

Abstract
Recent genome-wide association studies have identified a number of susceptibility loci for Alzheimer disease (AD). To understand the functional consequences and potential interactions of the associated loci, we explored large-scale data sets interrogating the human genome for evidence of positive natural selection. Our findings provide significant evidence for signatures of recent positive selection acting on several haplotypes carrying AD susceptibility alleles; interestingly, the genes found in these selected haplotypes can be assembled, independently, into a molecular complex via a protein-protein interaction (PPI) network approach. These results suggest a possible coevolution of genes encoding physically-interacting proteins that underlie AD susceptibility and are coexpressed in different tissues. In particular, PICALM, BIN1, CD2AP, and EPHA1 are interconnected through multiple interacting proteins and appear to have coordinated evidence of selection in the same human population, suggesting that they may be involved in the execution of a shared molecular function. This observation may be AD-specific, as the 12 loci associated with Parkinson disease do not demonstrate excess evidence of natural selection. The context for selection is probably unrelated to AD itself; it is likely that these genes interact in another context, such as in immune cells, where we observe cis-regulatory effects at several of the selected AD loci.
Main Text
Alzheimer disease (AD [MIM 104300]) is the most common neurodegenerative disease and is a leading cause of dementia.1 Sporadic late-onset AD has a genetic component that includes the well-known ε4 haplotype of APOE (MIM 107741) and other loci that harbor susceptibility alleles.2–6 However, the functional consequences and evolutionary history of these loci and their possible interactions remain largely unknown. Previous studies reporting evidence of selection at the APOE locus7,8 suggested the hypothesis that AD-associated pathways may have experienced selection in human populations, quite possibly due to selective pressure on a phenotype unrelated to AD, given that AD has little impact on an individual's reproductive fitness. To further explore this hypothesis, we integrated evidence for natural selection within validated AD loci with a pathway-based analysis of these loci and an examination of transcriptional patterns in immune cells. We identified several genes in AD susceptibility loci that appear to physically interact and that show evidence of having undergone natural selection. Additionally, several of these loci exhibit a cis-regulatory effect on the transcription levels of the selected genes in immune cells, suggesting one plausible mechanism by which an AD-related molecular pathway may have evolved in response to environmental pressures in early human history.
Given reports that the APOE ε4 haplotype exhibits evidence for natural selection,7,8 we assessed all validated and well-replicated AD susceptibility loci3,4 for evidence of recent (<60,000 years ago) positive selection, using linkage disequilibrium (LD)-based methods to detect genomic regions harboring genetic variants inferred to have recently and rapidly increased in frequency within human populations. We applied the integrated haplotype score (iHS)9 statistic, which measures the lengths of the haplotypes around a given SNP, to identify evidence of positive selection in human populations of African (Yoruba from Ibadan, Nigeria [YRI]), European (Centre d′Étude du Polymorphisme Humain samples from Utah residents of European descent [CEU]), and East Asian (Japanese subjects from Tokyo and Han Chinese subjects from Beijing representing Asian populations [ASI]) ancestry from phase II of the International HapMap Project.10 The iHS statistic was implemented with a mean of 0 and a variance of 1. To discover signatures of selection on the haplotype bearing AD susceptibility alleles, we searched for SNPs meeting a stringent threshold of |iHS| > 2 (corresponding to the most extreme 5% of iHS values across the genome among HapMap II SNPs with minor allele frequency > 0.05) within the LD block containing the index SNP of each locus from two large-scale AD genome-wide association studies (GWAS).3,4 Furthermore, we restricted our analysis to SNPs with r2 > 0.5 and/or D′ = 1, the published index SNP being associated with AD in each locus. Although we tested only 11 loci, we applied a stringent correction for genome-wide testing to identify only the most robust signals of selection. Using this approach, we found evidence for selection acting on haplotypes carrying AD-associated alleles in 3 of 11 loci, including PICALM (MIM 603025; rs561655), BIN1 (MIM 601248; rs7561528), and CD2AP (MIM 604241; rs9349407) (Table 1). All of these loci showed significant (with the use of a threshold for genome-wide testing) evidence for selection in the same HapMap population of East Asian descent, suggesting that the loci may have responded to the same selective pressure. Using a less stringent “suggestive” threshold of significance (|iHS| > 1.65, corresponding to the top 10% of iHS values across the genome), we also saw evidence of natural selection at MS4A2 (MIM 147138; rs610932) in the African population (Table 1). Except for the MS4A2 locus, the index SNP from the reported AD GWAS was not the SNP exhibiting the strongest evidence of selection. This is not surprising given that these index SNPs emerged from genome-wide screens and are most likely surrogate markers for the causal variant(s) in each locus. The evidence for selection may, in fact, help to pinpoint variants that are more likely to have a functional effect involved in driving the selection process and perhaps AD susceptibility.
Table 1.
Alzheimer Disease Susceptibility Loci with Evidence of Positive Selection
| Chr | Index SNP | Locus | Tag SNPa |
Integrated Haplotype Score (iHS) |
||
|---|---|---|---|---|---|---|
| ASI | CEU | YRI | ||||
| Loci with Evidence of Selection | ||||||
| 11q14 | rs561655 | PICALMb | rs659023 | 2.17 | 1.31 | −1.88 |
| 2q14 | rs7561528 | BIN1b | rs10200967 | 2.58 | 1.64 | 1.45 |
| 6p12 | rs9349407 | CD2APb | rs9395288 | 2.06 | −0.49 | −1.08 |
| 11q12 | rs610932 | MS4A2b | rs610932 | −0.88 | 0.73 | −1.92 |
| Other Loci | ||||||
| 7q35 | rs11767557 | EPHA1b | – | −0.74 | 0.27 | 0.59 |
| 1q32 | rs6701713 | CR1 | – | −0.11 | 0.26 | −0.71 |
| 19q13 | rs3865444 | CD33 | – | 0.43 | −0.87 | – |
| 19q13 | rs2075650 | APOE | – | 0.55 | 1.5 | – |
| 19q13 | rs4420638 | APOE | – | 0.45 | 0.7 | −0.16 |
| 8p21 | rs1532278 | CLU | – | – | – | – |
| 19p13 | rs3764650 | ABCA7 | – | – | – | – |
ASI, East Asian; YRI, African; CEU, European.
Tag SNPs: SNPs that best capture the selection signal on each AD susceptibility haplotype. Tag SNPs were chosen on the basis of their LD (r2 > 0.5; D′ = 1) with the published index SNP for each locus, evidence for selection signals, and AD GWAS association at p < 10−6. Absolute iHS > 2 and > 1.65 correspond to the most extreme 5% and 10% of iHS values across the genome, respectively.
Shows evidence for selection in gene-based analyses (Table S2).
Interestingly, each positively selected allele on the AD-associated haplotypes in the PICALM, BIN1, and CD2AP loci had a high positive iHS score in the East Asian subjects (Table 1). A positive iHS means that the haplotypes with the ancestral allele that humans share with chimpanzees are longer than those containing the derived allele; therefore, these results suggest that selection favored the ancestral allele at all three loci. Although a neighboring derived allele that is targeted by a selective force could drive selection at one ancestral allele, this is unlikely to be the case in all three loci. It is more likely that the three ancestral alleles are themselves the targets of selection. In all three cases, the selected ancestral alleles are those associated with diminished susceptibility to AD; the derived alleles are the risk alleles. This observation introduces the possibility that these three loci worked together in a shared pathway and were affected in a coordinated manner over the course of human evolution in the East Asian population that encountered a specific selective pressure. Furthermore, the three risk-associated alleles have all been selected against, suggesting that they may have converging effects on the same cellular function, one implicated in AD susceptibility but most likely important in other contexts as well.
To further confirm the robustness of the selection signals, we deployed alternative methods of selecting SNP sets with which to detect evidence for natural selection in AD loci and validate the results of the haplotype-based iHS analysis. In a region-based analysis, we determined the proportion of SNPs with |iHS| > 2 in a 50-SNP window centered on the index SNP. In a gene-based approach, we created a window of 50 SNPs centered on each gene closest to the index SNP; the genes in the upper 10% of the empirical distribution for number of significant SNPs were then considered to be candidate targets of selection (Table 2). Table 2 and Table S1 summarize the results of these analyses; they are consistent with the results of the haplotype-based analysis. In these analyses, we subsequently explored the possibility that APOE or genes associated with early-onset, Mendelian forms of AD (APP [MIM 104760], PSEN1 [MIM 104311], and PSEN2 [MIM 600759]) harbor evidence of natural selection; however, none of these loci returned |iHS| scores that met our suggestive or significant thresholds (data not shown).
Table 2.
Alzheimer-Disease-Associated Haplotypes, Regions, and Genes with Evidence of Positive Selection
| Locus | SNP | Gene |
Three Different Approaches to Assess for Evidence of Positive Selection |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Haplotype |
Region |
Gene |
|||||||||
| ASI | CEU | YRI | ASI | CEU | YRI | ASI | CEU | YRI | |||
| 6p12 | rs9349407 | CD2AP | + | + | + | ||||||
| 11q12 | rs610932 | MS4A2 | + | + | + | ||||||
| 11q14 | rs561655 | PICALM | + | + | + | + | + | ||||
| 2q14 | rs7561528 | BIN1 | + | + | + | ||||||
| 7q35 | rs11767557 | EPHA1 | + | ||||||||
Evidence of selection (indicated by “+”) based on the analyses from alternate methods of selecting SNP sets with which to evaluate evidence for selection.
The “haplotype” analysis is our primary analysis. In the haplotype analysis, we searched for SNPs in LD (r2 > 0.5; D′ = 1) with the index SNP in each locus (Table 1). We then considered haplotype blocks containing a linked SNP with |iHS| > 2 to be candidate targets of selection. We deployed secondary analyses to illustrate the robustness of our results. For our secondary regional analysis, we determined the number of SNPs with |iHS| > 2 in a 50-SNP window centered on the GWAS SNP. We then considered the regions in the upper 10% of the empirical distribution for proportion of SNPs with |iHS| > 2 to be candidate targets of selection. For gene-based analysis, we determined the proportion of SNPs with |iHS| > 2 in a 50-SNP window centered on the gene closest to the GWAS SNP. We then considered the genes in the upper 10% of the empirical distribution for number of significant SNPs to be candidate targets of selection. The symbol “+” indicates evidence for selection at each locus in three HapMap populations.
To test for enrichment of positive selection among the AD-associated loci, we compared the proportion of haplotypes with positively selected loci in the set of AD-validated loci with a similar list of Parkinson disease (PD [MIM 168600]) -validated loci (n = 12).11 In this targeted analysis, we imposed our suggestive |iHS| > 1.65 threshold and found that the proportion of AD-associated loci meeting this threshold of evidence for selection in at least one HapMap population (45%, 5 of 11 SNPs have |iHS| > 1.65; Table 1) was higher than the proportion of PD loci under selection (8%, 1 of 12 SNPs have |iHS| > 1.65; χ2AD versus PD = 4.774, pAD versus PD = 0.029). These results suggest enrichment for loci with evidence of selection among validated AD susceptibility loci. We obtained similar results when comparing the AD loci to random sets of SNPs with similar allele frequencies (data not shown).
On the basis of the hypothesis that evidence for natural selection might be a feature of other, as-yet undiscovered AD susceptibility loci, we extended these analyses to the list of loci that met a suggestive threshold of significance (p < 10−4) for association with AD susceptibility in a recent GWAS that provided a comprehensive list of such results.3 Out of an initial 447 suggestive SNPs in this study, we found 118 loci with independent effects (after LD pruning of SNPs with an r2 = 0.5 threshold). We found that 10 (8%) of these 118 loci have an |iHS| > 2 and therefore meet our threshold of genome-wide significance. Furthermore, 21 loci (18%) demonstrate suggestive evidence (|iHS| > 1.65) of natural selection, which is more than expected by chance (expected: 11.8 [10%]; p = 0.005). Thus, our observation in the validated AD loci may be a more general feature of AD susceptibility loci.
Given that 3 of 11 AD-associated loci show evidence of selection in the same human population, it is plausible that some of the genes they contain may have a correlated evolutionary history and are coevolving. Coevolution can occur when a heritable change in one gene establishes selective pressure for another gene.12 To detect such coevolutionary processes, we constructed protein-protein interaction (PPI) networks to identify interacting proteins, because two proteins are more likely to share correlated evolutionary history if they physically interact.13 This method of constructing networks does not leverage information regarding natural selection and is therefore independent of our earlier analyses. To construct a PPI network, we selected genes found within validated and suggested AD-associated loci (defined as the genomic segment bounded by SNPs with an r2 > 0.5 to the index SNP for a given locus) as input for the web-based tool Disease Association Protein-Protein Link Evaluator (DAPPLE), which uses high-confidence pairwise protein interactions and tissue-specific expression data to reconstruct a PPI network.14 The network is conservative, requiring that interacting proteins be known to be coexpressed in a given tissue. The resultant AD PPI network returned by DAPPLE is statistically significant for a network connectivity that allows a common interactor protein (not known to be associated with AD) between AD genes when compared to 1,000 random networks (permuted p = 0.043; Figure 1). This analysis is not significant (p = 0.367) when direct network connectivity between the AD genes is required. We were able to indirectly connect all but two of the proteins coded in known AD loci (ABCA7 [MIM 605414] and CR1 [MIM 120620]).
Figure 1.
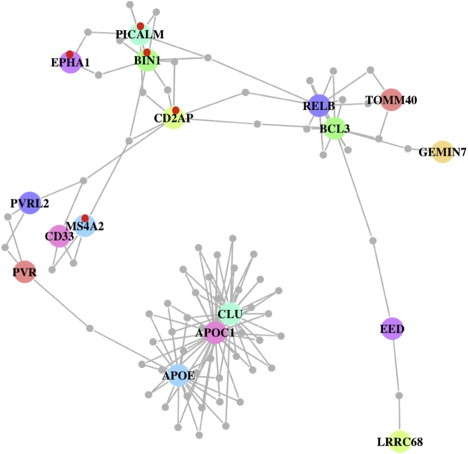
Protein-Protein Interaction Network Generated from Proteins Encoding for AD-Associated and -Suggested Genes
The 118 LD-pruned SNPs (p < 10-4) from the Alzheimer Disease Genetics Consortium (ADGC) GWAS3, which provided a complete list of suggestive results, were included as an input to DAPPLE. DAPPLE selects genes from a SNP list based on the region containing SNPs with an r2 > 0.5 to each index SNP; this region is then extended to the nearest recombination hot spot. AD-associated proteins are represented as nodes connected by an edge if there is in vitro evidence for high-confidence interaction. The large colored circles represent AD-associated and -suggestive proteins, and small circles in grey represent the connected proteins (not known to be associated with AD). Most of the AD-associated proteins are connected via common interactor proteins (gray) with which the associated proteins each share an edge. The red points indicate genes (CD2AP, PICALM, BIN1, EPHA1, and MS4A2) under positive selection in our gene-based analyses of HapMap II populations (Table S1). The PPI network is statistically significant for indirect connectivity (PPI network permuted p = 0.043).
When overlaying our AD PPI network allowing for a common interactor protein with the results of our analyses for evidence of natural selection, an intriguing subnetwork that includes the proteins encoded by PICALM, BIN1, CD2AP, and EPHA1 (MIM 179610) emerged. Integrating our gene-based PPI results with our gene-based selection analysis (Table S1), we saw that all four susceptibility loci showed evidence of positive selection in East Asians (Table 2 and Table S1). This evidence suggests a possible coevolution of these four genes, which, on the basis of the PPI analysis, may interact in a macromolecular complex (Figures 1 and 2). To assess the statistical significance of this subnetwork, we constructed a new PPI network by using these four genes as the seed regions, and found that the subnetwork was statistically significant for indirect connectivity (permuted p = 0.033; Figure 2). We observed that many of the interacting proteins connecting the AD-associated proteins also showed significant evidence for selection (Figure 2), and, interestingly, one of these proteins is encoded by a gene (GAB2 [MIM 606203]) that has been previously suggested to be associated with AD.15,16 Thus, these AD-associated and “connector” genes may have been under selection because of their participation in a single functional module over evolutionary time, and common function may also explain their individual associations with AD susceptibility. Given the late age-at-onset of AD, it is unlikely that this phenotype itself has been the target of selection over evolutionary time; rather, it is likely that these four genes, along with their interaction partners, compose a functional module that is important in other biological contexts.
Figure 2.
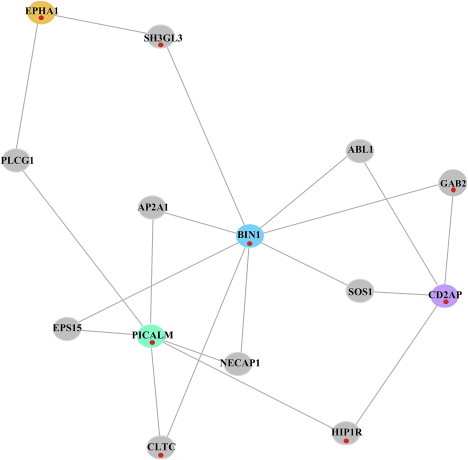
Protein-Protein Interaction Subnetwork of AD-Associated Genes under Positive Selection
The subnetwork is simply a highly interconnected subset of the larger network that emerges from DAPPLE and is illustrated in Figure 1. The subnetwork is statistically significant for indirect connectivity (PPI subnetwork permuted p = 0.033). As in Figure 1, the red dots mark genes with evidence for natural selection.
Some of the strongest selective pressures acting during human evolution have been attributed to human interactions with pathogens; we thus investigated the functional consequences of AD-associated variants with evidence of selection on gene expression levels in immune cells. This approach was additionally motivated by the observation that some AD-associated variants map close to genes with putative immune function (i.e., CD33 [MIM 159590], CR1, and MS4A2) and others, such as PICALM, BIN1, CD2AP, and EPHA1, are coexpressed in immune cells, particularly in the myeloid lineage.17 Thus, we assessed each of the 11 validated and well-replicated AD loci for evidence of an effect on RNA expression in cis; that is, we performed an expression quantitative trait locus (eQTL) analysis in each locus for genes in the vicinity of the index SNP (see Supplemental Material and Methods). We first investigated genetic variation and mRNA expression in an available data set derived from peripheral blood mononuclear cells (PBMCs) of 228 individuals of European ancestry with demyelinating disease,18 representing a set of subjects with an activated immune system. PBMCs are purified from peripheral blood and contain both myeloid and lymphoid cells. After gene-based permutation assessing significance of SNP-gene association p values, we identified five cis-regulatory effects in the 11 tested loci (Table S2). Specifically, we found three AD-associated index variants—rs610932, rs7561528, and rs3752246—with cis-regulatory effects in PBMCs: rs610932 influences the expression of neighboring MS4A2 (nominal p = 5.04 × 10−8), whereas rs7561528 and rs3752246 affect the expression of BIN1 (p = 4.76 × 10−4) and ABCA7 (p = 6.87 × 10−5), respectively. These cis-regulatory effects are all significant at a p < 0.05 permutation threshold; the same MS4A2 and BIN1 haplotypes harbor evidence of natural selection (Table 1).
In the PICALM locus, which has strong evidence of selection, the current best AD marker does not have a strong cis-regulatory effect on gene expression, but other SNPs that have evidence of association with AD (p < 5x10−8 in published studies) 3,4 and are in strong LD with the index SNP have replicated evidence of having a cis-regulatory effect on gene expression. Specifically, the AD risk allele rs659023G (pAD = 2.78 × 10−10)3 (r2 = 0.8 with index PICALM SNP rs561655) is significantly associated with decreased expression of PICALM in PBMC (p = 4.76 × 10−4; Figure S1). We replicate this cis-regulatory effect in CD4+ T lymphocytes (which are constituents of the PBMC cell mixture) of 40 healthy individuals of European ancestry (p = 2.48 × 10−4).
In the case of PICALM, the evidence for selection and the effect on gene expression converge (Figure 3; Table S3): the rs659023 SNP discussed above exhibits a strong correlation with PICALM RNA expression and has a high iHS (eQTL p = 4.76 × 10−4, |iHS| = 2.17). These results suggest that one or more potential functional variants that influence PICALM expression may have been selected for over the course of human history in this well-validated AD locus. This information can be leveraged to focus fine-mapping efforts onto a short list of candidate causal variants that can be targeted in well-powered AD susceptibility analyses.
Figure 3.
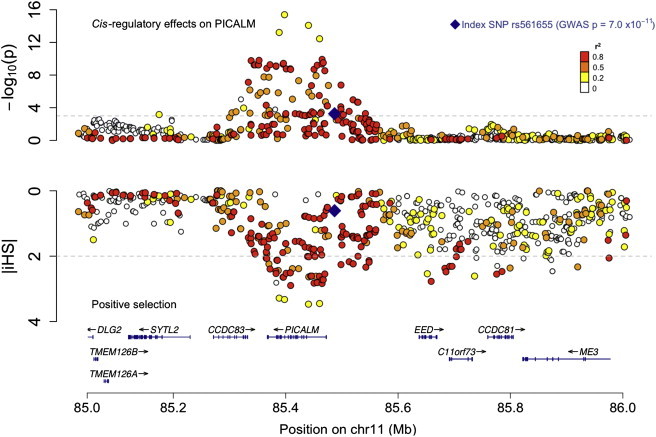
Colocalization of cis-Regulatory Effects and Positive Selection Signals in the PICALM Locus
The top panel reports the cis-regulatory effects of each SNP in the vicinity of the PICALM locus on PICALM RNA expression in PBMC; -log (p value) is reported on the y axis. The lower panel reports the evidence for positive selection at each SNP over the same chromosomal segment (1 Mb total); here we have inverted the y axis so that the most extreme iHS values are at the bottom of the scale. The RefSeq genes in the region are shown at the bottom of the figure. The LD (in r2) for each SNP with the index AD-associated PICALM SNP (rs561655) is illustrated with the use of colors, as indicated in the top right of the figure. The haplotype carrying GWAS index SNP rs561655 also contains other alleles that have the strongest selection signals and cis eQTL effects, suggesting that functional variants influencing the expression of PICALM may have been the target of recent natural selection. The SNPs with extreme iHS values are the same SNPs that have extreme p values in the cis eQTL analysis (Table S3).
In summary, we observe significant evidence for signatures of positive selection on haplotypes associated with late-onset Alzheimer disease. The most intriguing finding of our study is that multiple AD-associated genes (i.e., PICALM, BIN1, CD2AP, and EPHA1) with evidence for positive selection encode proteins that physically interact within an independently defined PPI network. Furthermore, some of the linking proteins in the network also show evidence of positive selection, and one of these, GAB2, has previously been suggested to be associated with AD.15,16 The convergence of results in these two complementary analyses suggests that these four interacting proteins encoding for AD susceptibility genes may have had a correlated evolutionary history, perhaps in response to a single evolutionary pressure that affected East Asian populations and, to a lesser degree, European populations (Table 2). Moreover, we found that several of these loci exhibit a cis-regulatory effect on the transcription of the selected genes in immune cells, suggesting that changes in gene expression may be one mechanism by which an AD-related molecular pathway evolved in early human history, most likely in a non-AD context.
The results presented here provide convincing support for recent positive selection of loci underlying late-onset AD, but what are the selective pressures underlying the signatures? It is difficult to identify the precise selective pressure that led to our observations. Selection may result from an array of forces including pathogen resistance as well as environmental and dietary changes as modern humans migrated out of Africa and spread throughout the world. For the genes with putative immune functions, we speculate that pathogens may have exerted selective pressures on populations and consequently influenced the frequency of AD-associated alleles. Regardless of what the selection pressure might have been, our results suggest that several different loci implicated in AD susceptibility may have worked together in another context to enhance survival over the course of recent human evolutionary history.
Evidence for natural selection at a given variant suggests that such a variant may have functional consequences, given that the selection process was mediated by an alteration of a biological process. One can think of selective pressures as natural, in vivo human experiments in which we can measure the response of human populations to unknown perturbations, and these alterations can inform the function of genes within a given locus. However, by itself, evidence of selection is not sufficient for identifying causal variants in susceptibility loci. Rather, it offers another key dimension of functional information to integrative analyses that include disease association, protein-protein interaction, and gene expression. Such analyses will powerfully explore the molecular mechanisms underlying associations between genetic variation and disease susceptibility. In the case of AD, we have highlighted a network of coexpressed, physically interacting susceptibility genes that is supported by evidence of selection; this observation lays the groundwork for future hypothesis-driven investigations into the function of the interactions of these susceptibility genes, which may be related to vesicular trafficking, given the known functions of PICALM, BIN1, CD2AP, and EPHA1. On the basis of our expression data, characterization of cell populations from peripheral blood may provide a reasonable substrate for functional investigations that seek to interrogate the coordinated functional consequences of these four susceptibility loci, and perhaps others.
Acknowledgments
This work is supported by the National Institutes of Health (NIH) (RC2 GM093080, R01 AG30146, R01 AG179917, R01 AG15819, K08 AG034290, P30 AG10161 and R01 AG11101). J.M.S. was additionally supported by the Burroughs Wellcome Fund. We thank the Brigham and Women's Hospital PhenoGenetic Project for providing mRNA samples from healthy subjects that were used in the CD4+ T lymphocyte transcriptional analysis for this study. We thank Michelle Lee for sample collection, Katherine Rothamel for data generation, and Scott Davis for mRNA expression quality control. We thank Christophe Benoist for his leadership on RC2 GM093080. These analyses were conducted under the auspices of a protocol approved by the institutional review board of Partners Healthcare.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Haplotter, http://haplotter.uchicago.edu/
HapMap FTP site, ftp://ftp.ncbi.nlm.nih.gov/hapmap/
iHS software, http://hgdp.uchicago.edu/Software/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Avramopoulos D. Genetics of Alzheimer's disease: recent advances. Genome Med. 2009;1:34. doi: 10.1186/gm34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naj A.C., Jun G., Beecham G.W., Wang L.S., Vardarajan B.N., Buros J., Gallins P.J., Buxbaum J.D., Jarvik G.P., Crane P.K. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J.C., Carrasquillo M.M., Abraham R., Hamshere M.L., Pahwa J.S., Moskvina V. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., European Alzheimer's Disease Initiative Investigators Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 6.Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M., Bis J.C., Smith A.V., Carassquillo M.M., Lambert J.C., CHARGE Consortium. GERAD1 Consortium. EADI1 Consortium Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drenos F., Kirkwood T.B. Selection on alleles affecting human longevity and late-life disease: the example of apolipoprotein E. PLoS ONE. 2010;5:e10022. doi: 10.1371/journal.pone.0010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vamathevan J.J., Hasan S., Emes R.D., Amrine-Madsen H., Rajagopalan D., Topp S.D., Kumar V., Word M., Simmons M.D., Foord S.M. The role of positive selection in determining the molecular cause of species differences in disease. BMC Evol. Biol. 2008;8:273. doi: 10.1186/1471-2148-8-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalls M.A., Plagnol V., Hernandez D.G., Sharma M., Sheerin U.M., Saad M., Simón-Sánchez J., Schulte C., Lesage S., Sveinbjörnsdóttir S., International Parkinson Disease Genomics Consortium Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser H.B., Hirsh A.E., Wall D.P., Eisen M.B. Coevolution of gene expression among interacting proteins. Proc. Natl. Acad. Sci. USA. 2004;101:9033–9038. doi: 10.1073/pnas.0402591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tillier E.R., Charlebois R.L. The human protein coevolution network. Genome Res. 2009;19:1861–1871. doi: 10.1101/gr.092452.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., Cotsapas C., Daly M.J., International Inflammatory Bowel Disease Genetics Constortium Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiman E.M., Webster J.A., Myers A.J., Hardy J., Dunckley T., Zismann V.L., Joshipura K.D., Pearson J.V., Hu-Lince D., Huentelman M.J. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikram M.A., Liu F., Oostra B.A., Hofman A., van Duijn C.M., Breteler M.M. The GAB2 gene and the risk of Alzheimer's disease: replication and meta-analysis. Biol. Psychiatry. 2009;65:995–999. doi: 10.1016/j.biopsych.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Jager P., Jia X., Wang J., de Bakker P., Ottoboni L., Aggarwal N., Piccio L., Raychaudhuri S., Tran D., Aubin C. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.