

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is a common hereditary myopathy causally linked to reduced numbers (≤8) of 3.3 kilobase D4Z4 tandem repeats at 4q35. However, because individuals carrying D4Z4-reduced alleles and no FSHD and patients with FSHD and no short allele have been observed, additional markers have been proposed to support an FSHD molecular diagnosis. In particular a reduction in the number of D4Z4 elements combined with the 4A(159/161/168)PAS haplotype (which provides the possibility of expressing DUX4) is currently used as the genetic signature uniquely associated with FSHD. Here, we analyzed these DNA elements in more than 800 Italian and Brazilian samples of normal individuals unrelated to any FSHD patients. We find that 3% of healthy subjects carry alleles with a reduced number (4–8) of D4Z4 repeats on chromosome 4q and that one-third of these alleles, 1.3%, occur in combination with the 4A161PAS haplotype. We also systematically characterized the 4q35 haplotype in 253 unrelated FSHD patients. We find that only 127 of them (50.1%) carry alleles with 1–8 D4Z4 repeats associated with 4A161PAS, whereas the remaining FSHD probands carry different haplotypes or alleles with a greater number of D4Z4 repeats. The present study shows that the current genetic signature of FSHD is a common polymorphism and that only half of FSHD probands carry this molecular signature. Our results suggest that the genetic basis of FSHD, which is remarkably heterogeneous, should be revisited, because this has important implications for genetic counseling and prenatal diagnosis of at-risk families.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD [MIM 158900]), a common myopathy, has a prevalence of 1 in 20,000.1,2 The disease is characterized by weakness of selective muscle groups and wide variability of clinical expression.1,3,4 The onset of the disease is in the second or third decade of life and usually involves the weakening of facial and limb-girdle muscles. The mode of inheritance of classical FSHD is considered to be autosomal dominant, with complete penetrance by age 20.4,5 No biochemical, histological, or instrumental markers are available to independently confirm a specific FSHD diagnosis that remains mainly clinical.
The FSHD genetic defect does not reside in any protein-coding gene.6 Instead, FSHD has been genetically linked to the reduction of an integral number of tandem 3.3-kb D4Z4 repeats located on chromosome 4q35.7,8 Although nearly identical D4Z4 sequences reside on chromosome 10q26,9 only subjects with a reduced number of D4Z4 repeats on chromosome 4, but not chromosome 10, develop FSHD.10–12 Based on these results, p13E-11 EcoRI alleles larger than 50 kb (≥11 D4Z4 repeats) originating from chromosome 4 have been considered normal, whereas alleles of 35 kb or less (≤8 D4Z4 repeats) have been considered diagnostic for the disease.8,13
Because there are individuals with reduced D4Z4 alleles that do not have clinical signs of FSHD,14,15 it has been proposed that additional DNA sequences flanking the D4Z4 repeat array are necessary for disease development.16–18 These studies concluded that D4Z4 reduction is pathogenic only in a few genetic backgrounds, which include a specific simple sequence length polymorphism (SSLP) proximal to the D4Z4 repeat and the 4qA polymorphism distal to the repeat (Figure 1). These haplotypes, named 4A159, 4A161, and 4A168, have been proposed to be uniquely associated with FSHD. Recently it has been shown that a single-nucleotide polymorphism (SNP) in the pLAM sequence of the 4qA alleles provides a polyadenylation signal (PAS; ATTAAA) for the DUX4 transcript from the most distal D4Z4 unit on 4qA chromosomes. Thus, the molecular signature, named 4A(159,161,168)PAS, has been proposed to define alleles causally related to FSHD. This signature results from the combination of (1) a reduction in the number of D4Z4 elements, (2) the presence of the 4qA allele, and (3) the PAS in the pLAM sequence. In this scenario, FSHD arises from a specific genetic setting enabling the normally silent double homeobox protein 4 gene (DUX4 [MIM 606009]) to be expressed.18 On this basis, healthy subjects carrying reduced D4Z4 alleles would be explained by the absence of the 4A(159,161,168)PAS.
Figure 1.

Schematic Representation of Polymorphisms at the 4q and 10q Subtelomeres
(A) Schematic representation of the method used to calculate D4Z4 repeat numbers from EcoRI fragment sizes. The D4Z4 repeat array is indicated with triangles. Seven and eight D4Z4 repeats (31–36 kb EcoRI fragment size) were defined to be the upper diagnostic range for FSHD. D4Z4 repeat units on chromosomes 4 and 10 can be distinguished because all repeats on 10q contain BlnI restriction sites (B within the black triangles), whereas all D4Z4 repeats on 4q contain XapI restriction sites (X within the white triangles).
(B) Schematic representation of the current view of pathogenic haplotypes.
(C) Elements examined in the present study. In addition to the number of D4Z4 repeats, elements that distinguish subjects include: (1) the chromosomal localization of the D4Z4 repeat, chromosome 4q35 or 10q26; (2) the SSLP, which is a combination of five variable number tandem repeats, an 8 bp insertion/deletion, and two SNPs localized 3.5 kb proximal to D4Z4; it varies in length between 157 and 182 bp; (3) the AT(T/C)AAA SNP in the pLAM region; (4) a large sequence variation (termed 4qA or B) that is distal to D4Z4. In the 4qB variant, the terminal 3.3 kb repeat contains only 570 bp of a complete repeat, whereas in the 4qA variant the terminal repeat is a divergent 3.3 kb repeat named pLAM. 4q chromosomes that do not hybridize to probes for (A) and (B) are termed “null,” and their sequences vary from case to case.
This model does not apply to all FSHD cases. For example, nonpenetrant carriers have been reported in FSHD families,14,15 and there are FSHD patients carrying full-length D4Z4 alleles (≥11 repeats) that are clinically indistinguishable from patients carrying D4Z4 alleles of reduced size (≤8 repeats).19 Rare exceptions could be explained by a variety of mechanisms that do not challenge the basic hypothesis. However, recently we found that 2.7% of cases in the Italian National Registry for FSHD (which contains over 1,100 unrelated FSHD patients) were compound heterozygotes carrying two D4Z4-reduced alleles (0.5% were homozygotes for the 4A161 haplotype). Based on this finding, we estimated that the population frequency of the 4A161PAS haplotype associated with a D4Z4-reduced allele could be higher than 1%.20
The correlation between genotype and phenotype in FSHD thus appears to be more complex than just the presence of the 4APAS signature. In order to confirm the high frequency of this signature in the normal population and reevaluate the allele distribution in FSHD patients, we performed a systematic unbiased clinical and molecular study of 801 normal control subjects from Italy and Brazil and 253 FSHD probands from the Italian Registry for FSHD. Our results establish that the 4APAS structure is a frequent genetic polymorphism that is neither sufficient nor necessary for the development of FSHD. This result is not incompatible with evidence implicating DUX4 or other factors as important mediators of the disease. However, it does demonstrate that the pathogenesis is more complex than currently thought and that the current genetic signature is insufficient for diagnosis.
Subjects and Methods
Control Population
The control group consisted of 801 unrelated healthy subjects with no family history of muscular dystrophy. Subjects were recruited from the Italian and Brazilian populations through advertisements. Italian controls subjects were equally distributed among Northern, Central, and Southern regions. The local ethics committee approved the study. All subjects enrolled in the study were clinically and molecularly characterized after giving informed consent to participate (see Table S1 available online).
FSHD Patients
Two-hundred-fifty-three unrelated FSHD patients were accrued through the Italian National Registry for FSHD. All subjects were clinically and molecularly characterized. In particular, we considered patients to have typical FSHD if: (1) disease onset occurred in facial or shoulder girdle muscles; (2) there was facial and/or scapular fixator weakness; and (3) there was absence of atypical signs suggesting an alternative diagnosis (including extraocular, masticatory, pharyngeal, or lingual muscle weakness and cardiomyopathy).21,22 Clinical data were collected with the FSHD clinical form. The clinical severity of the disease was measured according to the FSHD score, as previously described.23 Briefly, the FSHD score quantifies the degree of weakness and defines the level of disability affecting six separate muscle groups: facial (score 0–2), shoulder girdle (score 0–3), upper limbs (score 0–2), pelvic girdle (score 0–5), leg muscles (score 0–3), and Beevor's sign (score 0–1).23 The final clinical evaluation score, calculated by summing the single scores, ranged from 0, when no signs of muscle weakness are present, to 15, when all muscle groups tested are severely impaired.23 All selected subjects were evaluated using a standard protocol, and each subject received the standardized FSHD score previously described.23
Molecular Genetic Analysis
DNA was prepared from isolated lymphocytes according to standard procedures. In brief, restriction endonuclease digestion of DNA was performed in agarose plugs with the appropriate restriction enzyme: EcoRI, EcoRI/BlnI, XapI (p13E-11 probe), Hind III (4qA/4qB probes), and NotI (B31 probe). Digested DNA was separated by pulsed-field gel electrophoresis (PFGE) in 1% agarose gels. Allele sizes were estimated by southern hybridization with probe p13E-11 of 7 μg of EcoRI-, EcoRI/BlnI-, and XapI-digested genomic DNA extracted from peripheral blood lymphocytes, electrophoresed in a 0.4% agarose gel for 45–48 hr at 35 V, alongside an 8–48 kb marker (Bio-Rad). To assess the chromosomal origin of the two D4Z4-reduced alleles, DNA from each proband was analyzed by NotI digestion and hybridization with the B31 probe (Figure S1). Restriction fragments were detected by autoradiography or by using a Typhoon Trio system (GE Healthcare).
4qA/4qB allelic variants were defined using 7 μg of HindIII-digested DNA, PFGE electrophoresis, and Southern blot hybridization with radiolabeled 4qB and 4qA probes according to standard procedures. The 4qA/4qB variants were attributed to each chromosome based on the size of EcoRI restriction fragments (Figure S1). To define the SSLP and the pLAM SNP (AT(T/C)AAA) sequences flanking the D4Z4 repeat units, linear gel electrophoresis of EcoRI-digested DNA was used to isolate each D4Z4-reduced allele. The SSLP sequence was determined after PCR amplification using specific oligonucleotides (forward primer 5′-GGTGGAGTTCTGGTTTCAGC-3′ labeled with hexachlorofluorescein [HEX], reverse primer 5′-CCTGTGCTTCAGAGGCATTTG-3′) as previously reported.17,18 Analysis of the pLAM SNP was performed on PCR-amplified DNA using specific oligonucleotides (forward primer 5′-ACGCTGTCTAGGCAAACCTG-3′, reverse primer 5′-TGCACTCATCACACAAAAGATG-3′). SSLP size differences and pLAM sequences were analyzed using an ABI Prism 3130 Genetic Analyzer.17,18
Results
Presence of FSHD-Sized Alleles in the Healthy Population
Previous smaller investigations of the Dutch population suggested that 3% of healthy subjects had D4Z4 alleles of 35–38 kb in size, and one-third of these might present a potentially pathogenic 4A allele.11,24 An additional recent genetic criteria is that, in order to have FSHD, the reduction of D4Z4 repeats must be associated with a specific chromosomal background, 4A(159/161/168)PAS, allowing the expression of the DUX4 gene.18 However, the frequency of compound heterozygotes in patients with FSHD suggested that the frequency of D4Z4-reduced 24–35 kb alleles associated with the 4A161PAS in the Italian population would be >1%.20 Because this prediction has crucial implications for clinical practice, we searched for D4Z4-reduced alleles associated with the 4A161PAS haplotype in 801 healthy individuals, 560 from Italy and 241 from Brazil. Figure 2 shows that 25 of these 801 subjects carry D4Z4 alleles ranging from 21 to 35 kb (4 to 8 D4Z4 units); 17 of these 25 alleles are associated with 4qA (Figure 2, groups 1 and 2), and 11 carry the 4A161PAS haplotype (Figure 2, group 1; Figures S2–S7). Therefore, 3% (25 of 801) of normal controls carry D4Z4 alleles of reduced size, and ∼1.3% (11) have the supposedly pathogenic 4A161PAS haplotype. The age of all these healthy carriers ranged between 40 and 78 years, an age in which FSHD is considered to be fully penetrant. On this basis, we conclude that the haplotype 4A161PAS has the frequency of a common polymorphism and that it may be permissive but is not sufficient to cause autosomal-dominant disease.
Figure 2.
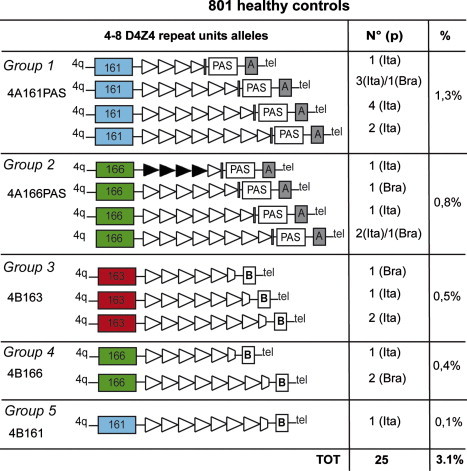
Molecular Haplotypes of 25 Healthy Subjects from Italian and Brazilian Control Populations
Haplotypes of 25 4q alleles with 4–8 D4Z4 repeats identified in 801 normal healthy individuals randomly selected from the general populations of Italy and Brazil. We characterized the sequence variants in the SSLP (colored rectangles), D4Z4 repeat number (triangles), distal variants A or B (shaded boxes), and the pLAM PAS (unshaded rectangles). The second and third columns present the number of reduced D4Z4 alleles detected (N°), the provenance (p) of the subjects (Ita for Italy and Bra for Brazil), and the prevalence of each haplotype among the 801 individuals examined (%). The supposedly pathogenic haplotype 4A161PAS is the most frequent and is present in 1.3% of healthy controls.
Multiple Haplotypes Associated with FSHD
Our observation that in the general population 1.3% of healthy subjects carry the FSHD “pathogenic” signature 4A161PAS, which enables the expression of DUX4, challenges the notion that FSHD is a fully-penetrant autosomal-dominant disorder caused by the reduction of D4Z4 repeat number associated with 4A161PAS haplotype. To test the DUX4 polyadenylation model more broadly, we systematically studied the 4q35 haplotype of 253 probands accrued through the Italian National Registry for FSHD (Table S2). The D4Z4 repeat size was systematically studied in all subjects. Table 1 shows that 204 of 253 probands (80.6%) carry D4Z4 alleles with 1–8 units, 19 (7.5%) have D4Z4 alleles with 9–10 repeats, and the remaining 30 (11.8%) show large D4Z4 alleles (≥11 repeats) on both copies of chromosome 4 (Table 1). We then analyzed the 223 FSHD patients with 10 or fewer D4Z4 repeats for the presence of the 4A/B, PAS, and SSLP (see Figure 1). Only 127 FSHD probands carry the 4A161PAS haplotype associated with alleles having 1–8 D4Z4 repeats (group 1 in Figure 3). Among the remaining probands, 52 have reduced alleles associated with the 4A166PAS haplotype previously considered to not be “permissive” for FSHD disease (group 2 in Figure 3), 13 carry the 4A162PAS, 5 carry the 4A164PAS, 2 carry the 4A167PAS, 1 carries the 4A163PAS (groups 7–10 in Figure 3), and 3 bear reduced D4Z4 alleles with the 4qB polymorphism, which lacks both the pLAM region and the PAS (groups 3–4 in Figure 3). Examples of each group are reported in Figures S8–S12. Collectively, our data reveal that in our cohort of FSHD probands the SSLP allelic variants associated with D4Z4-reduced alleles differ from those previously reported (compare groups 2–10 in Figure 3 with Figure 1).17,18 This genotypic difference is also supported by the fact that we did not find the 4A168 “permissive” haplotype associated with FSHD in our population. In contrast, haplotypes considered to not be “permissive” for FSHD disease were frequent. In particular, the 4A166PAS haplotype is present associated with almost one-quarter (23.3%) of D4Z4-reduced alleles detected in our FSHD probands. More importantly, 49 of 253 FSHD probands (19%) carry alleles with more than 8 D4Z4 repeats, and only 127 (50.1%) carry D4Z4-reduced alleles associated with the 4A161PAS, the expected molecular signature for FSHD.
Table 1.
Distribution of 253 Unrelated FSHD Patients versus D4Z4 Repeat Units
|
Number of Unrelated FSHD Patients | |||
|---|---|---|---|
| D4Z4 Repeat Units | De Novo Cases | Familial Cases | Percentage |
| 1–3 | 5 | 22 | 10.7% |
| 4–8 | 2 | 175 | 69.9% |
| 9–10 | 0 | 19 | 7.5% |
| >11 | 0 | 30 | 11.8% |
Figure 3.

Molecular Haplotypes of 223 Unrelated FSHD Patients
Overview of haplotypes of the D4Z4-reduced alleles (≤10 repeats) on chromosome 4 found in 223 unrelated FSHD patients. Alleles are separated into 1–3 (column 1), 4–8 (column 3), and 9–10 (column 5) D4Z4 repeat units. Within these columns, we group the observed haplotypes based on the type of SSLP and PAS. N° indicates the number of alleles found in each haplotype, and TOT(%) represents the total number of alleles found and the prevalence of each haplotype. The 4A haplotypes not previously observed in FSHD patients include 4A166 (group 2), 4A162 (group 7), 4A164 (group 8), 4A167 (group 9), and 4A163 (group 10), and the 4B haplotypes include 4B163 (group 3) and 4B166 (group 4). Frequencies of 4A161 (group 1, 63.7%) and 4A166 (group 2, 25.1%) are different than previously reported in other normal populations. Chromosomes with 4qB haplotypes (groups 3–4) lack the pLAM and PAS.
Discussion
The practice of medical genetics requires a clear, definite evaluation of the significance of mutations and/or variations of DNA sequences for diagnosis to provide prognostic information and genetic counseling. This is particularly important for a progressive disease with unpredictable onset and a high variability of clinical expression, such as FSHD.
The extensive use over the past 20 years of DNA analysis for studying Mendelian disorders has revealed many complex mechanisms in addition to single mutant genes that cause disease. Identical phenotypes may be produced by mutations in different genes,25 the same mutation can cause different phenotypes,26 and distinct mutations in the same gene may result in different disorders that segregate with diverse Mendelian or even multifactorial patterns.27 In addition, the incomplete penetrance of certain mutations argues for the importance of modifying loci or epigenetic mechanisms influencing the clinical expression in many Mendelian disorders.28 Thus, establishing the value of mutational events underlying genetic diseases may be complex even when there are simple patterns of inheritance in diseases with a well-characterized pathologic course.
FSHD seems to fall in this complex pattern, even though it is currently considered to be a fully penetrant disease with a wide variability in clinical spectrum, ranging from subjects with very mild muscle weakness to wheelchair-bound patients.5,13 The molecular test initially used for FSHD diagnosis was based on the observation that 95% of FSHD patients carry a reduction of integral numbers of D4Z4 repeats at 4q35 with full penetrance.10 However, the wide use of this test revealed several exceptions to the original model. Through the years, the threshold size of D4Z4 alleles has been increased from the original 28 kb7 (6 repeats) to 35 kb10 (8 repeats), with FSHD cases carrying D4Z4 alleles of 38–41 kb (9–11 repeats), considered borderline alleles.22,29 Additional genotype-phenotype studies led to the identification of subjects carrying D4Z4-reduced alleles with no sign of muscle weakness in FSHD families,14,15 as well as of healthy unrelated subjects without family history of FSHD.11,30 The present results from our systematic clinical and molecular analysis of FSHD patients from the Italian National Registry for FSHD, as well as a large number of healthy controls, challenge the current model for FSHD diagnosis.
Remarkably, our data establish as a general rule rather than an exception that detection of a D4Z4-reduced allele is not sufficient to diagnose FSHD. Although the majority of FSHD patients (70%) carry D4Z4 alleles with 4–8 units, this size range is carried by 3% of healthy subjects from the general population. Additionally, there is little predictive value of the 4qA161PAS haplotype in the absence of family history, because 1.3% of healthy subjects carry this haplotype, which therefore has the frequency of a common polymorphism (Figure 1) rather than a rare mutation. Finally, 49 of 253 probands (19%) do not carry D4Z4 alleles with 1–8 repeats, and only 50% of the probands carry the 4A161 permissive haplotype.
In summary, our study indicates that a profound rethinking of the genetic disease mechanism and modes of inheritance of FSHD are now required and that entirely new models and approaches are needed. Our results do not exclude an important pathogenic role for DUX4 or other candidate factors but do establish a complex mechanism beyond current understanding. Indeed, our data point at the possibility that in the heterozygous state a D4Z4 reduction might produce a subclinical sensitized condition that requires other epigenetic mechanisms or a contributing factor to cause overt myopathy. In some rare cases, that could be by becoming homozygous20 and doubling the dose of a dominant factor such as DUX4. In others, it might be by the simultaneous heterozygosity for a different and recessive myopathy, as suggested by many reports in which the FSHD contractions are found in association with a second molecular defect.31–44 This possibility is also consistent with previous reports of expression changes of candidate proteins such as CRYM that were associated with FSHD in some families but that were unchanged when other families were examined. Finally, it is also plausible that drugs or toxic agents might contribute to the disease onset and clinical variability. This would explain the observation of discordant monozygotic twins carrying the FSHD reduction.45,46 It is hoped that broadening the scope of investigations, including next-generation deep sequencing in particular in families with asymptomatic and clinically affected members carrying the same FSHD allele, may finally lead to an understanding of the molecular pathogenesis of this complex disease. These findings have important clinical implications for genetic counseling of patients and families with FSHD, with particular regard to the interpretation of data in prenatal diagnosis.
Acknowledgments
We are indebted to all FSHD patients and their families for participating in this study. The Associazione Amici del Centro Dino Ferrari-University of Milan is gratefully acknowledged. We thank Paul D. Kaufman and Michael R. Green for their in-depth critique of the manuscript. DNA from Brazilian controls was kindly provided by Naila Lourenço and Antonia Cerqueira (Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of São Paulo, Brazil). This work was supported by Telethon GUP08004 and GUP11009, by Association Française Contre les Myopathies 14339, by National Institute of Health-National Institutes of Neurological Disorders and Stroke grant RO1 NS047584, by Centros de Pesquisa, Inovação e Difusão/Fundação de Amparo à Pesquisa do Estado de São Paulo, by Institutos Nacionais de Ciência e Tecnologia, and by Conselho Nacional de Desenvolvimento Científico e Tecnológico.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Padberg, G.W. (1982). Facioscapulohumeral disease. PhD thesis, Leiden University, Leiden, Holland.
- 2.Mostacciuolo M.L., Pastorello E., Vazza G., Miorin M., Angelini C., Tomelleri G., Galluzzi G., Trevisan C.P. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin. Genet. 2009;75:550–555. doi: 10.1111/j.1399-0004.2009.01158.x. [DOI] [PubMed] [Google Scholar]
- 3.Flanigan K.M. Facioscapulohumeral muscular dystrophy and scapuloperoneal disorders. In: Engel A., Franzini-Armstrong C., editors. Myology. McGrow Hill Professional; New York: 2004. pp. 1123–1133. [Google Scholar]
- 4.Lunt P.W., Harper P.S. Genetic counselling in facioscapulohumeral muscular dystrophy. J. Med. Genet. 1991;28:655–664. doi: 10.1136/jmg.28.10.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tawil R., van der Maarel S., Padberg G.W., van Engelen B.G. 171st ENMC international workshop: Standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2010;20:471–475. doi: 10.1016/j.nmd.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Hewitt J.E., Lyle R., Clark L.N., Valleley E.M., Wright T.J., Wijmenga C., van Deutekom J.C., Francis F., Sharpe P.T., Hofker M. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 1994;3:1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 7.Wijmenga C., Hewitt J.E., Sandkuijl L.A., Clark L.N., Wright T.J., Dauwerse H.G., Gruter A.M., Hofker M.H., Moerer P., Williamson R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 8.van Deutekom J.C.T., Wijmenga C., van Tienhoven E.A., Gruter A.M., Hewitt J.E., Padberg G.W., van Ommen G.J., Hofker M.H., Frants R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 9.Deidda G., Cacurri S., Grisanti P., Vigneti E., Piazzo N., Felicetti L. Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur. J. Hum. Genet. 1995;3:155–167. doi: 10.1159/000472291. [DOI] [PubMed] [Google Scholar]
- 10.van Deutekom J.C.T., Bakker E., Lemmers R.J., van der Wielen M.J., Bik E., Hofker M.H., Padberg G.W., Frants R.R. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum. Mol. Genet. 1996;5:1997–2003. doi: 10.1093/hmg/5.12.1997. [DOI] [PubMed] [Google Scholar]
- 11.van Overveld P.G.M., Lemmers R.J., Deidda G., Sandkuijl L., Padberg G.W., Frants R.R., van der Maarel S.M. Interchromosomal repeat array interactions between chromosomes 4 and 10: a model for subtelomeric plasticity. Hum. Mol. Genet. 2000;9:2879–2884. doi: 10.1093/hmg/9.19.2879. [DOI] [PubMed] [Google Scholar]
- 12.Matsumura T., Goto K., Yamanaka G., Lee J.H., Zhang C., Hayashi Y.K., Arahata K. Chromosome 4q;10q translocations; comparison with different ethnic populations and FSHD patients. BMC Neurol. 2002;2:7. doi: 10.1186/1471-2377-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lunt P.W., Jardine P.E., Koch M.C., Maynard J., Osborn M., Williams M., Harper P.S., Upadhyaya M. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum. Mol. Genet. 1995;4:951–958. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 14.Ricci E., Galluzzi G., Deidda G., Cacurri S., Colantoni L., Merico B., Piazzo N., Servidei S., Vigneti E., Pasceri V. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann. Neurol. 1999;45:751–757. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 15.Tonini M.M., Passos-Bueno M.R., Cerqueira A., Matioli S.R., Pavanello R., Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul. Disord. 2004;14:33–38. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Lemmers R.J., Wohlgemuth M., Frants R.R., Padberg G.W., Morava E., van der Maarel S.M. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2004;75:1124–1130. doi: 10.1086/426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemmers R.J., Wohlgemuth M., van der Gaag K.J., van der Vliet P.J., van Teijlingen C.M., de Knijff P., Padberg G.W., Frants R.R., van der Maarel S.M. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007;81:884–894. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camaño P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Greef J.C., Lemmers R.J., Camaño P., Day J.W., Sacconi S., Dunand M., van Engelen B.G., Kiuru-Enari S., Padberg G.W., Rosa A.L. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–1554. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scionti I., Fabbri G., Fiorillo C., Ricci G., Greco F., D'Amico R., Termanini A., Vercelli L., Tomelleri G., Cao M. Facioscapulohumeral muscular dystrophy: new insights from compound heterozygotes and implication for prenatal genetic counselling. J. Med. Genet. 2012;49:171–178. doi: 10.1136/jmedgenet-2011-100454. [DOI] [PubMed] [Google Scholar]
- 21.Padberg G.W., Lunt P.W., Koch M., Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991;1:231–234. doi: 10.1016/0960-8966(91)90094-9. [DOI] [PubMed] [Google Scholar]
- 22.Butz M., Koch M.C., Müller-Felber W., Lemmers R.J., van der Maarel S.M., Schreiber H. Facioscapulohumeral muscular dystrophy. Phenotype-genotype correlation in patients with borderline D4Z4 repeat numbers. J. Neurol. 2003;250:932–937. doi: 10.1007/s00415-003-1116-y. [DOI] [PubMed] [Google Scholar]
- 23.Lamperti C., Fabbri G., Vercelli L., D'Amico R., Frusciante R., Bonifazi E., Fiorillo C., Borsato C., Cao M., Servida M. A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: The FSHD clinical score. Muscle Nerve. 2010;42:213–217. doi: 10.1002/mus.21671. [DOI] [PubMed] [Google Scholar]
- 24.Wohlgemuth M., Lemmers R.J., van der Kooi E.L., van der Wielen M.J., van Overveld P.G., Dauwerse H., Bakker E., Frants R.R., Padberg G.W., van der Maarel S.M. Possible phenotypic dosage effect in patients compound heterozygous for FSHD-sized 4q35 alleles. Neurology. 2003;61:909–913. doi: 10.1212/wnl.61.7.909. [DOI] [PubMed] [Google Scholar]
- 25.Casasnovas C., Cano L.M., Albertí A., Céspedes M., Rigo G. Charcot-Marie-tooth disease. Foot Ankle Spec. 2008;1(6, Spec.):350–354. doi: 10.1177/1938640008326247. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi M., Asai N., Iwashita T., Murakami H., Ito S. Mechanisms of development of multiple endocrine neoplasia type 2 and Hirschsprung's disease by ret mutations. Recent Results Cancer Res. 1998;154:229–236. doi: 10.1007/978-3-642-46870-4_14. [DOI] [PubMed] [Google Scholar]
- 27.Kanagawa M., Toda T. The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J. Hum. Genet. 2006;51:915–926. doi: 10.1007/s10038-006-0056-7. [DOI] [PubMed] [Google Scholar]
- 28.Chahwan R., Wontakal S.N., Roa S. The multidimensional nature of epigenetic information and its role in disease. Discov. Med. 2011;11:233–243. [PubMed] [Google Scholar]
- 29.Vitelli F., Villanova M., Malandrini A., Bruttini M., Piccini M., Merlini L., Guazzi G., Renieri A. Inheritance of a 38-kb fragment in apparently sporadic facioscapulohumeral muscular dystrophy. Muscle Nerve. 1999;22:1437–1441. doi: 10.1002/(sici)1097-4598(199910)22:10<1437::aid-mus15>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Weiffenbach B., Bagley R., Falls K., Hyser C., Storvick D., Jacobsen S.J., Schultz P., Mendell J., Willems van Dijk K., Milner E.C. Linkage analyses of five chromosome 4 markers localizes the facioscapulohumeral muscular dystrophy (FSHD) gene to distal 4q35. Am. J. Hum. Genet. 1992;51:416–423. [PMC free article] [PubMed] [Google Scholar]
- 31.Lecky B.R., MacKenzie J.M., Read A.P., Wilcox D.E. X-linked and FSH dystrophies in one family. Neuromuscul. Disord. 1991;1:275–278. doi: 10.1016/0960-8966(91)90101-w. [DOI] [PubMed] [Google Scholar]
- 32.Felice K.J., North W.A., Moore S.A., Mathews K.D. FSH dystrophy 4q35 deletion in patients presenting with facial-sparing scapular myopathy. Neurology. 2000;54:1927–1931. doi: 10.1212/wnl.54.10.1927. [DOI] [PubMed] [Google Scholar]
- 33.van der Kooi A.J., Visser M.C., Rosenberg N., van den Berg-Vos R., Wokke J.H., Bakker E., de Visser M. Extension of the clinical range of facioscapulohumeral dystrophy: report of six cases. J. Neurol. Neurosurg. Psychiatry. 2000;69:114–116. doi: 10.1136/jnnp.69.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krasnianski M., Eger K., Neudecker S., Jakubiczka S., Zierz S. Atypical phenotypes in patients with facioscapulohumeral muscular dystrophy 4q35 deletion. Arch. Neurol. 2003;60:1421–1425. doi: 10.1001/archneur.60.10.1421. [DOI] [PubMed] [Google Scholar]
- 35.Chuenkongkaew W.L., Lertrit P., Limwongse C., Nilanont Y., Boonyapisit K., Sangruchi T., Chirapapaisan N., Suphavilai R. An unusual family with Leber's hereditary optic neuropathy and facioscapulohumeral muscular dystrophy. Eur. J. Neurol. 2005;12:388–391. doi: 10.1111/j.1468-1331.2004.01060.x. [DOI] [PubMed] [Google Scholar]
- 36.Filosto M., Tonin P., Scarpelli M., Savio C., Greco F., Mancuso M., Vattemi G., Govoni V., Rizzuto N., Tupler R., Tomelleri G. Novel mitochondrial tRNA Leu(CUN) transition and D4Z4 partial deletion in a patient with a facioscapulohumeral phenotype. Neuromuscul. Disord. 2008;18:204–209. doi: 10.1016/j.nmd.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Rudnik-Schöneborn S., Weis J., Kress W., Häusler M., Zerres K. Becker's muscular dystrophy aggravating facioscapulohumeral muscular dystrophy—double trouble as an explanation for an atypical phenotype. Neuromuscul. Disord. 2008;18:881–885. doi: 10.1016/j.nmd.2008.06.387. [DOI] [PubMed] [Google Scholar]
- 38.Korngut L., Siu V.M., Venance S.L., Levin S., Ray P., Lemmers R.J., Keith J., Campbell C. Phenotype of combined Duchenne and facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2008;18:579–582. doi: 10.1016/j.nmd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Zouvelou V., Manta P., Kalfakis N., Evdokimidis I., Vassilopoulos D. Asymptomatic elevation of serum creatine kinase leading to the diagnosis of 4q35 facioscapulohumeral muscular dystrophy. J. Clin. Neurosci. 2009;16:1218–1219. doi: 10.1016/j.jocn.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Tsuji M., Kinoshita M., Imai Y., Kawamoto M., Kohara N. Facioscapulohumeral muscular dystrophy presenting with hypertrophic cardiomyopathy: a case study. Neuromuscul. Disord. 2009;19:140–142. doi: 10.1016/j.nmd.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Reilich P., Schramm N., Schoser B., Schneiderat P., Strigl-Pill N., Müller-Höcker J., Kress W., Ferbert A., Rudnik-Schöneborn S., Noth J. Facioscapulohumeral muscular dystrophy presenting with unusual phenotypes and atypical morphological features of vacuolar myopathy. J. Neurol. 2010;257:1108–1118. doi: 10.1007/s00415-010-5471-1. [DOI] [PubMed] [Google Scholar]
- 42.Jordan B., Eger K., Koesling S., Zierz S. Camptocormia phenotype of FSHD: a clinical and MRI study on six patients. J. Neurol. 2011;258:866–873. doi: 10.1007/s00415-010-5858-z. [DOI] [PubMed] [Google Scholar]
- 43.Tonini M.M., Passos-Bueno M.R., Cerqueira A., Pavanello R., Vainzof M., Dubowitz V., Zatz M. Facioscapulohumeral (FSHD1) and other forms of muscular dystrophy in the same family: is there more in muscular dystrophy than meets the eye? Neuromuscul. Disord. 2002;12:554–557. doi: 10.1016/s0960-8966(02)00014-7. [DOI] [PubMed] [Google Scholar]
- 44.Ricci G., Scionti I., Alì G., Volpi L., Zampa V., Fanin M., Angelini C., Politano L., Tupler R., Siciliano G. Rippling muscle disease and facioscapulohumeral dystrophy-like phenotype in a patient carrying a heterozygous CAV3 T78M mutation and a D4Z4 partial deletion: Further evidence for “double trouble” overlapping syndromes. Neuromuscul. Disord. 2012 doi: 10.1016/j.nmd.2011.12.001. in press. Published online January 13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griggs R.C., Tawil R., McDermott M., Forrester J., Figlewicz D., Weiffenbach B., FSH-DY Group Monozygotic twins with facioscapulohumeral dystrophy (FSHD): implications for genotype/phenotype correlation. Muscle Nerve. 1995;2:S50–S55. [PubMed] [Google Scholar]
- 46.Tupler R., Barbierato L., Memmi M., Sewry C.A., De Grandis D., Maraschio P., Tiepolo L., Ferlini A. Identical de novo mutation at the D4F104S1 locus in monozygotic male twins affected by facioscapulohumeral muscular dystrophy (FSHD) with different clinical expression. J. Med. Genet. 1998;35:778–783. doi: 10.1136/jmg.35.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.