

Abstract
Birth weight is an important indicator of both perinatal and adult health, but little is known about the genetic factors contributing to its variability. Intrauterine growth restriction is a leading cause of perinatal morbidity and mortality and is also associated with adult disease. A significant correlation has been reported between lower birth weight and increased expression of the maternal PHLDA2 allele in term placenta (the normal imprinting pattern was maintained). However, a mechanism that explains the transcriptional regulation of PHLDA2 on in utero growth has yet to be described. In this study, we sequenced the PHLDA2 promoter region in 263 fetal DNA samples to identify polymorphic variants. We used a luciferase reporter assay to identify in the PHLDA2 promoter a 15 bp repeat sequence (RS1) variant that significantly reduces PHLDA2-promoter efficiency. RS1 genotyping was then performed in three independent white European normal birth cohorts. Meta-analysis of all three (total n = 9,433) showed that maternal inheritance of RS1 resulted in a significant 93 g increase in birth weight (p = 0.01; 95% confidence interval [CI] = 22–163). Moreover, when the mother was homozygous for RS1, the influence on birth weight was 155 g (p = 0.04; 95% CI = 9–300), which is a similar magnitude to the reduction in birth weight caused by maternal smoking.
Main Text
Very low birth weight shows a strong association with perinatal mortality and morbidity and is linked to an increased risk of developing adulthood diseases, such as obesity and type 2 diabetes (MIM 125853).1,2 Fetal growth relies on an effective nutrient supply from the mother to the fetus via the placenta; this nutrient supply is influenced by a complex interrelationship between the environment and genetics. Of particular interest are imprinted genes, which show expression from only one allele in a parent-of-origin dependent manner. Genomic imprinting is found almost exclusively in placental mammals. Its evolution is probably best explained by the “conflict hypothesis,” which suggests that paternally expressed imprinted genes promote fetal growth and ensure inheritance of the paternal genome to successive generations, whereas maternally expressed imprinted genes limit growth in order for the mother to survive and reproduce again.3
PHLDA2 (MIM 602131) encodes the pleckstrin homology-like domain, family A, member 2 protein and is a maternally expressed imprinted gene found in one of the most extensively studied imprinting clusters in human chromosomal region 11p15.5. Consistent with the “conflict hypothesis,” Phlda2-null mice exhibit placenta overgrowth, whereas doubling the Phlda2 expression in transgenic mice results in placental stunting accompanied by a 13% reduction in fetal weight; both of these findings suggest that Phlda2 has a growth-suppressing role.4,5 In humans, PHLDA2 is expressed in a variety of tissues but is predominantly expressed in the villous cytotrophoblast of the placenta throughout gestation,6,7 and upregulation has been observed in intrauterine growth restriction (IUGR) placentas.8–10 This complements our previous finding that PHLDA2 expression is significantly higher in the term placenta of lower-birth-weight babies.11 However, sequence analysis of all informative samples in the “Moore cohort” of white European normal births confirmed that only maternal, monoallelic PHLDA2 expression was present.11 This indicates that loss of imprinting (LOI) was not responsible for the increased PHLDA2 expression and suggests that additional regulatory mechanisms, including the PHLDA2 promoter, other than imprinting must be involved.
In this study, we examined the PHLDA2 promoter region for genetic polymorphisms that might affect PHLDA2 transcriptional activity and therefore could affect birth weight. From the Moore cohort (n = 263), recruited from Queen Charlotte and Chelsea Hospital,11 we sequenced a ∼2 kb upstream region beginning at the transcription start site and overlapping the promoter CpG island. The UCSC Genome Browser (build GRCh37/hg19) listed 20 SNPs, encompassing rs12798267 to rs412300, in this region. However, none of these SNPs were identified in this cohort, suggesting that they either are rare in the white European population or have not been accurately validated. We only detected one variable sequence: a tandem 15 bp (5′-GGGGCGGGGAGGGGC- 3′; bp 4,934–4,967 of NG_009266.1) repeat sequence (RS) variant present 48 bp upstream of the PHLDA2 transcription start site (Figure S1, available online). The tandem repeat (RS2) is most common (it is present in 87% of chromosomes), and the minor allele is a single copy (RS1) that is found in the remaining 13% of chromosomes (Figure 1). In addition, RS1 was not found to be in linkage disequilibrium (LD) with nearby SNPs rs3847646 (located ∼3 kb upstream) or rs13390 or rs1056819 (present within PHLDA2 exons 1 and 2, respectively).
Figure 1.
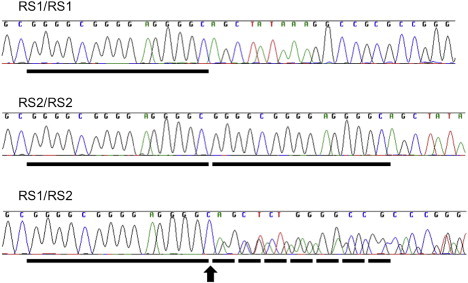
Sequence Electropherograms Showing the 15 bp RS in the PHLDA2 Promoter Region
RS1/RS1 homozygous, RS2/RS2 homozygous, and RS1/RS2 heterozygous sequences are shown. Each black bar represents the location of a single 15 bp copy. The start of the overlapping sequence in the heterozygous sample is indicated by the black arrow and dotted bars.
We investigated the effect of the PHLDA2 RS on the gene's promoter activity by transiently transfecting luciferase reporter constructs into the transformed human embryonic kidney (HEK) 293T cell line and the human trophoblast cell line 1 (TCL-1). We made the promoter constructs by cloning 300 bp (with the use of HindIII and Xho1) and 600 bp (with the use of HindIII and Sac1) DNA fragments upstream of the PHLDA2 start site and inserting them into the pGL3.1-Basic vector (Promega, UK). These sequences contained either RS1 or RS2. RS1 showed significantly lower PHLDA2 promoter activity for both the 300 bp (74% decrease; t test, p = 0.004) and 600 bp (42% decrease; t test, p = 0.001) constructs (Figure 2). The experiments were performed in duplicate for HEK293T cells and in triplicate for TCL-1 cells (Figure S2), and each assay included six replicates. TFSEARCH (Transcriptional Factor Search) shows that the RS2 allele potentially harbors four SP1 and two MZF1 binding sites. However, losing a 15 bp copy (RS1) removes three of these sites, suggesting the possibility that the number of available transcription-factor binding sites might be important for promoter efficiency.
Figure 2.
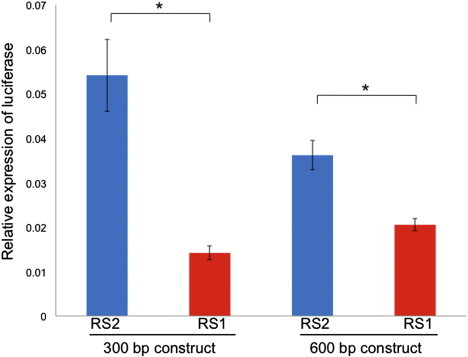
The Effects of RS1 and RS2 on PHLDA2 Promoter Efficiency in HEK 293T Cells
The bars indicate the firefly luciferase expression relative to Renilla luciferase activity in HEK 293T cells for the 300 bp and 600 bp constructs with either RS2 or RS1. Luciferase activity was measured 30 hr after transfection. This data shows the mean of six replicate samples ± SEM (standard error of the mean). Asterisks represent p < 0.05.
Given that high expression of PHLDA2 is associated with lower birth weight11 and that RS1 reduces the PHLDA2 promoter efficiency in vitro, we investigated whether RS1 could be associated with increased birth weight. Because PHLDA2 is maternally expressed and paternally silenced, RS1 homozygotes and heterozygotes with a maternally inherited RS1 were grouped and deemed the “RS1 effect group.” RS2 homozygotes and heterozygotes with a paternally inherited RS1 were named the “unaffected group” (Table S1). We assessed the parental origin of the RS1 allele in the heterozygous babies by genotyping their corresponding parental DNA samples. Uninformative cases were those in which both parents were heterozygotes.
We first genotyped the parental DNA samples corresponding to the heterozygous babies in the Moore cohort, and 28 babies were revealed to have maternally inherited RS1 (Table 1). To investigate the effect of maternally inherited RS1 on birth weight, we applied a linear-regression model and corrected for the following covariates: the gender of the baby, maternal weight, gestational age, parity and maternal diabetes, hypertension, and smoking habits (Table S2). A two-tailed test was used throughout; p values were based on Wald tests, and standard residual plots were examined and showed no evidence of departure from model assumptions. This analysis showed that babies in the RS1 effect group tended to have an average birth weight 122 g higher than that of the babies in the unaffected group (p = 0.15; 95% confidence interval [CI] = −43–286) (Table 1). We then carried out the same analysis on the UCL-FGS cohort (baby-parent trios, n = 385) from the University College London Fetal Growth Study.12 This produced a similar trend to the Moore cohort: babies in the RS1 effect group (n = 16) were on average 68 g heavier (p = 0.61; 95% CI = −196–332) (Table 1). The reproducibility of this trend suggests a potentially valid finding despite the fact that statistical confidence could not be achieved because too few individuals (approximately 13%) had maternal RS1 (Table 1).
Table 1.
Baby Genotypes and Influence on Birth Weight: Individual Studies and Meta-Analysis
| Study | RS2/RS2 | RS1/RS1 | RS2/RS1 | Pa | Mb | RS1 Effect Group | Effect Estimate (g) | 95% CI (g) | p value |
|---|---|---|---|---|---|---|---|---|---|
| Moore | 193 | 4 | 66 | 22 | 24 | 28 | 122 | −43–286 | 0.15 |
| UCL-FGS | 292 | 5 | 88 | 20 | 11 | 16 | 68 | −196–332 | 0.61 |
| ALSPAC | 6,649 | 128 | 2,008 | 465 | 51 | 179 | 88 | 6–170 | 0.03∗ |
| Combinedc | 7,134 | 137 | 2,162 | 507 | 86 | 223 | 93 | 22–163 | 0.01∗ |
The RS1 effect group consists of babies with maternally inherited RS1. RS1/RS2 heterozygous babies with heterozygous parents are uninformative for the parental origin of RS1 and were therefore removed from the analysis. All effect estimates (g) have been adjusted for the following covariates: gender, parity, maternal weight, gestational age, maternal smoking, diabetes, and hypertension. The observed genotype frequency had no evidence of deviation from the Hardy-Weinberg equilibrium. Asterisks represent p < 0.05. Three further alleles with different numbers of repeats were identified at the PHLDA2 RS locus in an extremely small number of individuals (n = 25) from the ALSPAC cohort and were thus excluded from the statistical analysis.
The number of heterozygous babies with paternally inherited RS1.
The number of heterozygous babies with maternally inherited RS1.
The meta-analysis of all three cohorts.
To address this, we then introduced a third and larger collection, the ALSPAC cohort (n = 8,785) from the Avon Longitudinal Study of Parents and Children study.13 Because this cohort only includes samples from the mother and child and because PHLDA2 is a maternally expressed transcript, the RS1 effect group (n = 179) consisted of homozygous RS1/RS1 babies and heterozygous babies with homozygous RS1/RS1 mothers (Table S3). Using the same analysis as previously described, we found that maternal inheritance of RS1 in this cohort results in an average 88 g increase in the baby's birth weight (p = 0.03; 95% CI = 6–170) (Table 1). We then performed a meta-analysis to combine the data from all three cohorts by using both fixed- and random-effects models. The results from both models showed that babies inheriting maternal RS1 have a 93 g heavier birth weight (p = 0.01; 95% CI = 22–163) (Figure 3). No evidence of heterogeneity was found across the studies (p = 0.92, I2 = 0%). In addition, no evidence of association was found between birth weight and paternally inherited RS1, consistent with the imprinting of PHLDA2. Medical records and clinical data for all three cohorts were obtained with informed consent, and the study was approved by the ALSPAC Law and Ethics Committee and the local research ethics committees of Hammersmith and Queen Charlotte's and Chelsea Hospital Trust and University College London.
Figure 3.
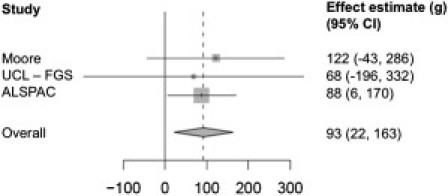
Meta-Analysis Showing the Relationship between Birth Weight and PHLDA2 Promoter RS1 Effect
The data is depicted in a Forest plot; the 95% CI for each study is represented by a horizontal line, and the estimated effect sizes are shown as gray squares. The weight of the study in the meta-analysis is represented by the size of the squares. The scale used is in grams (g). The diamond shape indicates the mean and 95% CI for the total estimate of the effect. Both random- and fixed-effect models produced the same results, and the plot represents the results from the fixed-effect model.
The meta-analysis indicated that the fetal genotypes had a direct influence on the babies′ birth weight; therefore, we further investigated the ALSPAC cohort to see whether the maternal genotypes would have an effect on the babies′ birth weight. Because we cannot determine the parental origin of the RS1 allele of the heterozygous mothers without the grandparents′ samples, we instead compared the effect of three maternal genotype groups on the babies′ birth weight by using the homozygous RS2/RS2 group (n = 465) as the baseline. To test this, we used a linear-regression model corrected for the same covariates described in the previous analysis. Our analysis showed that the heterozygous group (n = 529) had a low impact on the babies′ birth weight (+0.3 g; p = 0.99; 95% CI = −69–70); this result was expected because half of the babies should inherit a paternal RS1. However, when the mothers were homozygous for RS1 (n = 61), the babies were found to be 155 g heavier (p = 0.04; 95% CI = 9–300), indicating that maternal genotypes have an additional influence on fetal growth potentially through the intrauterine environment. This change is of similar magnitude to the reduction caused by maternal smoking (Table S2).
Notably, the effect of heterozygous mothers on birth weight was not midway between each homozygote, even though half would be expected to carry the maternal RS1. Instead, the homozygous RS1/RS1 group had considerably more than twice the effect on birth weight than did the heterozygous group. This suggests a three-generation cumulative effect, given that a homozygous RS1/RS1 mother also inherits maternal RS1 from her mother. Alternatively, homozygous RS1/RS1 mothers could also affect babies′ birth weight by influencing the circulating PHLDA2 protein/mRNA levels in the maternal blood, given that PHLDA2 shows biallelic expression in adult blood.14
Maternal inheritance of RS1 did not affect the placental weight (+2.5 g; p = 0.93; 95% CI = −62–67) but did have a small and statistically-significant influence on head circumference (+0.23 cm; p = 0.04; 95% CI = 0.01–0.45). Interestingly, although the RS1 sequence is conserved in monkeys, the duplicated RS2 allele seems to be exclusive to humans (Figure S1). This implies an evolutionary role in human reproductive success. Consistent with the conflict hypothesis, maternal PHLDA2 RS1 is associated with both increased growth of the baby and head circumference. Conversely, the net effect of the common (RS2/RS2) allele in humans is limited birth weight and head circumference, an effect which might provide an evolutionary advantage—protecting the mother and her birth canal.
Given the perinatal and life-long health complications associated with very low birth weight,1,2,15 a number of studies have investigated the genetic contribution of putative growth-regulating genes, including the imprinted genes IGF2 (MIM 147470) and H19 (MIM 103280).16–19 Genome-wide linkage or association studies have located several loci associated with birth weight,20–23 although none have yet been directly associated with actual gene function. PHLDA2 has not previously been detected in these screens, perhaps as a result of the complexity introduced by the parent-of-origin effect but also because PHLDA2 RS1 is not in linkage disequilibrium with nearby SNPs and is therefore not well-represented on the genotyping platforms used. In addition, our study maximizes the information content for this allele because we specifically genotyped all informative individuals for what is essentially the functional and presumably causal variant. The biochemical function of PHLDA2 remains unknown. It is a small cytoplasmic protein that binds to phosphoinositide lipids via its PH domain.24 A recent study showed a relationship between PHLDA2 expression and lower growth velocity of the fetal femur; this relationship suggests that PHLDA2 possibly plays a role in bone development.25 Although increased Phlda2 expression in transgenic mice resulted in a smaller placenta and a corresponding reduction in birth weight,5 we could not replicate this finding on the human placenta either in a comparative study with PHLDA2 expression11 or indirectly via association with the promoter RS genotype. Nevertheless, a profound effect on the babies′ birth weight was still detected, suggesting that placental weight was not the predominant regulatory factor. It also appears that this effect is controlled through the maternal expression of the gene, which is consistent with the conflict hypothesis and is mediated by the maternal genetic inheritance at the DNA level in the promoter. This provides the first example of a maternal genetic effect working together with a maternally driven epigenetic effect. We suspect it will be the first of many examples once further details of the interactions of the genome with the epigenome are unraveled. The PHLDA2 promoter RS and its expression might serve as a useful genetic biomarker that can be used to predict birth size. Further insight into the function of PHLDA2 along with other imprinted genes will help us understand the genetic basis of fetal growth as well as the common and serious complications—such as IUGR—of pregnancy.
Acknowledgments
We would like to thank M. Sweeney for her help at the sequencing facility at the Institute of Neurology and all the members of Professor Moore's Development and Growth research group for valuable suggestions and help. This research was funded by the Child Health Research Appeal Trust (the Institute of Child Health and the Great Ormond Street Hospital for Children [GOSH]), Overseas Research Studentship (M.I.), the Medical Research Council, Wellbeing of Women, March of Dimes, PARKS, and the GOSH Charity. P.S. is supported by the GOSH Charity. We are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting the families, and the whole ALSPAC (Avon Longitudinal Study of Parents and Children) team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. The UK Medical Research Council, the Wellcome Trust, and the University of Bristol provide core support for ALSPAC. We would also like to thank the University College London Fetal Growth Study cohort team for their collaborative work.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ALSPAC, http://www.bristol.ac.uk/alspac
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
TFSEARCH, http://www.cbrc.jp/research/db/TFSEARCH
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Barker D.J. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004;23(6, Suppl):588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 2.McIntire D.D., Bloom S.L., Casey B.M., Leveno K.J. Birth weight in relation to morbidity and mortality among newborn infants. N. Engl. J. Med. 1999;340:1234–1238. doi: 10.1056/NEJM199904223401603. [DOI] [PubMed] [Google Scholar]
- 3.Moore T., Haig D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 4.Frank D., Fortino W., Clark L., Musalo R., Wang W., Saxena A., Li C.M., Reik W., Ludwig T., Tycko B. Placental overgrowth in mice lacking the imprinted gene Ipl. Proc. Natl. Acad. Sci. USA. 2002;99:7490–7495. doi: 10.1073/pnas.122039999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tunster S.J., Tycko B., John R.M. The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol. Cell. Biol. 2010;30:295–306. doi: 10.1128/MCB.00662-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxena A., Frank D., Panichkul P., Van den Veyver I.B., Tycko B., Thaker H. The product of the imprinted gene IPL marks human villous cytotrophoblast and is lost in complete hydatidiform mole. Placenta. 2003;24:835–842. doi: 10.1016/s0143-4004(03)00130-9. [DOI] [PubMed] [Google Scholar]
- 7.Qian N., Frank D., O'Keefe D., Dao D., Zhao L., Yuan L., Wang Q., Keating M., Walsh C., Tycko B. The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum. Mol. Genet. 1997;6:2021–2029. doi: 10.1093/hmg/6.12.2021. [DOI] [PubMed] [Google Scholar]
- 8.McMinn J., Wei M., Schupf N., Cusmai J., Johnson E.B., Smith A.C., Weksberg R., Thaker H.M., Tycko B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006;27:540–549. doi: 10.1016/j.placenta.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Diplas A.I., Lambertini L., Lee M.-J., Sperling R., Lee Y.L., Wetmur J., Chen J. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics. 2009;4:235–240. doi: 10.4161/epi.9019. [DOI] [PubMed] [Google Scholar]
- 10.Kumar N., Leverence J., Bick D., Sampath V. Ontogeny of growth-regulating genes in the placenta. Placenta. 2012;33:94–99. doi: 10.1016/j.placenta.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Apostolidou S., Abu-Amero S., O'Donoghue K., Frost J., Olafsdottir O., Chavele K.M., Whittaker J.C., Loughna P., Stanier P., Moore G.E. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J. Mol. Med. 2007;85:379–387. doi: 10.1007/s00109-006-0131-8. [DOI] [PubMed] [Google Scholar]
- 12.Hindmarsh P.C., Geary M.P., Rodeck C.H., Kingdom J.C., Cole T.J. Intrauterine growth and its relationship to size and shape at birth. Pediatr. Res. 2002;52:263–268. doi: 10.1203/00006450-200208000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Jones R.W., Ring S., Tyfield L., Hamvas R., Simmons H., Pembrey M., Golding J., ALSPAC Study Team A new human genetic resource: A DNA bank established as part of the Avon longitudinal study of pregnancy and childhood (ALSPAC) Eur. J. Hum. Genet. 2000;8:653–660. doi: 10.1038/sj.ejhg.5200502. [DOI] [PubMed] [Google Scholar]
- 14.Müller S., van den Boom D., Zirkel D., Köster H., Berthold F., Schwab M., Westphal M., Zumkeller W. Retention of imprinting of the human apoptosis-related gene TSSC3 in human brain tumors. Hum. Mol. Genet. 2000;9:757–763. doi: 10.1093/hmg/9.5.757. [DOI] [PubMed] [Google Scholar]
- 15.Simmons R.A. Developmental origins of adult disease. Pediatr. Clin. North Am. 2009;56:449–466. doi: 10.1016/j.pcl.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adkins R.M., Somes G., Morrison J.C., Hill J.B., Watson E.M., Magann E.F., Krushkal J. Association of birth weight with polymorphisms in the IGF2, H19, and IGF2R genes. Pediatr. Res. 2010;68:429–434. doi: 10.1203/PDR.0b013e3181f1ca99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes M.V., Soares M.R., Pasqualim-Neto A., Marcondes C.R., Lôbo R.B., Ramos E.S. Association between birth weight, body mass index and IGF2/ApaI polymorphism. Growth Horm. IGF Res. 2005;15:360–362. doi: 10.1016/j.ghir.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 18.Petry C.J., Ong K.K., Barratt B.J., Wingate D., Cordell H.J., Ring S.M., Pembrey M.E., Reik W., Todd J.A., Dunger D.B., ALSPAC Study Team Common polymorphism in H19 associated with birthweight and cord blood IGF-II levels in humans. BMC Genet. 2005;6:22. doi: 10.1186/1471-2156-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petry C.J., Seear R.V., Wingate D.L., Acerini C.L., Ong K.K., Hughes I.A., Dunger D.B. Maternally transmitted foetal H19 variants and associations with birth weight. Hum. Genet. 2011;130:663–670. doi: 10.1007/s00439-011-1005-x. [DOI] [PubMed] [Google Scholar]
- 20.Andersson E.A., Pilgaard K., Pisinger C., Harder M.N., Grarup N., Faerch K., Poulsen P., Witte D.R., Jørgensen T., Vaag A. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 2010;53:1908–1916. doi: 10.1007/s00125-010-1790-0. [DOI] [PubMed] [Google Scholar]
- 21.Arya R., Demerath E., Jenkinson C.P., Göring H.H., Puppala S., Farook V., Fowler S., Schneider J., Granato R., Resendez R.G. A quantitative trait locus (QTL) on chromosome 6q influences birth weight in two independent family studies. Hum. Mol. Genet. 2006;15:1569–1579. doi: 10.1093/hmg/ddl076. [DOI] [PubMed] [Google Scholar]
- 22.Fradin D., Heath S., Lepercq J., Lathrop M., Bougnères P. Identification of distinct quantitative trait Loci affecting length or weight variability at birth in humans. J. Clin. Endocrinol. Metab. 2006;91:4164–4170. doi: 10.1210/jc.2006-0529. [DOI] [PubMed] [Google Scholar]
- 23.Freathy R.M., Mook-Kanamori D.O., Sovio U., Prokopenko I., Timpson N.J., Berry D.J., Warrington N.M., Widen E., Hottenga J.J., Kaakinen M., Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Meta-Analyses of Glucose and Insulin-related traits Consortium. Wellcome Trust Case Control Consortium. Early Growth Genetics (EGG) Consortium Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat. Genet. 2010;42:430–435. doi: 10.1038/ng.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxena A., Morozov P., Frank D., Musalo R., Lemmon M.A., Skolnik E.Y., Tycko B. Phosphoinositide binding by the pleckstrin homology domains of Ipl and Tih1. J. Biol. Chem. 2002;277:49935–49944. doi: 10.1074/jbc.M206497200. [DOI] [PubMed] [Google Scholar]
- 25.Lewis R.M., Cleal J.K., Ntani G., Crozier S.R., Mahon P.A., Robinson S.M., Harvey N.C., Cooper C., Inskip H.M., Godfrey K.M., Southampton Women's Survey Study Group Relationship between placental expression of the imprinted PHLDA2 gene, intrauterine skeletal growth and childhood bone mass. Bone. 2012;50:337–342. doi: 10.1016/j.bone.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.