

Abstract
CHIME syndrome is characterized by colobomas, heart defects, ichthyosiform dermatosis, mental retardation (intellectual disability), and ear anomalies, including conductive hearing loss. Whole-exome sequencing on five previously reported cases identified PIGL, the de-N-acetylase required for glycosylphosphatidylinositol (GPI) anchor formation, as a strong candidate. Furthermore, cell lines derived from these cases had significantly reduced levels of the two GPI anchor markers, CD59 and a GPI-binding toxin, aerolysin (FLAER), confirming the pathogenicity of the mutations.
Main Text
CHIME syndrome (MIM 280000), also known as Zunich neuroectodermal syndrome, is an extremely rare autosomal recessive multisystemic disorder clinically characterized by colobomas, congenital heart defects, early onset migratory ichthyosiform dermatosis, mental retardation (intellectual disability), and ear anomalies, including conductive hearing loss. Other clinical manifestations include distinctive facial features, abnormal growth, genitourinary abnormalities, seizures, and feeding difficulties.1 To date, eight cases have been reported, all having nearly identical phenotypes.
In 2010, Cantagrel et al.2 described a congenital disorder of glycosylation (CDG) in which individuals with pathological mutations in SRD5A3 (MIM 611715) presented with CHIME-like features (MIM 612379). Yet individuals with Classical CHIME as described by Zunich1 lacked mutations in SRD5A3. We hypothesized that CHIME syndrome could be a glycosylation disorder on the basis of the clinical similarity to those individuals identified by Cantagrel.2
To test this, we obtained DNA samples from six of the eight previously described cases from five unrelated families for the purpose of whole-exome sequencing (WES). All clinical samples were obtained with proper informed consent in accordance with the Sanford-Burnham Medical Research Institute's institutional review board consent guidelines.
To identify the genetic cause of CHIME syndrome, we performed WES on five of six previously described cases.3,4 Exome sequences were enriched with the Roche Nimblegen Seqcap EZ whole-exome Ver 2.0 on an Illumina HiSeq platform, and the raw data were aligned to hg18. Analysis employed Agilent's AVADIS NGS software. The five sequenced exomes had an average of 12,549 total variants and an average of 3,774 novel variants with 87% of the exome targets having at least 10× coverage (Table 1).
Table 1.
Summary of WES Statistics for Five CHIME Syndrome Cases
|
Individual |
Average | |||||
|---|---|---|---|---|---|---|
| 680-2-2 | 680-2-3 | 682-2-1 | 665-3-1 | 3988 | ||
| Total number of sequenced reads | 53,341,240 | 54,687,243 | 47,134,146 | 117,769,811 | 121,450,190 | 78,876,526 |
| Total number of unmapped reads (%) | 1,257,149 (2.4%) | 765,214 (1.4%) | 738,306 (1.6%) | 934,246 (0.8%) | 988,578 (0.8%) | 936,699 (1.2%) |
| Total number of mapped reads | 52,084,091 | 53,922,029 | 46,395,840 | 116,835,565 | 120,461,612 | 77,939,827 |
| Percentage Targets with 10× coverage | 78 | 88 | 85 | 88 | 95 | 87 |
| Percentage Targets with 20× coverage | 43 | 65 | 59 | 79 | 89 | 67 |
| Total NS-SS-Indels | 10,022 | 12,211 | 11,921 | 15,614 | 12,978 | 12,549 |
| Novel NS-SS-Indels | 2,338 | 3,211 | 3,181 | 7,173 | 2,966 | 3,774 |
A sixth case previously described by Tinschert et al.5 was the only sample analyzed by comparative genomic hybridization (CGH) array; it showed a 1 MB maternally inherited deletion on chromosome 17. DNA was isolated from whole blood with QIAGEN's DNA blood kit according to the manufacturer's protocol (QIAGEN, Hilden, Germany). Array CGH was performed on Agilent's SurePrint G3 Human CGH Microarray Kit 2x400K (Design ID 021850, Agilent, Santa Clara, CA, USA) according to the manufacturer's protocol, except that dyes were used inversely on sample and reference. An Agilent microarray scanner provided the raw data that were processed by Feature Extraction 9.5. Deleted and amplified regions were identified on Agilent's Genomic Workbench Standard Edition 5.0.14. Customized CGH array confirmed copy number variants and familial segregation. Agilent's eArray platform had a general probe density of 1 per 200 bp to 1 per 2.5 kb depending on the size of the variant. The coordinates of the array result were mapped to hg18.
We focused exome analysis on the 17 genes in this region (chromosome 17: 15,620,754–16,698,489 hg18) as likely candidates. We excluded synonymous changes, variants in dbSNP v133, and variants present in a limited (30) in-house exome library. All cases had compound heterozygous mutations in only PIGL [RefSeq NM_004278.3] within that region (Figure 1). Sanger sequencing confirmed all mutations in PIGL, and carrier status in each available parent or sibling (family 665 was not available) excluded de novo events.
Figure 1.
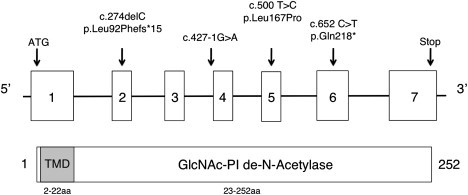
Organization of Human PIGL and Associated Mutations
Schematic representation showing both genomic and protein organization of human PIGL with corresponding mutations as well as functional protein domains including a 20aa transmembrane domain (TMD) and the core de-N-acetylase domain.
PIGL (NP_004269 [MIM 605947]) is an endoplasmic reticulum (ER)-localized enzyme that catalyzes the second step of glycosylphosphatidylinositol (GPI) biosynthesis, the de-N-acetylation of N-acetylglucosaminyl-phosphatidylinositol (GlcNAc-PI → GlcN-PI) that occurs on the cytoplasmic side of the ER.6 Following de-N-acetylation, glucosaminyl-phosphatidylinositol (GlcN-PI) flips to the luminal side of the ER where GlcN-PI undergoes further extensions prior to its transfer to acceptor proteins.7
Aside from a possible founder missense mutation, the other mutations identified in our six CHIME cases are predicted to be highly damaging (frameshift, nonsense, essential splice site, and entire gene deletion) (Table 2). The c.500T>C (p.Leu167Pro) mutation found in all six cases is at a highly conserved residue (Table 3) located in the catalytic domain and predicted by both PolyPhen and SIFT to be damaging.
Table 2.
Mutations Identified in Each of the Five CHIME Families
| Individual | DNA Level Mutations | Protein Level Alterations | Reference |
|---|---|---|---|
| 680-2-2 | c.274delC, c.500T>C | p.Leu92Phefs∗15, p.Leu167Pro | Shashi et al.3 |
| 680-2-3 | c.274delC, c.500T>C | p.Leu92Phefs∗15, p.Leu167Pro | Shashi et al.3 |
| 682-2-1 | c.500T>C, c.652C>T | p.Leu167Pro, p.Gln218∗ | Shashi et al.3 |
| 665-3-1 | c.500T>C | p.Leu167Proa | Schnur15 |
| 3988 | c.427-1G>A, c.500T>C | c.427-1G>A, p.Leu167Pro | Sidbury4 |
| 33300 | del17p12-p11.2, c.500T>C | del17p12-p11.2, p.Leu167Pro | Tinschert5 |
Second mutation not identified; parent DNA unavailable.
Table 3.
Evolutionary Conservation of Leu167
| Species | Ortholog of Human Leu167 |
|---|---|
| Homo sapiens | YAAVRA L HSEGK |
| Mus musculus | YKAVRA L HSGGK |
| Rattus norvegicus | YKAVRA L HSGGK |
| Danio rerio | YKTLSH L ASAGR |
| Saccharomyces cerevisiae | YAAVKK L VDDYA |
| Gallus gallus | YAAVRA L HSEGK |
| Drosophila melanogaster | YAAAS L CLANL |
Utilizing two large public databases, we found the heterozygous missense mutation c.500T>C in eight out of nearly 13,000 alleles. In the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project, the c.500T>C mutation appears at a frequency of 6:10,752 alleles (5,376 genomes; with all six heterozygotes being of European origin). In the 1000 Genomes database, it was present at 2:2,188 alleles (1,094 genomes). Given the relatively rare frequency (<0.1%) and the fact that all six CHIME cases are of European ancestry, we hypothesized that c.500T>C was due to a founder mutation.
We compared two microsatellite markers, 17xATT (trinucleotide marker) and 19xGT (dinucleotide marker), on chromosome 17: 15,620,754–16,698,489 (hg18) flanking PIGL. The c.500T>C missense mutation was tightly linked with the 17xATT in all CHIME cases with an ATT repeat size of 22, whereas European controls (16) had a repeat size of 17 repeats. Furthermore, all CHIME cases had a 19xGT repeat size of 13, whereas the European controls (15) had a repeat size of 19. These results support linkage disequilibrium between the c.500T>C allele and allele 22 of the 17xATT and allele 13 of the 19xGT marker (data not shown).
To confirm that the mutations in PIGL are pathological, we utilized a primary fibroblast cell line from individual 3988 and an Epstein-Barr virus (EBV)-transformed lymphoblast cell line from individual 33300 to measure two separate cell surface GPI-anchor-containing markers, CD59 and a GPI-binding toxin, aerolysin (FLAER). In agreement with other proven GPI deficiencies, cells available from CHIME syndrome cases are also deficient for both GPI anchor markers (Figure 2). It is important to note that individual 33300 carries the chromosome 17 deletion and c.500T>C mutation, making her hemizygous for the mutation and proving that it is pathogenic.
Figure 2.
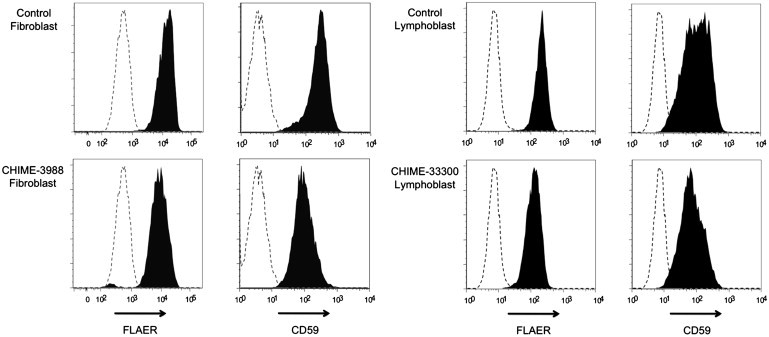
Cell Surface Expression of Total GPI Anchor and CD59
Fluorescence-activated cell sorting analysis for two separate GPI anchor markers, CD59 and FLAER, were used on a primary fibroblast line from individual 3988 and an EBV transformed lymphoblast line from individual 33300 to evaluate GPI anchor levels. In both instances, two normal controls were used. Shown is a representation of the two. Dotted lines indicate isotype controls.
Three inherited genetic disorders were previously identified in GPI biosynthetic genes: PIGM (MIM 610273), PIGN (MIM 606097), and PIGV (MIM 610274).8–10 Somatic mutations in PIGA (MIM 311770) cause paroxysmal nocturnal hemoglobinuria (MIM 300818), a hematologic disorder.11 Interestingly, epidermal-specific knockout of mouse PigA recreates features of human Harlequin ichthyosis.12 Mutations in PIGM, encoding the first mannosyltransferase, are associated with venous thrombosis and seizures (MIM 610293).8 Mutations in PIGN, which encodes the ethanolamine phosphate transferase, cause multiple congenital anomalies-hypotonia-seizures syndrome (MIM 614080).9 Deficiencies in PIGV, which encodes the second mannosyltransferase in GPI formation, are associated with the hyperphosphatasia mental retardation syndrome (MIM 239300).10
One clear difference between PIGL deficiency and the other GPI deficiencies is the magnitude of GPI marker decrease. Although we have consistently seen a significant decrease of 2- to 4-fold in both cell lines, the decrease was less dramatic than those seen in the other PIG deficiencies. One explanation is that the p.Leu167Pro alteration mildly affects the binding and de-N-acetylation of the GlcNAc-PI, but GlcN-PI can still be flipped into the luminal side of the ER. Furthermore, although we show a link between the p.Leu167Pro alteration and CHIME syndrome, we cannot exclude the possibility that additional mutations in PIGL could cause disorders other than CHIME syndrome.
GPI anchor deficiencies cause remarkable clinical diversity but that is typical of other glycosylation pathways such as the 38 Congenital Disorders of Glycosylation or 6 α-dystroglycanopathies.13 Hypomorphic alleles predominate, and the clinical impact often depends more on the severity of the mutation than on the specific mutated gene.14 In conclusion; we analyzed six previously described CHIME syndrome cases by using a combination of genetic and biochemical approaches, including CGH array and WES. We show that mutations in PIGL impair GPI biosynthesis and are the underlying cause of this disorder.
Acknowledgments
Supported by The Rocket Fund, a Sanford Professorship (H.H.F.) and R01 DK55615. K.H. was supported by the Bundesministerium für Bildung und Forschung network grant MR-NET 01GS08166.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Agilent eARRAY, https://earray.chem.agilent.com/earray/
NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, www.genome.ucsc.edu
References
- 1.Zunich J., Esterly N. In: CHIME syndrome (Zunich syndrome) Neurocutaneous Disorders Phakomatoses and Hamartoneoplastic Syndromes. Ruggieri M., Pascual-Castroviejo I., Di Rocco C., Weinheim G., editors. Springer-Verlag; Wien: 2008. pp. 949–955. [Google Scholar]
- 2.Cantagrel V., Lefeber D.J., Ng B.G., Guan Z., Silhavy J.L., Bielas S.L., Lehle L., Hombauer H., Adamowicz M., Swiezewska E. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142:203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shashi V., Zunich J., Kelly T.E., Fryburg J.S. Neuroectodermal (CHIME) syndrome: an additional case with long term follow up of all reported cases. J. Med. Genet. 1995;32:465–469. doi: 10.1136/jmg.32.6.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sidbury R., Paller A.S. What syndrome is this? CHIME syndrome. Pediatr. Dermatol. 2001;18:252–254. doi: 10.1046/j.1525-1470.2001.018003252.x. [DOI] [PubMed] [Google Scholar]
- 5.Tinschert S., Anton-Lamprecht I., Albrecht-Nebe H., Audring H. Zunich neuroectodermal syndrome: migratory ichthyosiform dermatosis, colobomas, and other abnormalities. Pediatr. Dermatol. 1996;13:363–371. doi: 10.1111/j.1525-1470.1996.tb00702.x. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe R., Ohishi K., Maeda Y., Nakamura N., Kinoshita T. Mammalian PIG-L and its yeast homologue Gpi12p are N-acetylglucosaminylphosphatidylinositol de-N-acetylases essential in glycosylphosphatidylinositol biosynthesis. Biochem. J. 1999;339:185–192. [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson M.A.J., Kinoshita T., Hart G.W. Glycosylphosphatidylinositol Anchors. In: Varki A., Cummings R.D., Esko J.D., Freeze H., Hart G., Marth J., editors. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2009. pp. 143–161. [Google Scholar]
- 8.Almeida A.M., Murakami Y., Layton D.M., Hillmen P., Sellick G.S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006;12:846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 9.Maydan G., Noyman I., Har-Zahav A., Neriah Z.B., Pasmanik-Chor M., Yeheskel A., Albin-Kaplanski A., Maya I., Magal N., Birk E. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011;48:383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 10.Krawitz P.M., Schweiger M.R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 11.Takeda J., Miyata T., Kawagoe K., Iida Y., Endo Y., Fujita T., Takahashi M., Kitani T., Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 12.Hara-Chikuma M., Takeda J., Tarutani M., Uchida Y., Holleran W.M., Endo Y., Elias P.M., Inoue S. Epidermal-specific defect of GPI anchor in Pig-a null mice results in Harlequin ichthyosis-like features. J. Invest. Dermatol. 2004;123:464–469. doi: 10.1111/j.0022-202X.2004.23227.x. [DOI] [PubMed] [Google Scholar]
- 13.Freeze H.H., Ng B.G. Golgi glycosylation and human inherited diseases. In: Warren G., Rothman J., editors. Cold Spring Harb Perspect Biol. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2011. pp. 35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godfrey C., Foley A.R., Clement E., Muntoni F. Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Schnur R.E., Greenbaum B.H., Heymann W.R., Christensen K., Buck A.S., Reid C.S. Acute lymphoblastic leukemia in a child with the CHIME neuroectodermal dysplasia syndrome. Am. J. Med. Genet. 1997;72:24–29. [PubMed] [Google Scholar]