

Abstract
Joubert syndrome (JBTS) is an autosomal-recessive disorder characterized by a distinctive mid-hindbrain malformation, developmental delay with hypotonia, ocular-motor apraxia, and breathing abnormalities. Although JBTS was first described more than 40 years ago in French Canadian siblings, the causal mutations have not yet been identified in this family nor in most French Canadian individuals subsequently described. We ascertained a cluster of 16 JBTS-affected individuals from 11 families living in the Lower St. Lawrence region. SNP genotyping excluded the presence of a common homozygous mutation that would explain the clustering of these individuals. Exome sequencing performed on 15 subjects showed that nine affected individuals from seven families (including the original JBTS family) carried rare compound-heterozygous mutations in C5ORF42. Two missense variants (c.4006C>T [p.Arg1336Trp] and c.4690G>A [p.Ala1564Thr]) and a splicing mutation (c.7400+1G>A), which causes exon skipping, were found in multiple subjects that were not known to be related, whereas three other truncating mutations (c.6407del [p.Pro2136Hisfs∗31], c.4804C>T [p.Arg1602∗], and c.7477C>T [p.Arg2493∗]) were identified in single individuals. None of the unaffected first-degree relatives were compound heterozygous for these mutations. Moreover, none of the six putative mutations were detected among 477 French Canadian controls. Our data suggest that mutations in C5ORF42 explain a large portion of French Canadian individuals with JBTS.
Main Text
Joubert syndrome (JBTS [MIM 213300]) is an autosomal-recessive disorder characterized by the presence of hypotonia, apnea or hyperpnea in infancy, oculomotor apraxia, and variable developmental delay or intellectual impairment (reviewed in Sattar et al.1). The diagnostic hallmark of JBTS is the presence of a complex malformation of the midbrain-hindbrain junction that comprises cerebellar vermis hypoplasia or aplasia, deepened interpeduncular fossa, and elongated superior cerebellar peduncles. This malformation appears like a molar tooth on an axial brain MRI (magnetic resonance imaging). In a subset of individuals, JBTS also involves other organs and results in cystic kidneys, retinopathy, or polydactyly. JBTS is a genetically heterogeneous condition for which 15 genes have been described to date.2–19 All of these genes appear to play a role in the development and/or function of nonmotile cilia. Although JBTS was first described in French Canadian siblings more than 40 years ago by Marie Joubert and colleagues, until now, the causal mutations have not yet been identified in the original family nor in most French Canadians subjects.20,21
There is a high prevalence of JBTS in the French Canadian population living in the Lower St. Lawrence (“Bas-du-Fleuve” in French) region of the province of Quebec (Figure 1). In total, we identified 16 living affected individuals (from 11 unrelated families) who have at least one grandparent originating from that region. Informed consent was obtained from all individuals or their legal guardians. This project was approved by our institutional ethics committee. We were initially able to collect blood-derived DNA from 15 of these individuals, including an affected individual (II-1 in family 394; individual BD in Joubert et al.20) from the original JBTS family described by Marie Joubert and colleagues in 1969. There was a striking cluster of seven families from the east end of the region (Matapedia region); one family is from Mont-Joli (population of 6,568), three families are from Amqui (population of 6,261), and three other families are from Sayabec (population of 1,877). Individual II-1 from family 394 did not undergo brain-imaging studies, but an MRI scan performed on her brother (II-2) showed the molar-tooth sign (MTS) (Figure 2B).21 All the other affected individuals showed the MTS and variable expression of the classical JBTS features. The cohort included three families with two affected siblings, and the parents were not affected in any family (consistent with a recessive mode of transmission).
Figure 1.
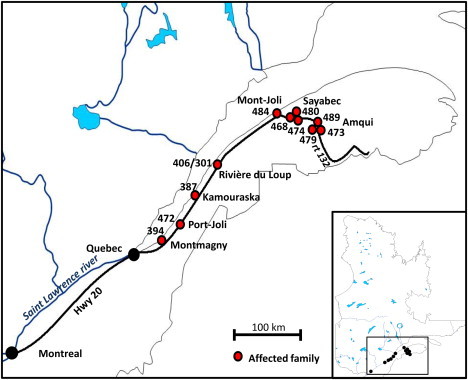
Distribution of Individuals with JBTS in the Lower St. Lawrence Region
Numbers refer to families (pedigrees in Figure 2). Note the cluster of families along Route 132, which follows the Matapedia River.
Figure 2.
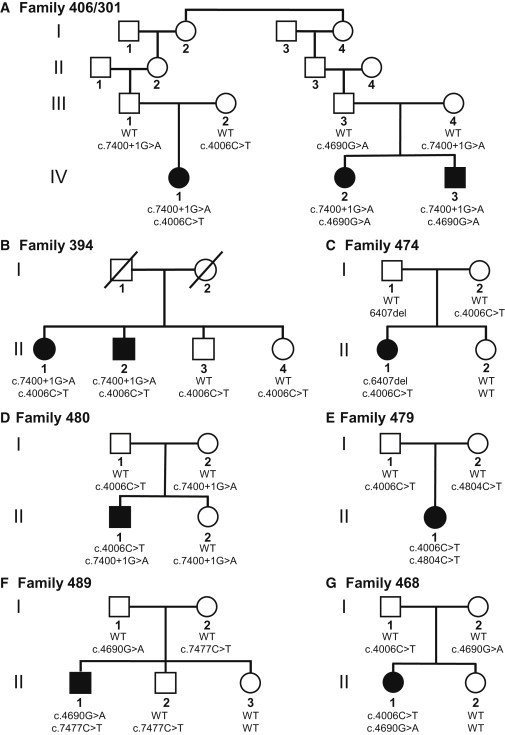
Segregation of C5ORF42 Mutations in Families Affected by JBTS
It was initially established that the population of the Lower St. Lawrence region was a result of both the immigration of a limited number of settlers (6,000 individuals) from Quebec City and its surrounding areas in the late 17th century and beginning of the 18th century and a rapid increase in settlers resulting from a high fertility rate.22 The establishment of settlers in the region followed a west-to-east pattern, and settlers later migrated to regions farther east. A small number of Acadians also contributed to the early population of the Matapedia region.23 The demographic growth of this population thus appears to be characterized by a series of bottlenecks that might have resulted in regional founder effects. We hypothesized that a founder effect could underlie the clustering of individuals with JBTS in the Lower St. Lawrence region, raising the possibility that a common homozygous mutation explains a large portion of them. We performed whole-genome SNP genotyping in all 15 individuals with JBTS by using the Illumina Human 610 Genotyping BeadChip panel, which interrogates 620,901 SNPs, and we used PLINK24 to search for homozygosity regions containing >30 consecutive SNPs and extending over >1Mb. We identified several overlapping regions of shared homozygosity, but these regions were not found in more than five families, were small (1 megabase or less), and contained genes that are unlikely to play a role in cilia development and/or function (Table S1, available online). Altogether, the genotyping data suggest the presence of allelic and/or genetic heterogeneity within our cohort.
Given the lack of hints from genotype-based mapping, we decided to sequence the protein-coding exomes of all our JBTS-affected subjects in the hopes of identifying a unique candidate gene harboring private pathogenic variants in a large fraction of the samples. Genomic DNA from each sample was captured with the Agilent SureSelect 50 Mb oligonucleotide library, and the captured DNA was sequenced with paired-end 100 bp reads on Illumina HiSeq2000. The result was an average of 14.7 Gb of raw sequence for each sample. Data were analyzed as previously described.25 After we used Picard (v. 1.48) to remove putative PCR-generated duplicate reads, we aligned the reads to human genome assembly hg19 by using a Burroughs-Wheeler algorithm (BWA v. 0.5.9). The median read depth of the bases in CCDS (consensus coding sequence) exons was 115 (determined with Broad Institute Genome Analysis Toolkit v. 1.0.4418).26 On average, 88% (±2.9%) of the bases in CCDS exons were covered by at least 20 reads. We called sequence variants by using custom scripts for Samtools (v. 0.1.17), Pileup, and varFilter, and we required at least three variant reads as well as >20% variant reads for each called position. Single-nucleotide variants (SNVs) had Phred-like quality scores of at least 20, and small insertions or deletions (indels) had scores of at least 50. We used Annovar to annotate variants according to the type of mutation, occurrence in dbSNP, SIFT score, and 1,000 Genomes allele frequency.27 To identify potentially pathogenic variants, we filtered out (1) synonymous variants or intronic variants other than those affecting the consensus splice sites, (2) variants seen in more than one of 261 exomes from individuals with rare, monogenic diseases unrelated to JBTS (these individuals were sequenced at the McGill University and Genome Quebec Innovation Centre), and (3) variants with a frequency greater than 0.5% in the 1,000 Genomes Browser (Tables 1 and 2).
Table 1.
Variant Prioritization Steps in the Analysis of Combined Exome Sequences from 13 Individuals with JBTS
| Filters Applied (Sequentially) | Number of Variants Retained |
|---|---|
| Nonsynonymous, splicing, and coding indel variants | 34,157a |
| After excluding variants present in >1 in-house exome | 7,075 |
| After excluding variants reported in 1,000 Genomes Browser (frequency > 0.5%) |
6,911 |
Total number of variants identified in the combined 13 exomes; redundant variants were counted only once.
Table 2.
Genes with Rare Homozygous or Multiple Heterozygous Variants from the Combined Exome Sequences from 13 Individuals with JBTS
| Number of Families with Mutations in the Same Gene | Number of Genes | Gene Identity |
|---|---|---|
| 1 family | 528 | C5ORF42, … |
| 2 families | 16 | C5ORF42, ACAN, ADAMTS18, C10orf68, FSIP2, LRP1B, MUC12, MUC16, MUC4, MYO16, PKD1L2, PKHD1L1, RGPD4, SHROOM4, TMEM231, ZNF717 |
| 3 families | 7 | C5ORF42, MUC5B, PLEC, FAT3, FLG, TTN, LAMA5 |
| 4 families | 1 | C5ORF42 |
| 5 families | 1 | C5ORF42 |
| >5 families | 0 | - |
We first examined the exome datasets to look for rare variants in the 15 genes already associated with JBTS (these genes are INPP5E [MIM 613037], TMEM216 [MIM 613277], AHI1 [MIM 608894], NPHP1 [MIM 607100], CEP290 [MIM 610142], TMEM67 [MIM 609884], RPGRIP1L [MIM 610937], ARL13B [MIM 608922], CC2D2A [MIM 612013], CXORF5 [MIM 300170], KIF7 [MIM 611254], TCTN1 [MIM 609863], TCTN2 [MIM 613885], TMEM237 [MIM 614424], and CEP41)2–19 as well as in the JBTS candidate gene, TTC21B (MIM 612014).28 Two individuals (II-1 from family 484 and II-2 from family 473, Figure S2) that are not known to be related were each found to be carrying two heterozygous missense variants (c.4667A>T [p.Asp1556Val] and c.3376G>A [p.Glu1126Lys]) in CC2D2A (RefSeq accession number NM_001080522.2).13,14 These amino acids are highly conserved, and both mutations are predicted to be deleterious according to SIFT (scores < 0.05)29 and Polyphen-2 (scores > 0.90)30 (Figure S1). The c.4667A>T (p.Asp1556Val) mutation has already been reported in individuals with JBTS.31 Segregation studies have indicated that the affected individuals but none of their unaffected first-degree relatives were compound heterozygous for these mutations (Figure S2). We conclude that these mutations are probably pathogenic. Both individuals have a mild phenotype. They have oculomotor apraxia and only mild motor delay (they walked at 18 [II-1 from family 484] and 19 [II-2 from family 473] months of age and do not have gait ataxia). The individual who is of school age performs well in a regular classroom. Four additional individuals were singly heterozygous for rare variants in the other known JBTS-associated genes; such variants are c.265C>T (p.Leu89Phe) in TMEM216 (M_001173991.2), c.3257A>G (p.Glu1086Gly) in AHI1 (NM_001134831), c.1600G>A (p.Glu534Lys) in CEP290 (NM_025114.3), and c.3032T>C (p.Met1011Thr) in TTC21B (NM_024753.4). Because each of these genes has previously been associated with recessive JBTS, these heterozygous variants are unlikely to fully explain the disorder that these individuals have.
We next looked at the whole-exome data for the other protein-coding genes containing homozygous or multiple heterozygous variants in the 13 affected individuals who did not have mutations in CC2D2A (Tables 1 and 2). Strikingly, five subjects, including a member of the initial JBTS family, carried two different heterozygous variants in an unstudied anonymous gene, C5ORF42 (NM_023073.3). Mutations in six other genes were found in affected individuals among sets of three families (Tables 1 and 2). Because these latter genes (MUC5B, PLEC, FAT3, FLG, TTN, and LAMA5) are known to accumulate mutations at a high rate, they are unlikely to be linked to the disease (Table S2). All five affected individuals with changes in C5ORF42 carried the same missense mutation, c.4006C>T (p.Arg1336Trp) (NM_023073.3), as well as one of three different mutations: one mutation that affects a consensus donor splice site, c.7400+1G>A (NM_023073.3), and two truncating mutations, c.6407del (p.Pro2136Hisfs∗31) and c.4804C>T (p.Arg1602∗) (NM_023073.3) (Figures 2 and 3 and Table 3). Sanger sequencing in the five affected individuals confirmed the presence of these variants. Segregation studies indicated that the affected individuals, but not their unaffected first-degree relatives, were compound heterozygotes for these variants (Figure 2 and Table 3). Subsequently, we were able to collect DNA from individual II-2 (individual M.D.19-20), the affected brother of II-1 in the initial JBTS family (family 394), and we found that he was compound heterozygous for the same C5ORF42 mutations identified in his affected sister (Figure 2B and Table 3). None of these four variants was detected in 261 in-house control exomes, which were derived from other projects including some French Canadian subjects, and in the 1,000 Genomes Browser. RT-PCR performed on RNA extracted from the blood of individuals II-2 (from family 394) and III-4 (from family 406/301), who both carry the c.7400+1G>A splicing mutation, showed that this mutation causes skipping of exon 35 in C5ORF42 (NM_023073.3) and results in the creation of a premature stop codon (Figure S3). The p.Arg1336Trp amino acid substitution is predicted to be damaging (SIFT = 0.00; Polyphen-2 = 0.99) and to affect a residue that is conserved across vertebrate species (Figure 3B).
Figure 3.
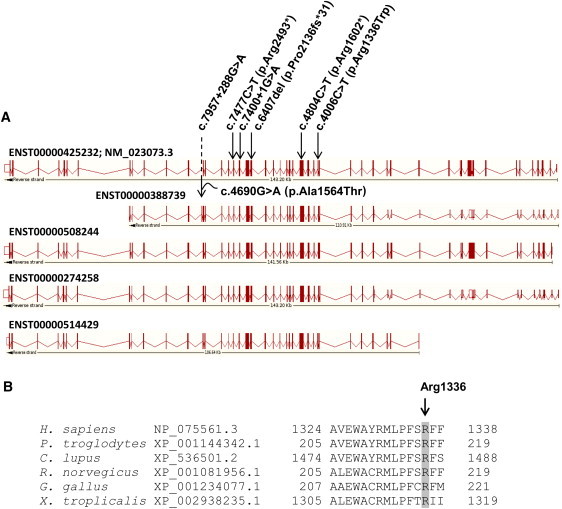
C5ORF42 Mutations Identified in Individuals with JBTS
(A) Scheme showing the positions of the mutations with respect to the different C5ORF42 Ensembl-annotated transcripts that are predicted to produce proteins. The numbering on top is based on the cDNA positions of ENST00000425232 (identical to RefSeq accession number NM_023073.3). Mutation c.7957+288G>A is annotated as part of a coding exon in ENST00000388739 and causes a missense change (p.Ala1564Thr).
(B) NCBI HomoloGene-generated amino acid alignment of C5ORF42. Its predicted orthologs show the conservation of the Arg1336 residue.
Table 3.
Clinical Description of JBTS Individuals with C5ORF42 Mutations
| Genotype |
Family 406/301 |
Family 394 |
Family 474 |
Family 480 |
Family 489 |
Family 479 |
Family 468 |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| IV-1 | IV-2 | IV-3 | II-1 | II-2 | II-1 | II-1 | II-1 | II-1 | II-1 | |
| c.4006C>T (p.Arg1336Trp) | + | − | − | + | + | + | + | − | + | + |
| c.7400+1G>A | + | + | + | + | + | − | + | − | − | − |
| c.6407del (p.Pro2136Hisfs∗31) | − | − | − | − | − | + | − | − | − | − |
| c.7477C>T (p.Arg2493∗) | − | − | − | − | − | − | − | + | − | − |
| c.4804C>T (p.Arg1602∗) | − | − | − | − | − | − | − | − | + | − |
| c.7957+288G>A (c.4690G>A [p.Ala1564Thr]) | − | + | + | − | − | − | − | + | − | + |
| Age (years) | 8 | 1.5 | 3 | 52 | 45 | 4 | 10 | 7 | 13 | 31 |
| Sex | F | M | F | F | M | F | M | M | F | F |
| Developmental delay | + | + | + | + | + | + | + | + | + | + |
| Oculomotor apraxia | − | + | + | + | + | + | + | + | + | + |
| Breathing abnormality | + | + | + | + | + | + | + | + | − | − |
| Limb abnormalitya | − | + | − | − | − | + | − | − | − | − |
| Brain MRI | MTS | MTS | MTS | ND | MTS | MTS | MTS | MTS | MTS | MTS |
| Retinal involvementb | − (f) | − (e) | − (e) | − (h) | − (h) | − (f) | − (e) | − (e) | − (f) | − (h) |
| Renal involvementc | − (us) | − (us) | − (us) | − (h) | − (h) | − (us) | − (us) | − (us) | − (us) | − (h) |
The nucleotide and amino acid positions are based on reference sequence NM_023073.3 except for c.4690G>A (p.Ala1564Thr), which is based on Ensembl transcript ENST00000509849. The following abbreviations are used: F, female; M, male; MRI, magnetic resonance imaging; MTS, molar tooth sign; ND, not done; f, fundoscopy; e, electroretinogram; h, history; and us, ultrasound.
Individual IV-2 from family 406/301 has a 3/4 syndactyly in the left hand and individual II.1 from family 474 has preaxial and postaxial polydactyly of the four limbs. Individual II-1 from family 394 did not undergo an MRI, but the MRI of her brother (individual II-2 from family 394) documented a MTS.
Lack of retinal involvement was determined by electroretinogram, fundoscopy, or history.
Lack of renal involvement was determined by renal ultrasound or history.
On the basis of the exome-sequencing data, four additional JBTS-affected individuals from three families (301, 468, and 489) were each carrying a single heterozygous C5ORF42 mutation, including the already described c.4006C>T (p.Arg1336Trp) and c.7400+1G>A mutations and the truncating mutation c.7477C>T (p.Arg2493∗) (NM_023073.3) (Figure 2 and Table 3). The c.7477C>T (p.Arg2493∗) mutation was absent from our 261 control exomes and the 1,000 Genomes Browser. Our SNP genotyping data suggest that these four individuals—but not the other individuals with JBTS in our cohort—are heterozygous for a unique 5 Mb haplotype that encompasses C5ORF42 (Figure S4). It seemed unlikely that this haplotype would be carrying three different rare mutations; therefore, this observation suggests that the four individuals might carry a second mutation linked to this haplotype. Upon further inspection of the exome data, we discovered that all four individuals are also heterozygous for another missense variant, c.4690G>A (p.Ala1564Thr) (based on the ENST00000388739 transcript annotated by the Ensemble Genome Browser). This allele was not included in our original filtered dataset because it is located in an internal coding exon (chr5: 37,157,522–37,157,415) not annotated by RefSeq for the longest isoform of the gene (NM_023073.3). Sanger sequencing confirmed the presence of the various mutations in the four affected individuals. Segregation studies showed that the four affected individuals but none of their unaffected first-degree relatives were compound heterozygous for c.4690G>A (p.Ala1564Thr) and for one of the three other mutations (c.4006C>T [p.Arg1336Trp], c.7400+1G>A, and c.7477C>T [p.Arg2493∗]) (Figure 2). The additional, alternative exon (which we designate exon 40a) with the c.4690G>A (p.Ala1564Thr) mutation occurs between RefSeq annotated exons 40 and 41 (NM_023073.3), is present in brain expressed sequence tag (EST) clones with GenBank accession numbers AK096581 and BC144070, and retains the large open reading frame of the gene. Using RNA-sequencing data made publicly available by Illumina's Body Map 2.0 (see Web Resources), we were able to confirm the expression of the exon. The assembly of raw data from 16 different tissues identified a large number of reads that mapped to that exon in both brain and testes samples; significantly fewer reads mapped to other tissues (Figure S5). Reads that covered both ends of the exon and spliced correctly to neighboring exons were found in either brain or testes samples. The c.4690G>A (p.Ala1564Thr) mutation was also absent from our 261 control exomes and from the 1,000 Genomes Browser. It was not possible to get accurate SIFT or Polyphen-2 predictions for this mutation because the corresponding exon was not annotated across species.
We further addressed the frequency of the six putative C5ORF42 mutations identified in our JBTS individuals in the French Canadian population. Genotyping 477 French Canadian controls, including 96 Acadians subjects and 96 subjects from the Gaspésie region located immediately east of Matapedia, did not identify a carrier of any of the six C5ORF42 mutations. However, some of these mutations are reported in the heterozygous state at very low frequencies in the National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project (ESP) dataset; these mutations are c.4006C>T (p.Arg1336Trp) (2/10,754; minor allele frequency [MAF] = 0.0186%), c.7477C>T (p.Arg2493∗) (1/10,755; MAF = 0.009%; rs139675596), and c.4690G>A (p.Ala1564Thr) (12/4,574; MAF = 0.262%; rs111294855). It should be noted that c.4006C>T and c.7477C>T correspond to CpG sites, which are associated with a higher mutation rate, possibly explaining the recurrence of these nonetheless rare mutations in different populations.
The presence of five potentially deleterious C5ORF42 mutations that segregate with the disease in seven presumably unrelated (though all French Canadian) families strongly suggests that disruption of this gene causes JBTS in our subjects. It remains uncertain whether c.4690G>A (p.Ala1564Thr) is pathogenic, considering that it is not clearly deleterious and that it is found at a higher frequency (0.26%) in the ESP dataset than are the other mutations. It is possible that this variant is linked to another mutation—not identified by our exome-sequencing approach—on the same haplotype.
Very little is known about C5ORF42 function. The RefSeq version of the full-length transcript (NM_023073.3; Ensemble accession number ENST00000425232) apparently derives from virtual assembly of overlapping mRNA and EST clones. The predicted major mRNA isoform comprises 11,199 bp and contains 52 exons; the putative encoded protein is similarly large and comprises 3,198 amino acids. With the exception of c.4690G>A (p.Ala1564Thr), all mutations reported herein are common to all annotated protein-coding transcripts (Figure 3A). The predicted protein sequence is well conserved across much of the gene length in other vertebrates. It does not appear to contain any specific known functional domains, although the Gene Ontology project suggests that it might be a transmembrane protein and ProtoNet predicts a coiled-coil structure within the protein. Proteomic studies have reported interactions among C5ORF42, the p21-activating kinase 1 (PAK1), and the small ubiquitin-like modifier 1 (SUMO1).32,33 Although the significance of these interactions remains to be validated and further investigated, it is noteworthy that these latter genes play a role in neural development.34,35 EST-expression (Unigene data), microarray profiling (Allen Brain Atlas), and BioGPS indicate that C5ORF42 is widely expressed in a variety of tissues, including the brain.
In terms of genotype-phenotype correlation, all JBTS individuals with mutations in C5ORF42 showed global developmental delay, and the onset of independent walking ranged between 30 months and 8 years of age (Table 3). Cognitive impairment was present in all individuals but was variable, ranging from borderline intelligence to mild intellectual disability. The majority of individuals also showed oculomotor apraxia and breathing abnormalities mainly characterized by episodes of hyperventilation. Two individuals showed limb abnormalities; one had preaxial and postaxial polydactyly, and another had syndactyly of the third and fourth finger on one hand. There was no evidence of retinal or kidney involvement. There was no clear correlation between the type of C5ORF42 mutation and the associated phenotype.
Surprisingly, we found that three mutations (c.4006C>T [p.Arg1336Trp], c.7400+1G>A, and c.4690G>A [p.Ala1564Thr]) in C5ORF42 were present in multiple individuals in our cohort. Haplotype studies indicate that each of these mutations is linked to a distinct haplotype in these families despite the lack of documented genealogical relationships among them (Figure S4). The higher frequency of these mutations in the population of the Lower St. Lawrence region could be explained by a founder effect with the coincidental occurrence of the three mutations in the same group of settlers or by multiple regional founder effects corresponding to sequential pioneer fronts. Although founder effects are typically associated with an increase in the frequency of a specific allele,33 which is often accompanied by other alleles that remain at their usual background frequency, they can also involve multiple common mutations.36,37
In summary, after the initial description of JBTS in a French Canadian family 40 years ago, we have shown that mutations in C5ORF42 explain this neurodevelopmental disorder in many affected individuals from the French Canadian population. We have also found that C5ORF42 is associated with a complex founder effect in this population. Although the function of C5ORF42 remains unknown, future studies will likely elucidate its role in cilia development and/or function.
Acknowledgments
Foremost, we thank the families who generously contributed their time and materials to this research study. This work was selected for study by the FORGE Canada Steering Committee, consisting of K. Boycott (University of Ottawa), J. Friedman (University of British Columbia), J. Michaud (Université de Montréal), F. Bernier (University of Calgary), M. Brudno (University Toronto), B. Fernandez (Memorial University), B. Knoppers (McGill University), M. Samuels (Université de Montréal), and S. Scherer (University of Toronto). We would like to thank Janet Marcadier (clinical coordinator) and Chandree Beaulieu (project manager) for their contribution to the infrastructure of the FORGE Canada Consortium. The authors also wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Québec Innovation Centre (Montréal, Canada). This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR), and the Ontario Genomics Institute (OGI-049). Additional funding was provided by Genome Québec and Genome British Columbia. K. Boycott is supported by a Clinical Investigatorship Award from the CIHR Institute of Genetics. J.L. Michaud is a National Scholar from the Fonds de la Recherche en Santé du Québec (FRSQ). M. Srour holds a training award from the FRSQ.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1,000 Genomes Browser, http://browser.1000genomes.org/index.html
Allen Brain Atlas, http://www.brain-map.org/
BioGPS, http://biogps.org
Ensemble Genome Browser, http://www.ensembl.org
ESP Exome Variant Server, http://evs.gs.washington.edu/EVS/
Gene Ontology, http://www.geneontology.org/
Illumina's Body Map 2.0 transcriptome, http://www.ebi.ac.uk/arrayexpress/browse.html?keywords = E-MTAB-513
NCBI HomoloGene, http://www.ncbi.nlm.nih.gov/homologene
NCBI Nucleotide Database, http://www.ncbi.nlm.nih.gov/nuccore
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Polyphen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
Unigene, http://www.ncbi.nlm.nih.gov/unigene
References
- 1.Sattar S., Gleeson J.G. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev. Med. Child Neurol. 2011;53:793–798. doi: 10.1111/j.1469-8749.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielas S.L., Silhavy J.L., Brancati F., Kisseleva M.V., Al-Gazali L., Sztriha L., Bayoumi R.A., Zaki M.S., Abdel-Aleem A., Rosti R.O. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edvardson S., Shaag A., Zenvirt S., Erlich Y., Hannon G.J., Shanske A.L., Gomori J.M., Ekstein J., Elpeleg O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet. 2010;86:93–97. doi: 10.1016/j.ajhg.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon-Salazar T., Silhavy J.L., Marsh S.E., Louie C.M., Scott L.C., Gururaj A., Al-Gazali L., Al-Tawari A.A., Kayserili H., Sztriha L., Gleeson J.G. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004;75:979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parisi M.A., Bennett C.L., Eckert M.L., Dobyns W.B., Gleeson J.G., Shaw D.W., McDonald R., Eddy A., Chance P.F., Glass I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L., International Joubert Syndrome Related Disorders Study Group Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 8.Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 9.Baala L., Romano S., Khaddour R., Saunier S., Smith U.M., Audollent S., Ozilou C., Faivre L., Laurent N., Foliguet B. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Märker T., Voesenek K., Kartono A. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 11.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 12.Cantagrel V., Silhavy J.L., Bielas S.L., Swistun D., Marsh S.E., Bertrand J.Y., Audollent S., Attié-Bitach T., Holden K.R., Dobyns W.B., International Joubert Syndrome Related Disorders Study Group Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noor A., Windpassinger C., Patel M., Stachowiak B., Mikhailov A., Azam M., Irfan M., Siddiqui Z.K., Naeem F., Paterson A.D. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am. J. Hum. Genet. 2008;82:1011–1018. doi: 10.1016/j.ajhg.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorden N.T., Arts H.H., Parisi M.A., Coene K.L., Letteboer S.J., van Beersum S.E., Mans D.A., Hikida A., Eckert M., Knutzen D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dafinger C., Liebau M.C., Elsayed S.M., Hellenbroich Y., Boltshauser E., Korenke G.C., Fabretti F., Janecke A.R., Ebermann I., Nürnberg G. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest. 2011;121:2662–2667. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L.J., Szymanska K., Jensen V.L., Janecke A.R., Innes A.M., Davis E.E., Frosk P., Li C.M., Willer J.R., Chodirker B.N. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J.E., Silhavy J.L., Zaki M.S., Schroth J., Bielas S.L., Marsh S.E., Olvera J., Brancati F., Iannicelli M., Ikegami K. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 2012;44:193–199. doi: 10.1038/ng.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joubert M., Eisenring J.J., Robb J.P., Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 21.Andermann F., Andermann E., Ptito A., Fontaine S., Joubert M. History of Joubert syndrome and a 30-year follow-up of the original proband. J. Child Neurol. 1999;14:565–569. doi: 10.1177/088307389901400903. [DOI] [PubMed] [Google Scholar]
- 22.Fortin J.C., Lechasseur A. Presses de l'Université Laval; Québec: 1999. Le Bas-Saint-Laurent. [Google Scholar]
- 23.Hébert P.M. Éditions de l′écho; Montréal: 1994. Les Acadiens du Québec. [Google Scholar]
- 24.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majewski J., Schwartzentruber J.A., Caqueret A., Patry L., Marcadier J., Fryns J.P., Boycott K.M., Ste-Marie L.G., McKiernan F.E., Marik I., FORGE Canada Consortium Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011;32:1114–1117. doi: 10.1002/humu.21546. [DOI] [PubMed] [Google Scholar]
- 26.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mougou-Zerelli S., Thomas S., Szenker E., Audollent S., Elkhartoufi N., Babarit C., Romano S., Salomon R., Amiel J., Esculpavit C. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum. Mutat. 2009;30:1574–1582. doi: 10.1002/humu.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bandyopadhyay S., Chiang C.Y., Srivastava J., Gersten M., White S., Bell R., Kurschner C., Martin C.H., Smoot M., Sahasrabudhe S. A human MAP kinase interactome. Nat. Methods. 2010;7:801–805. doi: 10.1038/nmeth.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganesan A.K., Kho Y., Kim S.C., Chen Y., Zhao Y., White M.A. Broad spectrum identification of SUMO substrates in melanoma cells. Proteomics. 2007;7:2216–2221. doi: 10.1002/pmic.200600971. [DOI] [PubMed] [Google Scholar]
- 34.Huang W., Zhou Z., Asrar S., Henkelman M., Xie W., Jia Z. p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol. Cell. Biol. 2011;31:388–403. doi: 10.1128/MCB.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilkinson K.A., Nakamura Y., Henley J.M. Targets and consequences of protein SUMOylation in neurons. Brain Res. Brain Res. Rev. 2010;64:195–212. doi: 10.1016/j.brainresrev.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yotova V., Labuda D., Zietkiewicz E., Gehl D., Lovell A., Lefebvre J.F., Bourgeois S., Lemieux-Blanchard E., Labuda M., Vézina H. Anatomy of a founder effect: Myotonic dystrophy in Northeastern Quebec. Hum. Genet. 2005;117:177–187. doi: 10.1007/s00439-005-1298-8. [DOI] [PubMed] [Google Scholar]
- 37.Roddier K., Thomas T., Marleau G., Gagnon A.M., Dicaire M.J., St-Denis A., Gosselin I., Sarrazin A.M., Larbrisseau A., Lambert M. Two mutations in the HSN2 gene explain the high prevalence of HSAN2 in French Canadians. Neurology. 2005;64:1762–1767. doi: 10.1212/01.WNL.0000161849.29944.43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.