

Abstract
Hypothalamic proopiomelanocortin (POMC) neurons and their peptide products mediate important aspects of energy balance, analgesia, and reward. In addition to peptide products, there is evidence that POMC neurons can also express the amino acid transmitters GABA and glutamate, suggesting these neurons may acutely inhibit or activate downstream neurons. However, the release of amino acid transmitters from POMC neurons has not been thoroughly investigated in an intact system. In the present study, the light-activated cation channel channelrhodopsin-2 (ChR2) was used to selectively evoke transmitter release from POMC neurons. Whole-cell electrophysiologic recordings were made in brain slices taken from POMC-Cre transgenic mice that had been injected with a viral vector containing a floxed ChR2 sequence. Brief pulses of blue light depolarized POMC-ChR2 neurons and induced the release of GABA and glutamate onto unidentified neurons within the arcuate nucleus, as well as onto other POMC neurons. To determine whether the release of GABA and glutamate from POMC terminals can be readily modulated, opioid and GABAB receptor agonists were applied. Agonists for μ- and κ-, but not δ-opioid receptors inhibited transmitter release from POMC neurons, as did the GABAB receptor agonist baclofen. This regulation indicates that opioids and GABA released from POMC neurons may act at presynaptic receptors on POMC terminals in an autoregulatory manner to limit continued transmission. The results show that, in addition to the relatively slow and long-lasting actions of peptides, POMC neurons can rapidly affect the activity of downstream neurons via GABA and glutamate release.
Introduction
Hypothalamic proopiomelanocortin (POMC) neurons are thought to exert their effects on energy balance, motivation, pain, and reward mainly through release of the mature POMC cleavage products α-MSH (α-melanocyte-stimulating hormone) and β-endorphin (Coll, 2007; Bertolini et al., 2009; Hegadoren et al., 2009). While these peptides are clearly important for proper function, classical amino acid transmitters may also play a significant role in POMC cell physiology (van den Pol, 2003; Meister, 2007). Previous reports have shown both GABA and glutamate release from POMC cells that have formed recurrent synapses in primary culture (Hentges et al., 2004, 2009). In addition, electron micrograph data has shown that POMC terminals make both symmetric and asymmetric synapses, suggesting both GABAergic and glutamatergic POMC terminals, respectively (Wang et al., 2001; Reyes et al., 2006). However, glutamate release from POMC neurons in an intact circuit has not been demonstrated and some studies have failed to detect GABAergic markers in POMC neurons using immunohistochemical or transgenic approaches (Ovesjö et al., 2001; Vong et al., 2011). A complete understanding of how POMC neurons affect the larger circuitry and behavior requires knowing the amino acid phenotype of these neurons. Thus, the present study was designed to determine whether POMC neurons release GABA and/or glutamate to affect neuronal activity in intact hypothalamic networks.
To examine transmitter release from POMC neuron terminals in an intact system, a combined optogenetics–electrophysiology approach was used. This approach is similar to that proposed for circuit mapping and functional studies (Petreanu et al., 2007; Atasoy et al., 2008; Kohl et al., 2011). Cell type-specific expression of the light-gated ion channel channelrhodopsin-2 (ChR2) in POMC cells was used to excite POMC neurons as a population with light while recording from downstream neurons in slice preparations. With this method, amino acid transmitter release could be observed through native connections. The results show that both GABA and glutamate are released from POMC neurons onto downstream cells within the arcuate nucleus (ARC), indicating not only the phenotype of amino acid transmitter release, but also that POMC cells can terminate within the ARC. In addition, evoked transmitter currents could be dynamically modulated through G-protein-coupled receptor activation, suggesting physiological regulation of GABA and glutamate release from POMC neurons may be another regulatory component of the POMC neuron system.
Materials and Methods
Animals.
Male and female transgenic mice expressing Cre recombinase specifically in POMC cells (POMC-Cre) were used for all experiments. Transgenic animals were produced by standard techniques (Xu et al., 2005a) and backcrossed >12 generations onto the C57BL/6 strain. Animals were 10–12 weeks of age when electrophysiological experiments were conducted. Mice were housed under controlled temperature (22–24°C) and a constant 12 h light/dark cycle. Mice were fed standard rodent chow and tap water ad libitum. All experiments met United States Public Health Service guidelines with the approval of the Colorado State University Institutional Animal Care and Use Committee.
Viral gene transfer in vivo.
POMC-Cre transgenic mice (8 weeks of age) were anesthetized with isoflurane and placed in a stereotaxic frame (David Kopf Instruments). A viral vector containing a floxed sequence for ChR2 with an mCherry tag [AAV2/9.EF1.dflox.hChR2(H134R)–mCherry.WPRE.hGH; obtained from the Penn Vector Core at the University of Pennsylvania School of Medicine, Philadelphia, PA] was unilaterally injected into the ARC of POMC-Cre mice. For each animal, a 300 nl injection (3.52E+12 GC/ml) was made using a Hamilton syringe and the following coordinates from bregma: X, −0.27 mm; Y, −1.35 mm; Z, −6.14 mm. After microinjection, the wound was sutured, and animals were left to recover for a minimum of 14, but no longer than 28 d before experiments to ensure a high level of ChR2 expression.
Immunofluorescence.
Eighteen days after AAV injection, mice were anesthetized and perfused transcardially with 4% paraformaldehyde, and brains were removed and postfixed as described previously (Hentges et al., 2009). Sagittal brain slices (50 μm) containing the ARC were prepared on a vibratome and were subsequently incubated in PBS with 3% normal goat serum and 0.3% Triton X-100. The POMC peptide adrenocorticotropin (ACTH) was detected using a rabbit anti-rat ACTH primary antibody (National Hormone and Peptide Program; 1:10,000; overnight at 4°C). Tissue was then washed in PBS and incubated with an Alexa Fluor 647-conjugated donkey anti-rabbit secondary antibody (Invitrogen; 1:400). ACTH immunoreactivity was visualized using a Zeiss 510 Meta laser-scanning confocal microscope based on the far-red fluor (633 nm excitation and 650 nm emission wavelengths), and ChR2-mCherry was visualized using 580 nm excitation and 600 nm emission filters. Colocalization of ACTH immunoreactivity and ChR2-mCherry expression was determined by counting the labeled cells in sections from two brains. Two investigators made independent counts of labeled cells in each brain to verify the results.
Electrophysiology.
Sagittal brain slices containing the ARC were prepared as previously described (Pennock and Hentges, 2011). Whole-cell voltage-clamp recordings were made at a temperature of 37°C with an internal recording solution containing the following (in mm): 57.5 KCl, 57.5 K-methyl sulfate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 0.1 EGTA, 2 ATP, 0.5 GTP, 10 phosphocreatine, pH 7.3. Recording pipettes had a tip resistance of 1.5–2.2 MΩ when filled with internal solution. Slices were maintained in artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 glucose (saturated with 95% O2 and 5% CO2). Whole-cell recordings were conducted through the use of an Axopatch 200B amplifier (Molecular Devices), and data were collected using AxographX software (Axograph) running on a Mac OS X operating system. POMC neurons expressing ChR2 were identified by the mCherry fluorophore expressed as a fusion protein on the channel. Cells were held at −60 mV for recording light-evoked postsynaptic currents (PSCs). When recording from unidentified cells within the ARC, the patched cells were within ∼100 μm of POMC-ChR2-expressing cells. Cells were excluded if the access resistance increased significantly from the initial access (6.04 ± 1.17 MΩ for unidentified cells, 4.6 ± 0.75 MΩ for ChR2-expressing POMC neurons, and 4.9 ± 0.71 MΩ for POMC neurons without ChR2). Postsynaptic currents were evoked using either a 25 or 100 ms blue light pulse delivered every 20 s to the slice from a mercury light source with a rapid shutter system (Lumen Pro 200; Prior Scientific). The light was adjusted to the minimum intensity required to evoke a consistent PSC, which was generally between 5 and 11 mW/mm2. PSCs were evoked for at least 5 min under baseline conditions to ensure a steady recording. PSCs were collected at 10 kHz and digitally filtered at 1 kHz. Data were analyzed by comparing an average of 3 min of baseline to an average of 2 min of steady-state response to drug. EPSCs and IPSCs were identified pharmacologically by blocking GABAA-mediated currents with bicuculline methiodide (BMI) (10 μm; Tocris) or AMPA- and NMDA-mediated currents with 6,7-dinitroquinoxaline-2,3(1H,4H) (DNQX) (10 μm; Sigma-Aldrich) and (+)-MK-801 (15 μm;Sigma-Aldrich). Representative recordings were plotted by minute, averaging the peak amplitude of three sweeps spaced 20 s apart.
Drugs.
Stock solutions of DNQX, (+)-MK-801, and (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593) (BIOMOL International) were prepared with DMSO at least 10,000× more concentrated than the final solution. Stock solutions of [d-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin (DAMGO) (Sigma-Aldrich), d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) (Tocris Bioscience), nor-binaltorphimine (nor-BNI) (Sigma-Aldrich), [d-Pen2,5]enkephalin (DPDPE) (Bachem), bicuculline (Tocris Bioscience), [Met5]enkephalin (Sigma-Aldrich), baclofen (Sigma-Aldrich), dynorphin A (Tocris Bioscience), naloxone (Sigma-Aldrich), (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP 55845) (Tocris Bioscience), and BMI were prepared with distilled water. All drugs were diluted to working concentrations in aCSF and applied via bath perfusion except (+)-MK-801, which was applied to the collected slices before being moved to the recording chamber.
Data analysis.
All data are presented as mean ± SEM. Comparisons between groups were made using t tests. A value of p < 0.05 was considered a significant difference.
Results
Functional expression of ChR2 in POMC cells
The selective expression of ChR2 in POMC neurons was verified in slices prepared from POMC-Cre mice that had been injected with the viral vector containing the floxed ChR2-mCherry ≥2 weeks earlier. Approximately 88% of cells expressing ChR2-mCherry also displayed ACTH immunoreactivity (ACTH-IR). Cells expressing mCherry without detectable ACTH-IR most likely represent POMC neurons with low ACTH expression since previous reports show proper expression of POMC-Cre by other methods (Choudhury et al., 2005; Xu et al., 2005a,b). Injection of the viral vector into the ARC of wild-type C57BL/6 mice yielded no detectable mCherry fluorescence (data not shown), indicating that expression is limited to cells that express Cre recombinase. Approximately 79% of ACTH-IR cells in the injected side of the ARC expressed mCherry (Fig. 1A). ChR2-mCherry was notable throughout the membranes of cell body and neuronal projections, indicating strong expression and widespread trafficking of the channel. These findings indicate specific targeting of strong ChR2 expression to POMC neurons.
Figure 1.
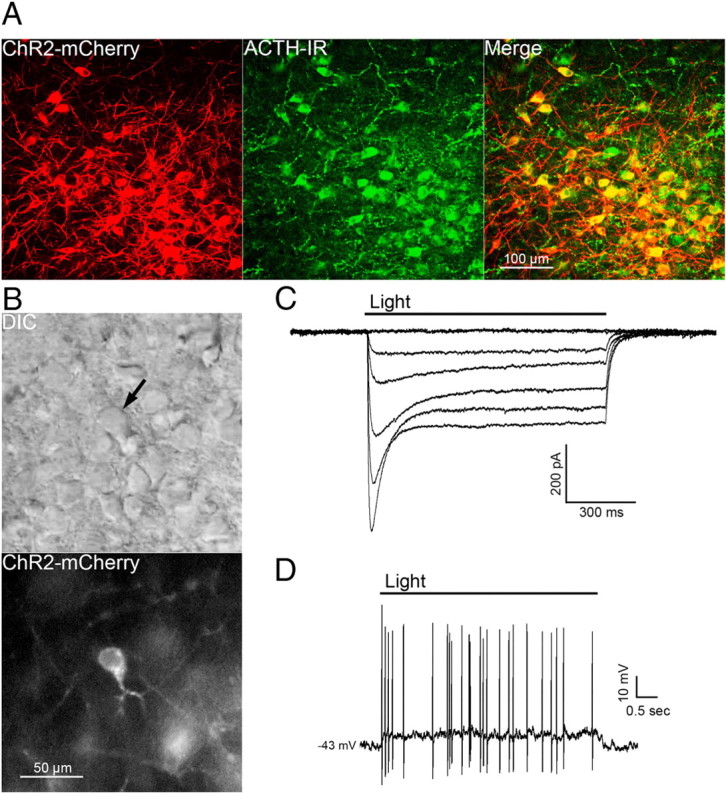
POMC cell-specific expression of functional ChR2. A, Confocal z-stack image of a brain slice containing cells expressing ChR2-mCherry 18 d after AAV injection (left panel) and cells immunoreactive for ACTH (center panel). A high degree of colocalization can been seen in the merged image (right panel). B, Images of the hypothalamus in a live brain slice from an AAV-injected mouse shown in DIC (top panel) and fluorescence (bottom panel). The arrow indicates a neuron with a high level of ChR2 expression within the focal plane. C, Whole-cell currents from a ChR2-expressing neuron elicited by pulses of blue light at increasing intensities (0 at top trace; 28 mW/mm2 elicited the largest current, bottom-most trace). D, Light-induced depolarization of a ChR2-expressing neuron caused action potential firing in current-clamp recordings.
Individual cells expressing high amounts of ChR2-mCherry were easily detected in live brain slices prepared from injected mice (Fig. 1B). POMC cells with ChR2 had similar basal whole-cell properties as POMC neurons that do not express ChR2. The input resistance was 1.3 ± 0.1 GΩ in ChR2-positive cells versus 1.6 ± 0.2 GΩ in ChR2-negative POMC neurons (n = 9; p = 0.31). Whole-cell capacitance was also not different between ChR2-expressing and -nonexpressing POMC neurons (29 ± 2.6 and 25.5 ± 1.4 pF, respectively; n = 9; p = 0.24). Whole-cell voltage-clamp recordings made in POMC-ChR2 neurons indicate that ChR2 mediates an inward current in response to blue light (∼470 nm; Fig. 1C). Peak and steady-state current responses were stereotypical of currents mediated by ChR2(H134R) (Lin et al., 2009). In current clamp, blue light depolarized POMC-ChR2 neurons by 12.4 ± 1.7 mV (n = 6; baseline membrane potential, −40 ± 1.8 mV) and caused action potential firing (Fig. 1D).
GABA and glutamate release from POMC neurons
Activation of POMC neurons via ChR2 was used to determine whether POMC cells release the amino acid transmitters glutamate and/or GABA in an intact neuronal network. Whole-cell voltage-clamp recordings were made in unidentified cells (not expressing detectable mCherry) in the ARC near POMC neurons. The unidentified cells had basal properties similar to POMC neurons (input resistance, 1.1 ± 0.15 GΩ; capacitance, 23.3 ± 1.54 pF; n = 15; p > 0.05 for both properties compared with POMC neurons with or without ChR2). The likelihood that a pulse of light evoked neurotransmitter release from POMC-ChR2 cells varied from 50 to ∼8% depending on the accuracy of the injection and the number of neurons expressing ChR2. When light pulses evoked transmitter release from POMC terminals, the currents in the postsynaptic cell were mediated by both GABAA and AMPA receptors, indicating GABA and glutamate release from POMC neurons. Light-evoked IPSCs were recorded in the presence of the AMPA receptor blocker DNQX (10 μm) (Fig. 2Ai) and were completely inhibited by the GABAA receptor antagonist BMI (10 μm) (Fig. 2Aii). Upon washout of BMI, the current returned (Fig. 2Aiii). Reversing the order of antagonist application showed the glutamatergic nature of some light-evoked currents. Currents evoked in the presence of BMI (Fig. 2Bi) were blocked by the addition of DNQX to the bath (Fig. 2Bii) and returned upon washout of DNQX (Fig. 2Biii). In the presence of BMI, the onset of evoked currents was often substantially delayed from the initial light stimulus (as seen in Fig. 2B), which may indicate a polysynaptic response. However, direct innervation was indicated in some instances, including where a brief light pulse evoked PSCs that were abolished only when both GABAA and AMPA receptors were blocked (Fig. 2Ci–Civ). Together, when a light-evoked PSC was detected in a non-POMC cell, it was most often GABAergic in nature (27 of 39 cells showed GABA-mediated PSCs) (Fig. 2D). Glutamate-mediated PSCs accounted for 23% of the PSCs observed (9 of 39 PSCs), and 3 cells appeared to receive input from both GABAergic and glutamatergic POMC cells (Fig. 2D).
Figure 2.

POMC neurons release GABA and glutamate onto cells within the arcuate nucleus. Light-evoked IPSCs observed in the presence of DNQX (10 μm) (Ai) were abolished with the addition of BMI (10 μm) (Aii). Light-evoked EPSCs observed in the presence of BMI (Bi) were abolished with the addition of DNQX (Bii). C, An example of a cell with light-evoked IPSCs and EPSCs. Upon washout of DNQX and/or BMI evoked currents returned (Aiii, Biii, Civ). The images in A and B consist of three overlaid sweeps, whereas those in C are each an average of three sweeps. The transmitter(s) mediating evoked currents was determined in all cases in which a PSC was observed and is presented in D. E, A light-evoked IPSC in a POMC cell that also displays a direct ChR2 current (top trace). The evoked IPSC is ablated by BMI (bottom trace). F, Currents evoked onto an unidentified cell are completely inhibited by treatment with TTX (1 μm).
The majority of light-evoked PSC recordings were made in non-POMC neurons, since the direct inward current carried through ChR2 can obscure the current induced by GABA or glutamate release. However, some POMC cells expressing ChR2 displayed both a direct ChR2-mediated inward current and a slightly delayed light-evoked GABA-mediated IPSC (n = 4; Fig. 2E). This result indicates that POMC neurons can innervate one another.
All light-evoked currents in cells downstream of POMC neurons could be completely blocked by BMI and/or DNQX, indicating that GABA and glutamate were the only transmitters being released in response to the acute depolarization of POMC neurons. The light-evoked release of transmitter from POMC neurons appears to be dependent on action potential propagation and not direct stimulation of terminal release based on the observation that the addition of tetrodotoxin (TTX) (1 μm) completely abolished light-evoked PSCs (98.68 ± 0.94% reduction; n = 4; p < 0.001; Fig. 2F).
Opioids inhibit neurotransmitter release from POMC terminals via μ and κ receptors
The ability of opioid receptor agonists to modulate the release of GABA and glutamate from POMC terminals was evaluated, since opioids reliably inhibit amino acid transmitter release from unidentified terminals within the ARC (Emmerson and Miller, 1999; Pennock and Hentges, 2011). The μ-opioid receptor (MOR) and δ-opioid receptor (DOR) agonist ME (10 μm) caused a dramatic decrease in light-evoked IPSCs (72.46 ± 4.00% reduction; n = 9; p < 0.001; Fig. 3A,B) and EPSCs (72.45 ± 4.64% reduction; n = 4; p < 0.001; Fig. 3C,D). This reduction in current was readily reversed by washing out of drug (103.73 ± 10.03% of baseline; n = 5; p = 0.73; Fig. 3C) or by coapplying the MOR-selective antagonist CTAP (500 nm; 97.22 ± 9.42% of baseline; n = 8; p = 0.74; Fig. 3A).
Figure 3.

Light-evoked IPSCs and EPSCs are inhibited by opioid receptor activation. A, Plot of light-evoked IPSC amplitudes over time shows an inhibition of light-evoked IPSCs by the nonspecific opioid agonist ME (10 μm). The IPSC inhibition is reversed by application of the MOR-selective antagonist CTAP (500 nm). B, Representative traces of IPSCs in control conditions and in the presence of ME. C, ME inhibits light-evoked EPSC amplitude and EPSC amplitude returns to baseline upon washout of ME. D, Representative traces of EPSCs in control conditions and in the presence of ME. The arrowheads in B and D indicate the timing of the light pulse.
The DOR-selective agonist DPDPE (100 nm) had no effect on PSC amplitude in any cell tested (100.12 ± 3.41% of baseline; n = 4; p = 0.49), whereas the MOR-selective agonist DAMGO (10 μm) reliably inhibited IPSC amplitude (72.70 ± 7.80% reduction; n = 4; p = 0.001; Fig. 4A,B). The inhibition was reversed by CTAP (500 nm) (88.65 ± 5.95% of baseline; n = 4; p = 0.15; Fig. 4A). Thus, μ but not δ receptor agonists can inhibit amino acid transmitter release from POMC neurons.
Figure 4.

μ- and κ-opioid receptors mediate inhibition of evoked release from POMC terminals. A, Plot of light-evoked IPSC amplitudes over time shows no effect of the DOR-selective agonist DPDPE (100 nm) and inhibition of IPSC amplitude by the MOR-selective agonist DAMGO (10 μm). The DAMGO-induced inhibition is reversed by application of the MOR-specific antagonist CTAP (500 nm). B, Representative traces taken during control and agonist treatment. C, The specific KOR agonist U69593 (500 nm) inhibited evoked IPSC amplitude and was reversed by the addition of nor-BNI (100 nm). D, Representative traces taken during control conditions and during U69593 treatment.
κ-Opioid receptor (KOR) agonists inhibit IPSCs regulating POMC neurons (Pennock and Hentges, 2011). Although POMC neurons are not directly hyperpolarized by KOR activation (Pennock and Hentges, 2011), the possibility that POMC neurons express KORs presynaptically to inhibit transmitter release was examined. The KOR-selective agonist U69593 (500 nm) inhibited light-evoked IPSCs in unidentified downstream neurons (63.39 ± 8.33% reduction; n = 6; p < 0.001) and the KOR antagonist nor-BNI (100 nm) reversed the inhibition (95.68 ± 8.57% of baseline; n = 6; p = 0.64; Fig. 4C,D). U69593 did not cause a significant outward current in the unidentified neurons that received POMC inputs (0.8 ± 0.5 pA; p > 0.05 compared with baseline; n = 6). Thus, KORs expressed in POMC neurons inhibit amino acid release from POMC neurons even though they do not mediate a detectable outward postsynaptic current in POMC neurons or neighboring unidentified neurons.
GABAB receptor activation inhibits transmitter release from POMC cells
In addition to opioid receptors, GABAB receptors can inhibit transmitter release from unidentified terminals in the ARC. To determine whether GABAB receptors could modulate transmitter release from POMC neuron terminals in particular, light-evoked release was monitored in the presence of the GABAB receptor agonist baclofen. Baclofen (30 μm) caused a significant inhibition of light-evoked IPSCs in neurons downstream of POMC-ChR2 neurons (89.04 ± 3.20% reduction; n = 6; p < 0.001; Fig. 5A,B). In four cells, baclofen (30 μm) completely ablated the light-evoked IPSC. Baclofen-induced inhibition of the IPSC was fully reversed by washout or by coapplying the GABAB receptor antagonist CGP 55845 (1 μm) (92.44 ± 4.19% of baseline; n = 6; p = 0.13). This result indicates that POMC neurons express GABAB receptors, and these receptors can inhibit presynaptic GABA release from POMC terminals.
Figure 5.

GABAB receptor activation inhibits GABA release from POMC terminals. A, Light-evoked IPSC amplitudes are inhibited by baclofen (30 μm). The inhibition is reversed by washout of drug. B, Representative traces taken during control conditions and during baclofen treatment.
Discussion
The results of the present study show that POMC neurons are not only peptidergic but release the classical amino acid transmitters GABA and glutamate. Considerable release of these transmitters occurs within the ARC and can be modulated dynamically by agents such as opioids and GABA. Although GABAergic and glutamatergic phenotypes had previously been suggested for POMC neurons based on immunolabeling and studies of transmitter release from cultured POMC neurons (Hentges et al., 2004, 2009; Meister, 2007), the finding that GABA and glutamate release can be evoked from POMC cells in live brain slices indicates that these transmitters are likely to play an important role in the intact circuitry.
Advantages of the optogenetic approach
The difficulty of determining transmitter phenotype in hypothalamic neurons using traditional means stems from (1) the fact that this structure lacks lamination precluding the use of paired recordings to detect released transmitters, and (2) the inability to directly detect GABA and glutamate reliably using immunohistochemical methods in this tightly packed heterogeneous brain region. The optogenetic approach used in the present study allows for the activation of an entire population of identified neurons, which greatly increases the likelihood of detecting transmitter release onto downstream target neurons. Using this approach, Atasoy et al. (2008) recently demonstrated that POMC neurons could release GABA onto neurons in the paraventricular nucleus. The present data show that POMC neurons can also release glutamate and that these neurons release both inhibitory and excitatory amino acid transmitters onto neurons within the arcuate nucleus. Thus, optogenetic activation of POMC neurons is a useful means of mapping distal projections and studying local innervation.
The use of combined optogenetic activation and electrophysiologic recording to detect transmitter release has many advantages over immunodetection and marker-based methods, including increased sensitivity and real-time assessment of release. Based on various labeling approaches, the amino acid phenotype of POMC neurons had been disputable. The inability to detect GABA immunoreactivity in POMC neurons (Ovesjö et al., 2001), and the presence of glutamatergic markers in a small population of POMC neurons (Collin et al., 2003; Kiss et al., 2005) suggested that POMC neurons are not GABAergic. However, when the GAD67 promoter was used to drive expression of the green fluorescent protein in transgenic mice, labeling could be observed in a subpopulation of POMC neurons (Hentges et al., 2009; Yee et al., 2009), suggesting that some POMC neurons may be GABAergic. In addition, approximately one-third of POMC neurons express GAD mRNA (Hentges et al., 2004). The GABAergic nature of some POMC neurons was further indicated in studies showing evoked GABA release from POMC neurons in primary cultures (Hentges et al., 2004, 2009). Despite the evidence suggesting that POMC neurons can be either GABAergic or glutamatergic based on phenotypic markers and GABA and glutamate release from cultured POMC neurons (Hentges et al., 2009), the ability of these neurons to release both transmitters in brain slices had not been demonstrated until the present study.
Interestingly, although the present data clearly show evoked GABA release from POMC terminals, a recent study failed to detect evidence of vesicular GABA transporter (vGAT) in POMC neurons using a vGAT-Cre mouse cross in a transgenic reporter system (Vong et al., 2011). Vesicular GABA transporter is the only protein identified to date that packages inhibitory transmitters into vesicles (McIntire et al., 1997; Gasnier, 2004). Whether the apparent lack of vGAT expression in POMC neurons is due to the transgenic expression system used, or reflects that very low levels of vGAT are sufficient to package GABA in POMC neurons remains to be determined. Nonetheless, the expression of GAD67-gfp (Hentges et al., 2009) and GAD mRNA (Hentges et al., 2009) in POMC neurons as well as the release of GABA from POMC neurons in culture (Hentges et al., 2004, 2009) and in brain slices (Atasoy et al., 2008; present study) indicate that at least a subpopulation of POMC neurons is indeed GABAergic. Further studies are needed to determine whether POMC neurons release GABA in a vGAT-independent manner or if vGAT expression is too low to be readily detected. Either way, vGAT may not a reliable indicator of the GABAergic nature of POMC neurons.
GABA and glutamate release from POMC neurons is consistent with synaptic morphology
The present data localize a portion of POMC cell amino acid transmitter release to the ARC. The release of GABA is consistent with ultrastructural studies demonstrating POMC terminals make symmetrical (GABAergic) synapses onto neurons within the ARC (Chen and Pelletier, 1983; Kiss and Williams, 1983). Asymmetrical (glutamatergic) POMC synapses within the ARC have not been described, although glutamate release was detected locally in the present study. The low percentage of PSCs that were mediated by glutamate and the relatively low number of POMC neurons expressing vGLUT2 (Vong et al., 2011) may explain why, morphologically, glutamatergic POMC terminals have not been observed in the ARC. Interestingly, POMC neurons have been shown to form both symmetrical and asymmetrical synapses onto neurons in distal target sites including the dorsal raphe nucleus (Wang et al., 2001) and the locus ceruleus (Reyes et al., 2006). Thus, GABA and glutamate release from POMC neurons may be important for regulation both locally and at distal target sites.
Amino acid transmitters and energy homeostasis
The studies of amino acid transmitters in hypothalamic circuits to date indicate a significant contribution of these transmitters in the regulation of food intake and metabolism. For example, glutamate release from neurons in the ventromedial hypothalamus is an important step in preventing hypoglycemia (Tong et al., 2007), the release of GABA from NPY/AGRP neurons in the ARC is also required for the normal regulation of energy balance (Tong et al., 2008), and GABA release from NPY/AGRP neurons into the parabrachial nucleus prevents starvation (Wu et al., 2009). It is likely that amino acid transmitters from POMC cells have important analogous functions, although perhaps with more diversity, given the release of both GABA and glutamate from POMC neurons.
Presynaptic regulation of transmitter release
In the present study, the ability of GABAB and opioid receptor agonists to regulate amino acid transmitter release from POMC neurons was demonstrated. These studies were undertaken primarily to determine whether amino acid release from POMC neurons could be dynamically modulated as would be expected if these transmitters play an important regulatory role. GABAB and opioid receptors were chosen, as both have been shown to potently modulate transmitter release from terminals within the hypothalamus (Mouginot et al., 1998; Emmerson and Miller, 1999; Pennock and Hentges, 2011). Since transmitter release was evoked only from POMC neurons and inhibited by GABAB, MOR, and KORs, it is now clear that these receptors can inhibit release specifically from POMC terminals. The results raise the possibility that GABA and/or opioids released from POMC terminals could activate presynaptic receptors to inhibit further transmitter release.
Conclusion
The data collected here show that POMC neurons are not only peptidergic but release the classical amino acid transmitters GABA and glutamate. Release of these transmitters can be dynamically modulated by agents such as opioids and GABA. Thus, in addition to the roles attributed to peptides, POMC neurons can cause rapid inhibition or excitation of downstream neurons.
Footnotes
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK0798749 (S.T.H.). We thank Ms. Connie King for technical assistance in managing the mouse colony and stereotaxic injections.
The authors declare no competing financial interests.
References
- Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini A, Tacchi R, Vergoni AV. Brain effects of melanocortins. Pharmacol Res. 2009;59:13–47. doi: 10.1016/j.phrs.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Chen YY, Pelletier G. Demonstration of contacts between proopiomelanocortin neurons in the rat hypothalamus. Neurosci Lett. 1983;43:271–276. doi: 10.1016/0304-3940(83)90200-8. [DOI] [PubMed] [Google Scholar]
- Choudhury AI, Heffron H, Smith MA, Al-Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, Bedford DC, Hisadome K, Diakonov I, Moosajee V, Bell JD, Speakman JR, Batterham RL, Barsh GS, Ashford ML, Withers DJ. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest. 2005;115:940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll AP. Effects of pro-opiomelanocortin (POMC) on food intake and body weight: mechanisms and therapeutic potential? Clin Sci (Lond) 2007;113:171–182. doi: 10.1042/CS20070105. [DOI] [PubMed] [Google Scholar]
- Collin M, Bäckberg M, Ovesjö ML, Fisone G, Edwards RH, Fujiyama F, Meister B. Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur J Neurosci. 2003;18:1265–1278. doi: 10.1046/j.1460-9568.2003.02840.x. [DOI] [PubMed] [Google Scholar]
- Emmerson PJ, Miller RJ. Pre- and postsynaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. J Physiol. 1999;517:431–445. doi: 10.1111/j.1469-7793.1999.0431t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasnier B. The SLC32 transporter, a key protein for the synaptic release of inhibitory amino acids. Pflugers Arch. 2004;447:756–759. doi: 10.1007/s00424-003-1091-2. [DOI] [PubMed] [Google Scholar]
- Hegadoren KM, O'Donnell T, Lanius R, Coupland NJ, Lacaze-Masmonteil N. The role of beta-endorphin in the pathophysiology of major depression. Neuropeptides. 2009;43:341–353. doi: 10.1016/j.npep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci. 2004;24:1578–1583. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci. 2009;29:13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss J, Csaba Z, Csáki A, Halász B. Glutamatergic innervation of neuropeptide Y and pro-opiomelanocortin-containing neurons in the hypothalamic arcuate nucleus of the rat. Eur J Neurosci. 2005;21:2111–2119. doi: 10.1111/j.1460-9568.2005.04012.x. [DOI] [PubMed] [Google Scholar]
- Kiss JZ, Williams TH. ACTH-immunoreactive boutons form synaptic contacts in the hypothalamic arcuate nucleus of rat: evidence for local opiocortin connections. Brain Res. 1983;263:142–146. doi: 10.1016/0006-8993(83)91211-8. [DOI] [PubMed] [Google Scholar]
- Kohl MM, Shipton OA, Deacon RM, Rawlins JN, Deisseroth K, Paulsen O. Hemisphere-specific optogenetic stimulation reveals left-right asymmetry of hippocampal plasticity. Nat Neurosci. 2011;14:1413–1415. doi: 10.1038/nn.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Meister B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol Behav. 2007;92:263–271. doi: 10.1016/j.physbeh.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Mouginot D, Kombian SB, Pittman QJ. Activation of presynaptic GABAB receptors inhibits evoked IPSCs in rat magnocellular neurons in vitro. J Neurophysiol. 1998;79:1508–1517. doi: 10.1152/jn.1998.79.3.1508. [DOI] [PubMed] [Google Scholar]
- Ovesjö ML, Gamstedt M, Collin M, Meister B. GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J Neuroendocrinol. 2001;13:505–516. doi: 10.1046/j.1365-2826.2001.00662.x. [DOI] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci. 2011;31:281–288. doi: 10.1523/JNEUROSCI.4654-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- Reyes BA, Glaser JD, Magtoto R, Van Bockstaele EJ. Pro-opiomelanocortin colocalizes with corticotropin-releasing factor in axon terminals of the noradrenergic nucleus locus coeruleus. Eur J Neurosci. 2006;23:2067–2077. doi: 10.1111/j.1460-9568.2006.04744.x. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. Weighing the role of hypothalamic feeding neurotransmitters. Neuron. 2003;40:1059–1061. doi: 10.1016/s0896-6273(03)00809-2. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QP, Guan JL, Shioda S. Immunoelectron microscopic study of beta-endorphinergic synaptic innervation of GABAergic neurons in the dorsal raphe nucleus. Synapse. 2001;42:234–241. doi: 10.1002/syn.10008. [DOI] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 2009;137:1225–1234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005a;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS. Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol. 2005b;3:e415. doi: 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee CL, Wang Y, Anderson S, Ekker M, Rubenstein JL. Arcuate nucleus expression of NKX2.1 and DLX and lineages expressing these transcription factors in neuropeptide Y(+), proopiomelanocortin(+), and tyrosine hydroxylase(+) neurons in neonatal and adult mice. J Comp Neurol. 2009;517:37–50. doi: 10.1002/cne.22132. [DOI] [PMC free article] [PubMed] [Google Scholar]