

Abstract
Acrodysostosis is a rare autosomal-dominant condition characterized by facial dysostosis, severe brachydactyly with cone-shaped epiphyses, and short stature. Moderate intellectual disability and resistance to multiple hormones might also be present. Recently, a recurrent mutation (c.1102C>T [p.Arg368∗]) in PRKAR1A has been identified in three individuals with acrodysostosis and resistance to multiple hormones. After studying ten unrelated acrodysostosis cases, we report here de novo PRKAR1A mutations in five out of the ten individuals (we found c.1102C>T [p.Arg368∗] in four of the ten and c.1117T>C [p.Tyr373His] in one of the ten). We performed exome sequencing in two of the five remaining individuals and selected phosphodiesterase 4D (PDE4D) as a candidate gene. PDE4D encodes a class IV cyclic AMP (cAMP)-specific phosphodiesterase that regulates cAMP concentration. Exome analysis detected heterozygous PDE4D mutations (c.673C>A [p.Pro225Thr] and c.677T>C [p.Phe226Ser]) in these two individuals. Screening of PDE4D identified heterozygous mutations (c.568T>G [p.Ser190Ala] and c.1759A>C [p.Thr587Pro]) in two additional acrodysostosis cases. These mutations occurred de novo in all four cases. The four individuals with PDE4D mutations shared common clinical features, namely characteristic midface and nasal hypoplasia and moderate intellectual disability. Metabolic screening was normal in three of these four individuals. However, resistance to parathyroid hormone and thyrotropin was consistently observed in the five cases with PRKAR1A mutations. Finally, our study further supports the key role of the cAMP signaling pathway in skeletogenesis.
Main Text
Acrodysostosis (MIM 101800) is a dominantly inherited condition consisting of (1) skeletal dysplasia characterized by facial dysostosis with nasal hypoplasia (a depressed nasal bridge and prominent mandible), severe brachydactyly with short broad metatarsals, metacarpals, and phalanges, cone-shaped epiphyses, advanced bone maturation, spinal stenosis, and short stature; (2) resistance to multiple hormones, including parathyroid hormone and thyrotropin; and (3) possible neurological involvement (moderate to mild intellectual disability).1,2 Differential diagnoses include Albright hereditary osteodystrophy (MIM 103580) and pseudopseudohypoparathyroidism (MIM 612463), which are both due to loss-of-function mutations in GNAS (α-stimulary subunit of the G protein) (MIM 139320) and are characterized by less severe hand and foot involvement.3
A recurrent c.1102C>T mutation in PRKAR1A (MIM 188830) has been recently identified in three cases of acrodysostosis with resistance to multiple hormones.4 This gene encodes the cyclic AMP (cAMP)-dependent regulatory subunit of protein kinase A. The mutated subunit impairs the protein-kinase-A response to cAMP and accounts for hormone resistance and skeletal abnormalities resembling those observed in Albright hereditary osteodystrophy.
After studying ten unrelated individuals with acrodysostosis, we found PRKAR1A mutations in five out of the ten, and we show that most of the remaining cases were accounted for by mutations in phosphodiesterase, 4D (PDE4D [MIM 600129]), which is also involved in cAMP metabolism.
Ten unrelated cases were included in this study. There was no family history, and each individual was the only affected member in his family. Inclusion criteria were the following: (1) the presence of severe generalized brachydactyly affecting metacarpals and phalanges and associated with cone-shaped epiphyses and (2) the exclusion of Albright hereditary osteodystrophy on the basis of normal bioactivity of the Gs alpha subunit and normal GNAS sequencing.
We performed a complete screening of phosphocalcic metabolism and blood levels of creatinine, calcium, phosphorus, thyroxin, thyrotropin, 25-hydroxyvitaminD, 1,25-dihydroxyvitaminD, parathyroid hormone (PTH), and fibroblast growth factor 23, as well as urinary levels of creatinine, calcium, and phosphorus. The clinical radiological and biochemical details are summarized in Table 1 and Figure 1.
Table 1.
Clinical, Radiological, and Biochemical Data of the Ten Acrodysostosis Individuals Reported
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | female | male | female | female | female | male | male | male | male | female |
| PRKAR1A mutation | c.1102C>T | c.1102C>T | c.1102C>T | c.1117T>C | c.1102C>T | − | − | − | − | − |
| PDE4D mutation | − | − | − | − | − | c.673C>A | c.677T>C | c.568T>G | c.1759A>C | − |
| IUGR | no | no | no | yes | no | yes | no | no | no | no |
| Postnatal growth retardation (<−2 SDs) | yes (26 years old) | no (8 years old) | yes (13 years old) | yes (22 years old) | yes (34 years old) | no (7 years old) | no (4 years old) | no (4 years old) | no (3 years old) | yes (38 years old) |
| Advanced bone age | − | yes | yes | − | − | yes | yes | yes | yes | − |
| Facial dysostosis | ||||||||||
| Nasal hypoplasia | no | no | no | no | no | yes | yes | yes | yes | no |
| Depressed nasal bridge | no | no | yes | no | yes | yes | yes | yes | yes | no |
| Prominent mandible | no | no | no | no | yes | no | no | yes | no | yes |
| Peripheral dysostosis | ||||||||||
| Severe brachydactyly | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Short metatarsals, metacarpals, and phalanges | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Cone-shaped epiphyses | yes (childhood) | yes | yes | yes (childhood) | yes (childhood) | yes | yes | yes | yes | nd |
| Hormonal screening | ||||||||||
| PTH (n = 10–46 ng/l) | 95 | 79 | 116 | 84 | 142 | 76 | 39 | 24 | 19 | normal |
| Calcemia (n = 2.2–2.7 mmol/l) | 2.35 | 2.47 | 2.4 | 2.57 | 2.37 | 2.18 | 2.47 | 2.4 | 2.5 | 2.38 |
| Phosphoremia (n = 1.3–1.85 mmol/l) | 1.23 | 1.56 | 1.76 | nd | 1.3 | 1.54 | 1.68 | 1.81 | 1.7 | 1.8 |
| 25-OHvitD (n = 30–80 ng/ml) | nd | 26 | 16 | nd | 22 | 26 | 25 | 45 | 30 | nd |
| 1,25-diOHvitD (pg/ml) | nd | 39 | 106 | 65 | 53 | nd | nd | nd | ||
| FGF23 (n = 1–120 UI/ml) | nd | nd | nd | nd | 145 | 90 | 112 | 171.2 | 60.9 | nd |
| Free T4 (n = 7.5–15 pmol/l) | 8.65 | hypothyroidism | hypothyroidism | hypothyroidism | 16.71 treatment | 10.4 | 10 | 14.3 | 17 | 17 |
| TSH (n = 0.34–5.6 mUI/l) | 2.67 | 13.41 | 15.42 | increased | 0.16 | 2.59 | 2.51 | 3.58 | 2.77 | 1.8l |
| Calciuria (n = 1.5–6 mmol/l) | nd | <0.2 | nd | nd | nd | 0.86 | 1.28 | 1.44 | 2.27 | nd |
| Phosphaturia (n = 10–50 mmol/l) | nd | 14.8 | nd | nd | nd | 13.76 | 7.12 | 36.7 | 4.70 | nd |
| Creatininuria (mmol/l) | nd | 4.5 | nd | nd | nd | 6.3 | 2.08 | 11.7 | 4.15 | nd |
| Neurology | ||||||||||
| Intellectual disability | no | no | no | no | no | yes; speech delay requiring orthophony; fine-motor-skill impairment | yes; speech delay requiring orthophony; fine-motor-skill impairment | yes; speech delay; psychomotor delay (walked at 17 months of age) | yes; speech delay; psychomotor delay requiring physiotherapy | no |
| Other | spinal stenosis; carpal tunnel | intracranial hypertension with jugular stenosis requiring derivation | intracranial hypertension with thrombophlebitis of the transverse sinus and jugular (treated by acetazolamide, anticoagulant, and derivation) | spinal stenosis | ||||||
The following abbreviations are used: IUGR, intrauterine growth retardation; SDs, standard deviations; nd, not done; PTH, parathyroid hormone; 25-OHvitD, 25-hydroxyvitaminD; 1,25-diOHvitD, 1,25-dihydroxyvitaminD; FGF23, fibroblast growth factor 23; T4, thyroxin; and TSH, thyrotropin.
Figure 1.
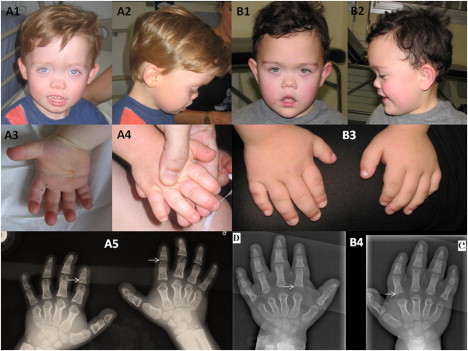
Pictures and X-Rays of Individuals 6 and 8 with PDE4D Mutations
(A1 and B1) Full-face pictures of individuals 6 (A) and 8 (B) showing facial dysostosis with a flat nasal bridge and nasal hypoplasia.
(A2 and B2) Profile pictures show malar hypoplasia.
(A3) Palmar face of right hand.
(A4 and B3) Dorsal face of hands, which are broad and shortened.
(A5 and B4) Standard X-rays of both hands show severe brachydactyly with short, broad metacarpals and phalanges, cone-shaped epiphyses (arrows), and advanced carpal maturation.
Informed consent for participation, sample collection, and photograph publication was obtained via protocols approved by the Necker Hospital ethics committee.
We sequenced PRKAR1A (RefSeq accession number NM_002734.3) by using specific primers (available upon request) in the ten individuals. De novo PRKAR1A mutations, including the recurrent mutation,4 were identified in five out of the ten individuals (c.1102C>T [p.Arg368∗] was found in four of the five, and c.1117T>C [p.Tyr373His] was found in one of the five) (Table 1). This missense mutation was predicted to be damaging by PolyPhen, was found to alter a conserved amino acid located in the catalytic domain, and was not identified in alleles from 200 ethnically matched controls.
The exclusion of PRKAR1A in five acrodysostosis cases prompted us to perform exome sequencing in two of these five individuals. Exome capture was performed with the SureSelect Human All Exon kit (Agilent Technologies).5 Single-end sequencing was performed on an Illumina Genome Analyzer IIx (Illumina) and generated 72 bp reads. For sequence alignment, variant calling, and annotation, we aligned the sequences to the human genome reference sequence (hg18 build) by using the Burrows-Wheeler Aligner.6 Downstream processing was carried out with the Genome Analysis Toolkit (GATK7), SAMtools,8 and Picard tools. Substitution calls were made with GATK Unified Genotyper, whereas indel calls were made with a GATK IndelGenotyperV2. All calls with a read coverage ≤2× and a Phred-scaled SNP quality of ≤20 were filtered out. All the variants were annotated with an in-house -developed annotation software system. We first focused our analyses on nonsynonymous variants, splice-acceptor and donor-site mutations, and coding indels because we anticipated that synonymous variants would be far less likely to cause disease (Table S1, available online). We also defined variants as previously unidentified if they were absent from control populations and from all datasets, including dbSNP129, the 1000 Genomes Project, and in-house exome data.
On the basis of the dominant mode of inheritance of acrodysostosis, we selected eight candidate genes that all harbor heterozygous mutations (Table S2). Given the involvement of PRKAR1A, a cAMP-activated protein kinase A, in some acrodysostosis cases,4 we then only considered gene(s) that encode proteins involved in the cAMP signaling pathway. Therefore, we regarded PDE4D (RefSeq accession number NM_001104631) as the best candidate gene. Indeed, PDE4D encodes a class IV cAMP-specific phosphodiesterase that regulates cAMP concentration. Exome analysis detected two PDE4D mutations (c.673C>A [p.Pro225Thr] and c.677T>C [p.Phe226Ser]) in the two individuals. These results were confirmed by Sanger sequencing. Subsequent screening of the 15 PDE4D coding exons in the three remaining cases led to the identification of two distinct heterozygous missense mutations (c.568T>G [p.Ser190Ala] and c.1759A>C [p.Thr587Pro]) in two additional cases. These mutations were not observed in the parents of acrodysostosis-affected individuals, confirming that they occurred de novo.
We identified a total of four distinct heterozygous PDE4D mutations in four individuals (Table 1). Among them, the p.Ser190Ala substitution affected a serine residue predicted to be phosphorylated (Uniprot database), and the p.Thr587Pro substitution disturbed the conserved catalytic PDEase_I domain (pfam database), which confers the 3′5′-cyclic nucleotide phosphodiesterase activity. The two remaining alterations (p.Pro225Thr and p.Phe226Ser) affected conserved residues across species. All four mutations were considered to be pathogenic by PolyPhen and were absent from alleles in 200 ethnically matched controls.
Here, we report PDE4D mutations in four unrelated cases of acrodysostosis and PRKAR1A mutations in five cases. All mutations occurred de novo, providing further evidence that acrodysostosis has a dominant mode of inheritance.
After we divided up the acrodysostosis-affected individuals and grouped them according to the mutations they had, our study revealed interesting genotype-phenotype correlations. Indeed, the four individuals with PDE4D mutations shared characteristic facial features, namely midface hypoplasia with the canonical nasal hypoplasia initially reported in acrodysostosis and moderate intellectual disability with speech delay.1,2 The characteristic facial dysostosis and intellectual disability were neither observed in our individuals with PRKAR1A mutations nor mentioned in the three previously reported cases.4 Along the same lines, hormone resistance was observed in only one person with PDE4D mutations—case 6 had increased PTH levels and normal serum-phosphate levels—whereas hormone resistance was consistently observed in individuals carrying PRKAR1A mutations (all five suffered from chronic resistance to parathyroid hormone, and four of the five had peripheral hypothyroidism). Although our study does not allow generalized conclusions, our findings might suggest that individuals with facial dysostosis and moderate intellectual disability should be screened for PDE4D mutations, whereas individuals with less characteristic facial features, no intellectual disability, and hormone resistance should be screened for mutations in PRKAR1A.
The five individuals harboring PRKAR1A mutations presented with growth retardation (<−2 standard deviations [SDs]) and decreased growth speed in late childhood (between 7 and 13 years of age). The adult individuals had a final height <−3 SDs. Alternatively, the cases harboring PDE4D mutations presently have normal growth charts, but they are only 3–7 years old, and predicting final adult height is therefore impossible. It is worth noting that two out of the four PDE4D cases presented with an acute intracranial hypertension due to sinus thrombosis; both of these individuals required derivation surgery and medical treatment. This observation should prompt the careful investigation of headache complaints in such cases. This feature, hitherto unreported in PRKAR1A-mutation-positive individuals, might be another distinctive characteristic specific to the clinical spectrum of symptoms associated with PDE4D mutations. Finally, neither PDE4D nor PRKAR1A mutations were found in one adult individual who had characteristic skeletal features but no hormone resistance or facial dysostosis. One cannot exclude a molecular defect not detectable by Sanger sequencing, but it is also conceivable that other genes might account for acrodysostosis.
Considering PRKAR1A mutations, we confirm that c.1102C>T is a recurrent mutation observed in seven of the eight patients reported so far, whereas only one missense mutation that changes a conserved amino acid located in the cAMP binding domain has been identified. Interestingly, the Arg388∗ substitution is considered a gain-of-function mutation because it decreases protein-kinase-A sensitivity to cAMP.4 In contrast, germ-line loss-of-function mutations resulting in constitutive activation of protein kinase A are responsible for Carney complex (MIM 160980), an autosomal-dominant multiple-neoplasia syndrome characterized by cardiac, endocrine, cutaneous, and neural myxomatous tumors and pigmented lesions of the skin and mucosae.9
All mutations that we have identified in PDE4D are heterozygous missense mutations and are presumably responsible for impaired phosphodiesterase activity. PDE4D belongs to the cAMP-hydrolyzing phosphodiesterase family, which is directly involved in the rate of cAMP degradation. Considering the crucial role of cAMP in intracellular signaling in response to a number of membrane-impermeable hormones, a dysregulation of cAMP levels could be the underlying mechanism of the acrodysostosis that results from PDE4D mutations.
The cAMP-specific PDE4 family is widely expressed, and PDE4 isoforms have similar catalytic functions, but they have distinct cellular functions because of differences in specific intracellular trafficking and signaling-complex formation.10,11 PDE4D uses different promoters to generate multiple alternatively spliced transcript variants (at least nine) that encode functional proteins; this might explain the phenotype variability observed in the four reported cases.
Of note, mice deficient in PDE4D exhibited delayed growth and female infertility due to impaired ovulation;12 these two symptoms have also been described in acrodysostosis cases.13 Mouse models have also revealed that PDE4D plays a critical role in the memory and hippocampal neurogenesis mediated by cAMP signaling.14 Flies deficient in the PDE4D homolog, dunce, also display impaired central-nervous-system and reproductive function.15 All together, these data support the involvement of PDE4D impairment in the regulation of cAMP signaling, especially in growth and central-nervous-system development.
Finally, our findings further support the key role of the cAMP signaling pathway in skeletogenesis, as previously shown for Albright hereditary osteodystrophy due to GNAS mutations. Ongoing studies will highlight the specific link between PRKAR1A and PDE4D, which are both involved in cAMP signaling and responsible for acrodysostosis.
Acknowledgments
We thank all individuals and their families for their contribution to this work.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Picard Tools, http://picard.sourceforge.net
Polyphen, http://genetics.bwh.harvard.edu/pph/
Uniprot, http://www.uniprot.org/
References
- 1.Maroteaux P., Malamut G. Acrodysostosis. Presse Med. 1968;76:2189–2192. [PubMed] [Google Scholar]
- 2.Robinow M., Pfeiffer R.A., Gorlin R.J., McKusick V.A., Renuart A.W., Johnson G.F., Summitt R.L. Acrodysostosis. A syndrome of peripheral dysostosis, nasal hypoplasia, and mental retardation. Am. J. Dis. Child. 1971;121:195–203. [PubMed] [Google Scholar]
- 3.Bastepe M., Jüppner H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005;63:65–74. doi: 10.1159/000083895. [DOI] [PubMed] [Google Scholar]
- 4.Linglart A., Menguy C., Couvineau A., Auzan C., Gunes Y., Cancel M., Motte E., Pinto G., Chanson P., Bougnères P. Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N. Engl. J. Med. 2011;364:2218–2226. doi: 10.1056/NEJMoa1012717. [DOI] [PubMed] [Google Scholar]
- 5.Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirschner L.S., Carney J.A., Pack S.D., Taymans S.E., Giatzakis C., Cho Y.S., Cho-Chung Y.S., Stratakis C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 10.Rall T.W., Sutherland E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958;232:1065–1076. [PubMed] [Google Scholar]
- 11.Sutherland E.W., Rall T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958;232:1077–1091. [PubMed] [Google Scholar]
- 12.Jin S.L., Richard F.J., Kuo W.P., D'Ercole A.J., Conti M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci. USA. 1999;96:11998–12003. doi: 10.1073/pnas.96.21.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham J.M., Jr., Krakow D., Tolo V.T., Smith A.K., Lachman R.S. Radiographic findings and Gs-alpha bioactivity studies and mutation screening in acrodysostosis indicate a different etiology from pseudohypoparathyroidism. Pediatr. Radiol. 2001;31:2–9. doi: 10.1007/s002470000355. [DOI] [PubMed] [Google Scholar]
- 14.Li Y.F., Cheng Y.F., Huang Y., Conti M., Wilson S.P., O'Donnell J.M., Zhang H.T. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J. Neurosci. 2011;31:172–183. doi: 10.1523/JNEUROSCI.5236-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudai Y., Jan Y.N., Byers D., Quinn W.G., Benzer S. dunce, a mutant of Drosophila deficient in learning. Proc. Natl. Acad. Sci. USA. 1976;73:1684–1688. doi: 10.1073/pnas.73.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.