

Cancer development is an evolutionary process involving replication, variation, and selection within the body of an organism (1). Therefore, just as species are shaped by the surrounding habitat, the properties of cancer cells should mirror their somatic environment. Based on this Darwinian perspective, and a large amount of circumstantial evidence, G. Gaudernack and I (2) recently proposed a general explanation for the elevated mutation rate that drives carcinogenesis. Bardelli et al. (3) have now tested this hypothesis in an elegant set of experiments, and in this issue of PNAS they present evidence for direct evolutionary relationships between carcinogenic environments and specific types of genetic instability.
Genetic instability designates genomewide elevation of mutation rate in cancer cells and mutants of single-cell organisms. Two distinct phenotypes have been characterized, and most cancers can be classified in one of these categories (4). Chromosomal instability (CIN) is the dominating phenotype of cancer cells and is recognized as numerical and structural aberrations of the genome. CIN has been related to genetic repair and mitotic control pathways, but the underlying mechanisms appear complex and remain to be clarified. Microsatellite instability (MIN), on the other hand, is a direct consequence of defects in nucleotide mismatch repair (MMR). The MMR machinery removes misincorporated nucleotides from the DNA molecule, and MIN is recognized as alterations at the nucleotide level, particularly in repetitive sequences. These mutation patterns are found in subsets of several cancer types, and germ-line defects in MMR underlie a cancer syndrome, hereditary nonpolyposis colon cancer, characterized by tumors with MIN.
Genetic instability appears early in tumorigenesis and is believed to play a critical role in the malignant process. Cells with CIN are found to activate or increase the number of copies of oncogenes and lose tumor-suppressor genes, whereas cells with MIN accomplish the same through mutations in repetitive DNA sequences (5). This relationship is well documented, and the effects of genetic instability in cancer development are straightforward in terms of Darwinian evolution: Genetic instability provides a repertoire of mutants from which the environment selects favorable variants (6). The “Darwinian problem” with genetic instability has been to explain why and how these phenotypes evolve in the first place (7). Why do mutants that ignore DNA damage outgrow the normal cells of the body, and how is this related to broiled meat and smoking and other factors that cause cancer? One shortcut has been to assume that environmental mutagens cause genetic instability by chance, which in turn provides favorable mutations (8). The fundamental problem with this model, however, is that an elevated mutation rate will destroy favorable genes by the same mechanisms that generate them and that stochastic mutations are much more likely to be deleterious than growth promoting. Elevated mutation rate, per se, therefore can not explain the rise and expansion of cells with genetic instability. The observation that practically all cancer cells express some form of genetic instability therefore demands an alternative explanation.
Another puzzle with genetic instability is that CIN and MIN are related to cancers occurring in different anatomical locations. This is particularly evident for colorectal cancer, where MIN tumors are confined to the proximal segments of the bowel, whereas CIN tumors are most abundant in the distal colon and rectum (9). This curious phenomenon strongly suggests that genetic instability is not a random event and led G. Gaudernack and I (10) to investigate and organize a broad range of data related to colorectal carcinogenesis. Of particular interest was the hypothesis by Karran and Bignami (11) that explains MIN as an adaptation to methylating agents. This relationship later was confirmed both in vitro (12) and in vivo (13) and is directly related to the MMR machinery. In general, it may be viewed in terms of costs and benefits of DNA repair, and the bottom line is that stopping for repairs can be a fatal strategy in hostile environments (Fig. 1).
Figure 1.

The cell cycle Grand Prix and effects of opposing repair strategies in different environments. Team I (green) always stops for repairs when a problem is indicated, whereas team II (red) ignores all warning lights. Team I wins under ordinary conditions (A) because it always has a faultless vehicle, whereas team II accumulates errors. In the harsher environment (B) the vehicles accumulate damages more quickly than can be repaired, and team I gets trapped in the checkpoint. Team II, on the other hand, jerks along in its faulty vehicle with a fair chance of making the finish line. This simple assessment of repair strategies thereby provides an explanation for the paradox that mutagenic environments favor repair deficiency.
Such strategic considerations led us to propose a general relationship between genetic instability and mutagenic agents and to search for environmental factors that could favor the CIN phenotype (10). A combination of diet-epidemiology, mutagenic “footprints,” and DNA repair mechanisms pointed to the miscellaneous group of agents that cause large alterations in the structure of DNA. Such bulky-adduct-forming (BAF) mutagens (14), comprising dietary components, pollutants, and intrinsic metabolites, therefore appeared as primary candidates for a selection pressure that promotes CIN. A causal relationship between the CIN phenotype and BAF mutagens has been missing, but is now demonstrated in an amusingly explicit model system (3).
HCT 116 is a colon cancer cell line of the MIN phenotype, reflecting mutations of the hMLH1 gene. Koi et al. (15) previously have designed a genetically stable mutant of this cell line (H3) by introducing a chromosome 3 with a normal hMLH1 allele, and this stabilized cell line formed the starting point for the experiments. The heterocyclic amine and well-characterized carcinogenic component of broiled meat, PhIP (2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine), was chosen as the BAF selection pressure (16), and clones that survived cytotoxic levels of PhIP were verified as resistant to the mutagen. Bardelli et al. (3) then analyzed them for chromosomal aberrations in the resistant clones and found a striking level of CIN in every one of them. To complete the testing of the hypothesis they performed the same procedure with a methylating selection pressure and confirmed the rise of methylation-tolerant and MMR-deficient cells of the MIN phenotype (Fig. 2). This conceptually simple experiment thus indeed shows that CIN and MIN reflect resistance to different carcinogens and that genomic instability in cancers may mirror the mutagenic environments in which they evolve.
Figure 2.
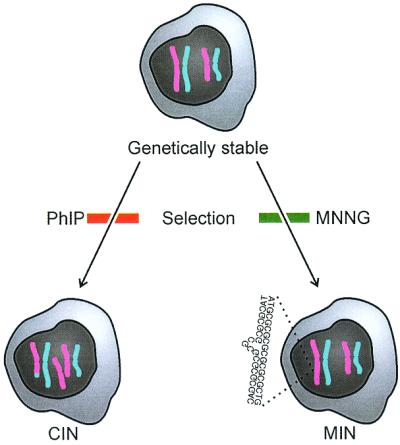
Mutagenic selection of genetic instability. Bardelli et al. (3) started out with the genetically stable cell line H3 (HCT 116 artificially complemented with chromosome 3). This cell line was first exposed to selection pressure involving cytotoxic levels of PhIP. The selected cells expressed CIN, illustrated as a trisomy and a translocation. The same cell line (H3) then was exposed to selection pressure involving cytotoxic levels of MNNG (N-methyl-N′-nitro-N-nitrosoguanine). This time the selected cells expressed MIN, illustrated as slippage in a repetitive sequence. The experiment thus supports the hypothesis that the CIN and MIN phenotypes reflect Darwinian adaptations to different mutagenic environments, methylating and BAF agents, respectively.
Over the last decades there has been substantial research on linking specific factors in the environment to mutagenic footprints in cancer cells (17). The Darwinian relationship between genes and environment now presented by Bardelli et al. (3) brings this field of molecular epidemiology to a new level. Cancer-promoting agents do not only leave footprints on DNA; they shape the somatic evolution of the entire genome of cancer cells. By combining molecular mechanisms, Darwinian models, and high-throughput technologies it should therefore be possible to extract considerable amounts of etiological information from the individual tumor.
The etiological implications of these findings are thus significant, and so are the therapeutic. Most cancer drugs are themselves mutagens (18) and must be expected to affect the evolution of cancers just like environmental agents. If the effect of PhIP reflects a general phenomenon, it will therefore be irrational to treat cancers of the CIN phenotype with any type of BAF agents, and MIN tumors certainly should not be treated with methylating agents. Cancer therapy is about manipulating the somatic environment in a way that disfavors the evolution of neoplastic cells, and this Darwinian perspective has immediate implications for clinical practice.
It has previously been shown that CIN is associated with disruption of the mitotic spindle checkpoint, involving dominant mutation of the hBUB1 gene (19). Bardelli et al. (3) therefore went on to explore the relationship between CIN and BAF agents the other way around. The starting point was again a colon cancer cell line of the MIN phenotype, but these cells (BUB-DLD1) had been engineered to allow regulated expression of a mutant hBUB1 gene. Cells not expressing the mutant gene were chromosomally stable, whereas expression induced the CIN phenotype. Bardelli et al. then tested for resistance to PhIP. The CIN cells were resistant to the cytotoxic effects of the mutagen, whereas the chromosomally stable cells were not.
This experiment thus confirms the relationship between PhIP and CIN and points to defects in chromosomal segregation as an underlying mechanism. Bardelli et al. were unable to identify such defects in the clones that evolved CIN through carcinogen selection, and there is apparently more to CIN than disrupted segregation of chromosomes. In particular, CIN also involves instability within the chromosomes, and segmental losses and amplifications, as well as translocations, are part of the phenotype. Such chromosomal shuffling, especially if involving centromeric regions, should be expected to activate apoptotic mechanisms. Loss of function of the mitotic spindle checkpoint therefore may be secondary to rearrangements, and it is interesting to speculate that nucleotide excision repair (NER) (20), which repairs bulky-adducts and causes chromosomal rearrangements, is an important component of CIN. In theory, NER could explain CIN the same way that MMR is related to MIN, but that does not appear to be the case because NER deficiency is rare in most cancers. An alternative is therefore to look for defects in the coupling between NER and cell cycle control (21, 22). This would be the equivalent of making repairs without stopping. It is destabilizing and risky, but in a mutagenic “war zone” it might be the only option (Fig. 1).
Several puzzles concerning genetic instability thus remain, and its relation to instability of DNA methylation patterns represents another focus of attention (23). Such patterns of epigenetic information also are replicated, mutated, and selected in the somatic environment and should accordingly evolve by Darwinian mechanism. In direct analogy to the findings of Bardelli et al. (3) it is therefore interesting to speculate that epigenetic instability represents evolutionary adaptations to carcinogenic agents that disturb DNA methylation.
Acknowledgments
I thank Gustav Gaudernack for insightful comments. My research is supported by the Research Council of Norway.
Footnotes
See companion article on page 5770.
References
- 1.Klein G. Adv Cancer Res. 1998;72:1–23. doi: 10.1016/s0065-230x(08)60698-3. [DOI] [PubMed] [Google Scholar]
- 2.Breivik J, Gaudernack G. Semin Cancer Biol. 1999;9:245–254. doi: 10.1006/scbi.1999.0123. [DOI] [PubMed] [Google Scholar]
- 3.Bardelli A, Cahill D P, Lederer G, Speicher M R, Kinzler K W, Vogelstein B, Lengauer C. Proc Natl Acad Sci USA. 2001;98:5770–5775. doi: 10.1073/pnas.081082898. . (First Published April 10, 2001; 10.1073/pnas.081082898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lengauer C, Kinzler K W, Vogelstein B. Nature (London) 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 5.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed J C, Perucho M. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 6.Cahill D P, Kinzler K W, Vogelstein B, Lengauer C. Trends Cell Biol. 1999;9:M57–M60. [PubMed] [Google Scholar]
- 7.Tomlinson I, Bodmer W. Nat Med. 1999;5:11–12. doi: 10.1038/4687. [DOI] [PubMed] [Google Scholar]
- 8.Janin N. Adv Cancer Res. 2000;77:189–221. doi: 10.1016/s0065-230x(08)60788-5. [DOI] [PubMed] [Google Scholar]
- 9.Lindblom A. Curr Opin Oncol. 2001;13:63–69. doi: 10.1097/00001622-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 10.Breivik J, Gaudernack G. Adv Cancer Res. 1998;76:187–212. doi: 10.1016/s0065-230x(08)60777-0. [DOI] [PubMed] [Google Scholar]
- 11.Karran P, Bignami M. BioEssays. 1994;16:833–839. doi: 10.1002/bies.950161110. [DOI] [PubMed] [Google Scholar]
- 12.Branch P, Hampson R, Karran P. Cancer Res. 1995;55:2304–2309. [PubMed] [Google Scholar]
- 13.Kawate H, Sakumi K, Tsuzuki T, Nakatsuru Y, Ishikawa T, Takahashi S, Takano H, Noda T, Sekiguchi M. Proc Natl Acad Sci USA. 1998;95:5116–5120. doi: 10.1073/pnas.95.9.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawley P D. IARC Sci Publ. 1994;125:3–22. [PubMed] [Google Scholar]
- 15.Koi M, Umar A, Chauhan D P, Cherian S P, Carethers J M, Kunkel T A, Boland C R. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 16.Keating G A, Layton D W, Felton J S. Mutat Res. 1999;443:149–156. doi: 10.1016/s1383-5742(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 17.Sram R J, Binkova B. Environ Health Perspect. 2000;108:57–70. doi: 10.1289/ehp.108-1637778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawley P D, Phillips D H. Mutat Res. 1996;355:13–40. doi: 10.1016/0027-5107(96)00020-6. [DOI] [PubMed] [Google Scholar]
- 19.Chahill D P, Lengauer C, Yu J, Riggins G J, Willson J K V, Markowitz S D, Kinzler K W, Vogelstein B. Nature (London) 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 20.Sancar A, Tang M S. Photochem Photobiol. 1993;57:905–921. doi: 10.1111/j.1751-1097.1993.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 21.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown J P, Sedivy J M, Kinzler K W, Vogelstein B. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 22.Zhou B B, Elledge S J. Nature (London) 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 23.Issa J P. Ann NY Acad Sci. 2000;910:140–153. doi: 10.1111/j.1749-6632.2000.tb06706.x. [DOI] [PubMed] [Google Scholar]