubMed] [Google Scholar]
- 46.Kitajima K, Tanaka M, Zheng J, et al. Redirecting differentiation of hematopoietic progenitors by a transcription factor, GATA-2. Blood. 2006;107(5):1857–1863. doi: 10.1182/blood-2005-06-2527. [DOI] [PubMed] [Google Scholar]
- 47.Galloway JL, Wingert RA, Thisse C, Thisse B, Zon LI. Loss of gata1 but not gata2 converts erythropoiesis to myelopoiesis in zebrafish embryos. Dev Cell. 2005;8(1):109–116. doi: 10.1016/j.devcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.