

Many genes, long-known or novel, have been identified as either regulators of cell differentiation or cell function in the skeleton. This identification certainly has improved our understanding of skeleton biology as a whole by providing multiple new pieces to the embryologic and physiologic puzzle (1, 2). However, so far most of these pieces remain unconnected. To date, what has been lacking in skeleton biology is the definition of genetic cascades linking these genes together in a linear fashion to explain complex processes like patterning, cell differentiation, or cell function. A complete explanation also would include the transition steps between the major processes. The work of Motyckova et al. (3) published in this issue of PNAS establishes precisely this type of genetic cascade for the late events of osteoclast cell lineage differentiation.
The osteoclasts are the cells responsible for bone resorption in vertebrates (see ref. 2 for review). They are multinucleated cells generated by the fusion of mononuclear progenitors of myelomonocytic origin (4). Once differentiated, osteoclasts become polarized by forming a ruffled membrane on the bone surface where they are able to resorb the mineralized bone extracellular matrix. This degradation process comprises two phases: first, the inorganic phase of the bone matrix is chemically dissolved, and then the protein component of the bone extracellular matrix, i.e., mostly type I collagen, is enzymatically degraded (5). Bone demineralization results from secretion of HCl by the osteoclasts in the microenvironment delineated by their ruffled borders and the bone surface. The HCl secretion establishes a local pH of ≈4.5 (6). This acidic milieu mobilizes the bone mineral, allowing the digestion of the proteic component of the bone matrix by a lysosomal protease called Cathepsin K (7, 8).
As the only skeleton cell type able to digest the mineralized bone matrix, the osteoclasts play a crucial role in bone physiology by allowing both bone modeling during growth and bone remodeling during adulthood. Thus, defects in osteoclast differentiation or function are associated with multiple genetically inherited or acquired diseases, all characterized by an arrest of bone resorption. The study of osteoclast biology has been facilitated by the characteristic phenotypic consequences of their mutations. Indeed, an arrest in osteoclast differentiation or a loss of function in the differentiated cells results in an osteopetrotic phenotype called marble bone disease. In this disease cartilage remnants are present within the bone metaphyses. The bone marrow is progressively replaced by the bone extracellular matrix that is continuously deposed by the osteoblasts, the bone-forming cells. This phenotype is easily detectable by x-rays because the bone matrix invades the bones, conferring to them a much denser appearance than normal (Fig. 1). Several diseases in human have characteristics very similar to osteopetrosis. Among them is pycrondisostosis, the disease whose physiopathology is studied by Motyckova et al. (3).
Figure 1.
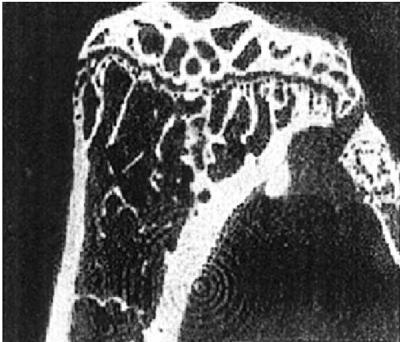
X-ray microtomography of a mouse tibia showing marbleized appearance characteristic of osteopetrosis. [Reproduced with permission from ref. 26 (Copyright 1999, National Academy of Sciences).]
Osteoclast molecular and developmental biology is probably one of the best-understood aspects of skeleton biology. Indeed, in the last 5–10 years the identity of many of the growth factors regulating osteoclast differentiation, their receptors, and to a large extent the signal transduction pathways they use have been elucidated (2). In addition to these secreted molecules a large number of transcription factors have been shown, genetically, to control osteoclast differentiation. These transcription factors are PU.1 (9), NFKB (10), c-fos (11), and the Mitf gene (12, 13). The Mitf gene is mutated in microphatalmia (mi) mice, a classical mouse mutant characterized by coat color defects, deafness, and osteopetrosis (12, 13).
Mitf is a member of the basic helix–loop–helix leucine zipper family of transcription factors. It is most closely related with three other members, TFEB, TFE3, and TFEC with which it can form heterodimers that bind to the E-box consensus sequence CA[C/T]GTG (14). Both Mitf and TFE3 are expressed at high levels by osteoclasts (15). Interestingly a loss-of-function mutation at the Mitf locus does not affect osteoclast differentiation (14, 16). It is only the dominant negative mutations in Mitf, such as mi/mi, that lead to osteopetrosis. In these cases, Mitf DNA binding is impaired whereas the dimerization helix–loop–helix-zipper motif is left intact, allowing the mutant molecules to dimerize with wild-type factors and sequester them in inactive complexes. It is this inactivating heterodimerization with related proteins such as TFE3, TFEB, and TFEC that affect osteoclast differentiation and result in a moderate osteopetrotic phenotype (14). In contrast with PU.1 or c-fos deficiency (9, 11), which affect the rate of progenitor cell maturation and lead to a decreased number of osteoclast, the defect in osteoclast differentiation in mi/mi mice is a late event. In these mice the osteoclasts are present but these cells do not fuse. Rather they remain mononucleated, do not form ruffled borders, and resorb bone extracellular matrix poorly (17–19).
In addition to these regulatory proteins, mutation in several structural proteins can cause osteopetrosis or osteopetrosis-like phenotype (7, 8, 20). One of these proteins, Cathepsin K, whose regulation of expression is the topic of the Motyckova et al. paper (3), is a cysteine protease from the papain family of proteases (21). Cathepsin K plays an important role during bone resorption by degrading type I collagen, the most abundant constituent of the bone extracellular matrix (8). Congenital Cathepsin K deficiency in humans causes pycnodisostosis, also called Toulouse-Lautrec disease, a condition very similar to osteopetrosis (22). Cathepsin K-deficient mice develop the same phenotypic abnormalities (23–25). Osteoclasts from Cathepsin K mutant mice are more differentiated than those from mi/mi mice in the sense that they are multinucleated cells, yet they cannot resorb the bone matrix. This loss of function of multinucleated osteoclasts is consistent with the hypothesis that Cathepsin K is a downstream target gene of Mitf, thereby explaining the milder phenotype of mice (and humans) with inactivating mutations in Cathepsin K compared with what is observed in the mi/mi mutant mice. Indeed, the phenotype observed in the latter mutant mice would be the result not only of Cathepsin K deficiency but also of the absence of other factors whose expression also would be transcriptionally controlled by Mitf. Consistent with this hypothesis, a recent study has shown that expression of the tartrate-resistant alkaline phosphatase (TRAP) gene, a gene highly expressed by osteoclasts and whose product is involved in the enzymatic process of bone resorption, also is regulated by Mitf (25).
An arrest in osteoclast differentiation or loss of function in the differentiated cells results in an osteopetrotic phenotype, also called marble bone disease.
The manuscript of Motyckova et al. (3) provides multiple convincing evidence of a direct regulation of Cathepsin K gene expression by Mitf: (i) Mitf regulates Cathepsin K promoter activity through its binding to three E-boxes, the 6-bp core binding elements of all basic helix–loop–helix transcription factors. (ii) Cathepsin K mRNA and protein are deficient in Mitf-deficient osteoclasts. (iii) Overexpression of wild-type Mitf up-regulates markedly the expression of endogenous Cathepsin K in cultured osteoclasts. (iv) Cathepsin K promoter activity is disrupted by dominant negative but not by loss-of-function mouse mutant allele of Mitf. By establishing these genetic and biochemical links, the study also explains the phenotypic analogies and differences between pycnodysostosis and osteopetrosis as seen in mice and humans.
Footnotes
See companion article on page 5798.
References
- 1.Karsenty G. Genes Dev. 1999;13:3037–3051. doi: 10.1101/gad.13.23.3037. [DOI] [PubMed] [Google Scholar]
- 2.Teitelbaum S. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 3.Motyckova G, Weilbaecher K N, Horstmann M, Rieman D J, Fisher D Z, Fisher D E. Proc Natl Acad Sci USA. 2001;98:5798–5803. doi: 10.1073/pnas.091479298. . (First Published May 1, 2001; 10.1073/pnas.091479298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin T J, Suda T. Proc Natl Acad Sci USA. 1990;87:7260–7264. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blair H C, Kahn A J, Crouch E C, Jeffrey J, Teitelbaum S L. J Cell Biol. 1986;102:1164–1172. doi: 10.1083/jcb.102.4.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silver I A, Mrrills R J, Etherington D J. Exp Cell Res. 1988;175:266–276. doi: 10.1016/0014-4827(88)90191-7. [DOI] [PubMed] [Google Scholar]
- 7.Li Y P, Chen W, Liang Y, Li E, Stashenko P. Nat Genet. 2000;23:447–449. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 8.Gowen M. J Bone Miner Res. 1999;14:1654–1663. doi: 10.1359/jbmr.1999.14.10.1654. [DOI] [PubMed] [Google Scholar]
- 9.Tondravi M M. Nature (London) 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- 10.Franzoso G. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grigoriadis A E, Wang Z Q, Cecchini M G, Hofsteter W, Felix R, Fleisch H A, Wagner E F. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 12.Hertwig P. Z Indukt Abstammungs-Vererbungsl. 1942;80:220–246. [Google Scholar]
- 13.Hodgkinson C A, Moore K J, Nakayama A, Steingrimsson E, Copeland N G, Jenkins N A, Arnheithre H. Cell. 1993;74:395–404. doi: 10.1016/0092-8674(93)90429-t. [DOI] [PubMed] [Google Scholar]
- 14.Hemesath T J, Steingrimsson E, McGill G, Hansen M J, Vaught J, Hodgkinson C A, Arnheiter H, Copeland N G, Jenkins N A, Fisher D E. Genes Dev. 1994;8:2880–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 15.Weilbaecher K N, Hershey C L, Takemoto C M, Horstmann M A, Hemesath T J, Tashjian A H, Fisher D E. J Exp Med. 1998;187:775–785. doi: 10.1084/jem.187.5.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steingrimsson E, More K J, Lamoreux M L, Ferre-D'Amare A R, Burley S, Zimring K, Skow D C, Hodgkinson L C, Arnheiter C A, Copeland N G, et al. Nat Genet. 1994;9:256–263. doi: 10.1038/ng1194-256. [DOI] [PubMed] [Google Scholar]
- 17.Walker D G. Science. 1975;190:784–785. doi: 10.1126/science.1105786. [DOI] [PubMed] [Google Scholar]
- 18.Thesingh C W, Scherft J P. Bone. 1985;6:43–52. doi: 10.1016/8756-3282(85)90406-5. [DOI] [PubMed] [Google Scholar]
- 19.Murphy H M. Calcif Tissue Res. 1973;13:19–26. doi: 10.1007/BF02015392. [DOI] [PubMed] [Google Scholar]
- 20.Sly W S, Hewtt-Emmett D, Whyte M P, Yu Y-S, Tashian E. Proc Natl Acad Sci USA. 1983;80:2752–2756. doi: 10.1073/pnas.80.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bromme D, Okamoto K. Biol Chem Hoppe Seyler. 1995;376:379–384. doi: 10.1515/bchm3.1995.376.6.379. [DOI] [PubMed] [Google Scholar]
- 22.Gelb B D, Shi G P, Chapman H A, Desnick R J. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- 23.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz J D, Schu P, von Figura K. Proc Natl Acad Sci USA. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gowen M, Lazner F, Dodds R, Kapadia R, Field J, Tavaria M, Bertoncello I, Drake F, Zavarselk S, Telis I, et al. J Bone Miner Res. 1999;14:1654–1663. doi: 10.1359/jbmr.1999.14.10.1654. [DOI] [PubMed] [Google Scholar]
- 25.Luchin A, Purdom G, Murphy K, Clark M-Y, Angel N, Cassady A I, Hume D A, Ostrowski M C. J Bone Miner Res. 2000;15:451–460. doi: 10.1359/jbmr.2000.15.3.451. [DOI] [PubMed] [Google Scholar]
- 26.Yoshitake H, Rittling S R, Denhardt D T, Noda M. Proc Natl Acad Sci USA. 1999;96:8156–8160. doi: 10.1073/pnas.96.14.8156. [DOI] [PMC free article] [PubMed] [Google Scholar]