

Abstract
Kohlschutter–Tonz syndrome (KTS) is a rare autosomal-recessive disorder of childhood onset, and it is characterized by global developmental delay, spasticity, epilepsy, and amelogenesis imperfecta. In 12 KTS-affected individuals from a Druze village in northern Israel, homozygosity mapping localized the gene linked to the disease to a 586,513 bp region (with a LOD score of 6.4) in chromosomal region 16p13.3. Sequencing of genes (from genomic DNA of an affected individual) in the linked region revealed chr16: 4,848,632 G>A, which corresponds to ROGDI c.469C>T (p.Arg157∗). The nonsense mutation was homozygous in all affected individuals, heterozygous in 10 of 100 unaffected individuals from the same Druze community, and absent from Druze controls from elsewhere. Wild-type ROGDI localizes to the nuclear envelope; ROGDI was not detectable in cells of affected individuals. All affected individuals suffered seizures, were unable to speak, and had amelogenesis imperfecta. However, age of onset and the severity of mental and motor handicaps and that of convulsions varied among affected individuals homozygous for the same nonsense allele.
Main Text
Kohlschutter–Tonz syndrome (KTS) (MIM 226750) is described as a rare autosomal-recessive neurodegenerative disorder characterized by progressive dementia, spasticity, and epilepsy.1,2 A clinical marker of KTS is a generalized enamel defect, amelogenesis imperfecta, which is most obvious as yellowed teeth. KTS was first identified in families from Switzerland,1,2 Sicily,3 the Druze community of northern Israel,4 and, subsequently, other locations in western Europe.5–9 To date, only 21 affected individuals have been reported. Seizures, intellectual impairment, and amelogenesis imperfecta were reported in all families, and other clinical features varied among reports. The goal of this project was to identify the gene and the mutation responsible for KTS in the highly consanguineous Druze community.
We have compiled 14 new KTS cases pertaining to five families, all of which originate from the same small Druze village in northern Israel. The index case (II-1 in family 1), who was referred to us at the age of 13 months for the evaluation of seizures, was noted to have amelogenesis imperfecta. Individual II-1 and her parents and siblings, as well as informative relatives of four other families with affected children, were enrolled (Figure S1, available online). A consanguineous liaison is evident for families 3, 4, and 5, although the parents of the affected children in family 4 are not known to be related. Family 2, unrelated to families 3–5, is consanguineous too (Figure S1). The study was approved by the institutional review board at Rambam Health Care Campus, Haifa, and after signed informed consent (self and parental), a blood sample was drawn for DNA extraction from all available family members (both affected and healthy individuals). All affected individuals were clinically evaluated by a pediatric clinical geneticist, medical records were reviewed, and parents were interviewed.
The clinical characteristics of 14 KTS-affected individuals (seven males and seven females; ages 2–24 years) are depicted in Table 1. Born at term after normal gestation, they all appeared normal at birth and had no apparent dysmorphology. Although all KTS cases ultimately displayed an unequivocal phenotype heralded by seizures and “yellow teeth,” they varied widely with regard to the severity of the manifestations, even within the same nuclear family.
Table 1.
Clinical Characteristics of 14 Individuals Affected by KTS
|
Family 1 |
Family 2 |
Family 3 |
Family 4 |
Family 5 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | II-6 | II-7 | IV-1 | IV-2 | IV-4 | V-1 | V-3 | V-4 | V-5 | V-6 | V-7 | V-8 | V-12 | |
| Gender | female | female | male | female | male | male | male | female | female | male | female | female | male | male |
| Birth weight (grams) | 3,500 | 3,500 | 3,500 | 3,250 | 3,250 | 3,500 | 2,500 | 2,400 | 2,300 | 3,140 | 2,800 | 3,500 | 3,800 | 3,500 |
| Deliverya | NVD | NVD | NVD | NVD | NVD | NVD | CS | CS | NVD | NVD | NVD | NVD | NVD | NVD |
| Current age (years) | 6 | 24 | 16 | 15.5 | 16.5 | 13.5 | 14.5 | 16.5 | deceased at age 2 | 16.5 | 19.5 | 15 | 3.5 | 4.5 |
| Clinical characteristics | ||||||||||||||
| Age of first convulsion (months) | 13 | 12 | 9 | 12 | 6.5 | 42 | 9 | 9 | birth | 10 | 9 | 9 | 9 | 11 |
| Seizure intensityb | + | +++ | ++ | + | +++ | + | +++ | + | +++ | ++ | +++ | + | + | +++ |
| Resistance to therapyc | + | +++ | ++ | + | +++ | + | +++ | + | +++ | ++ | +++ | + | + | +++ |
| Amelogenesis imperfecta | yes | yes | yes | yes | yes | yes | yes | yes | N/A | yes | yes | yes | yes | yes |
| Intellectual impairmentd | +++ | ++++ | +++ | +++ | +++ | ++ | +++ | ++ | N/A | +++ | ++++ | ++ | +++ | +++ |
| Speech | no | no | no | no | no | mumbling | no | mumbling | N/A | no | no | mumbling | no | no |
| Ambulant | yes | no | yes | yes | yes | yes | no | yes | N/A | yes | no | yes | yes | yes |
| Laboratory evaluation | ||||||||||||||
| EEGe | N | N/A | N | N/A | abnormal | abnormal | N/A | N/A | N/A | abnormal | N/A | N | abnormal | N |
| Brain MRIf | abnormal | abnormal | abnormal | N/A | N | N/A | N/A | N/A | N/A | abnormal | N/A | N/A | N | N |
The following abbreviations were used: NVD, normal vaginal delivery; CS, cesarean section; N/A, not available; EEG, electroencephalogram; N, normal; and MRI, magnetic resonance imaging.
All individuals were born at term.
Seizure intensity: +, occasional; ++, frequent; and +++, severe.
Therapy resistance: +, good response to treatment; ++, control by treatment; and +++, refractory.
Intellectual impairment: +, mild; ++, moderate; +++, severe; and ++++, profound.
An abnormal EEG revealed a pattern of slow, short epileptiform spikes and waves during light sleep and was normal thereafter.
An abnormal MRI revealed dilation of cerebellar sulci and third and lateral ventricles.
Family 4 has five affected children (V-4 to V-8) who all had epileptic episodes that varied in age of onset, intensity, frequency, and response to treatment. The firstborn child (V-4), who died at the age of 2 years, was vegetative, failed to thrive, and suffered from intractable convulsions and microcephaly. Their second-born affected child (V-6) displayed impaired psychomotor development from the first months of life and convulsive episodes, starting at 9 months of age, that were refractory to treatment. She was nonverbal and nonambulant. It was noted that her brother (V-5) lagged developmentally starting at 6 months of age, and he is regularly maintained on anticonvulsants (he has had a partial response). At 16.5 years of age, he is awkwardly ambulant and nonverbal, performs mostly by shouting and yelling, and is irritable and self-mutilating. His 15-year-old sister (V-7) suffers from a convulsive disorder that responds well to treatment, and, although intellectually disabled, she manages to communicate with her mother, utters a few words, and is ambulant. The youngest sibling (V-8), currently 3.5 years old, has a convulsive disorder that is only partially controlled, and he is hyperactive, nonverbal, relatively ambulant, and mentally disabled.
A wide interfamilial variability was again noted in families 1, 2, and 3. Family 2 has three affected children (IV-1, IV-2, and IV-4). The first affected boy (IV-2), who was hypotonic and sucked poorly as a neonate, experienced intractable seizures beginning at 6.5 months of age. The youngest boy (IV-4) was considered normal until, at the age of three years, he experienced a convulsive episode. Accordingly, IV-4 performs better than his older siblings. Affected children from family 1 (II-1, II-6, and II-7) and family 3 (V-1 and V-3) all presented with seizures at around one year of age (between 9 and 13 months). The severity of the epileptic events ranged from short convulsive episodes that resolved under antiepileptic treatment (such as with individuals II-1 and V-3) to generalized convulsions partially controlled by anticonvulsants (II-7) to frequent convulsive episodes resistant to therapy (II-6 and V-1). Intellectual disability was noted to correlate with the severity of the epileptic events. The phenotype displayed by our KTS cases is consistent but variable. All cases displayed amelogenesis imperfecta, seizures, severe developmental delay, and lack of speech. The magnitude of the intellectual disability, although severe to profound, is still directly related to the severity of the convulsive disorder as manifested by age of onset and response to treatment. Affected individuals lost their gross motor skills in either early or late adolescence because they became spastic. The most afflicted individuals were bedridden early in life. Metabolic workup was negative as a rule. Electroencephalograms (EEGs) undertaken shortly after a convulsive episode were interpreted, in most cases, as normal. Magnetic resonance images (MRIs) were available for seven individuals. In four of these individuals, dilatation of cerebellar sulci and third and lateral ventricles was evident and should probably be regarded as a correlate of cerebral atrophy.
With this in mind, the term “dementia” previously used for defining a major characteristic of KTS is not valid for the cases described here. KTS-affected children have global developmental delay that is cortical in nature (intellectual disability, spasticity, and seizures), and the progressive nature of their neurological decline might be attributed to, among other things, the intractability of their seizures and, as such, epileptic encephalopathy.
Homozygosity mapping was carried out with the assumptions that the disease follows a recessive model and that a single responsible allele is shared by all affected individuals. First, genomic DNA from ten affected individuals, one healthy sibling, and one obligate carrier parent was genotyped with the Illumina 6000 SNP array (BioRap Technologies at the Rappaport Institute); thereafter, five affected individuals were genotyped with the Affimetrix GeneChip Human Mapping 250K Nsp microarray (Biological Services Unit at the Weizmann Institute). Homozygosity-by-descent analysis was carried out manually and explored identical homozygous intervals in all affected individuals. Candidate homozygous loci were genotyped with microsatellite markers derived from Marshfield maps or with markers designed by us (our markers were based on the Tandem Repeats Finder program and the UCSC Human Genome Database). Haplotypes of family members were manually constructed and analyzed with SUPERLINK online. In the 6000 SNP array, the analysis identified a candidate segment, in chromosomal region 16p13.3, spanning 2,670,304 bp between rs2075852 and rs1012259; only one homozygous SNP (rs85930) shared homozygosity in all ten affected individuals, whereas the healthy mother and sibling were heterozygous. Evaluating this interval in the 250K SNP array, we identified a homozygous segment spanning only 873,963 bp between rs11865087 and rs3760030; this segment was shared by four of the five genotyped individuals. The fifth affected individual was clinically reevaluated and excluded from the research as a non-KTS case (data not shown). We developed and genotyped the following three microsatellite markers in this interval: GT32 at chr16: 4,533,109–4,533,282 (marker 1); TG19 at chr16: 4,854,835–4,855,072 (marker 2); and GT31 at chr16: 5,105,840–5,106,041 (marker 3). We depict haplotypes in Figure 1 to show the segregation of markers and SNPs in affected individuals and healthy family members; there are three affected and seven healthy individuals in family 1, three affected and two healthy individuals in family 2, and six affected and eight healthy individuals in families 3–5 (Figure 1A). Individual V-4 (family 4), who died at 2 years of age, and individual V-8 (family 4), who has not yet been diagnosed with KTS at this stage, were excluded from the analysis. All affected individuals shared the same homozygous haplotype for markers 2 and 3, and a 586,513 bp segment between microsatellite marker 1 (chr16: 4,533,109) and rs3760030 (chr16: 5,119,872) was defined as the linkage locus for KTS (Figure 1A). Under a recessive model of full penetrance, the LOD score for linkage of this region to KTS was 6.4.
Figure 1.
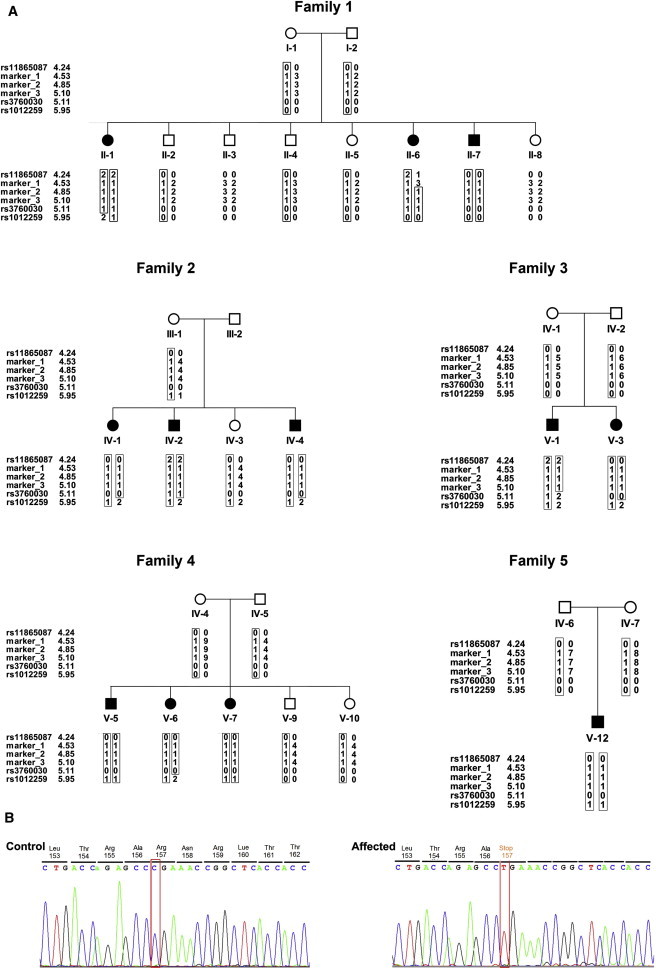
Families 1–5 Haplotypes and the c.469C>T Mutation in Exon 7 of ROGDI
(A) Disease-associated haplotypes are shown in boxes. Markers 1 and rs3760030 define the minimal homozygosity locus associated with the disease (allele 0: not genotyped). The numbers flanking the genotyped markers indicate the distance from 16pter (GRCh37/hg19 assembly).
(B) The ROGDI c.469C>T (p.Arg157∗) mutation in genomic DNA of a KTS-affected individual compared to a control.
Chromosomal region 16p13.3 harbors 17 genes. We performed Sanger sequencing of coding regions and flanking intron-exon boundaries on genomic DNA from one affected individual (II-1 in family 1) by using the primers that we designed. The genomic sequences were retrieved from the UCSC Genome Browser (GRCh37/hg19 assembly [Feb. 2009]). In the first ten genes, no potentially damaging variants were found. In contrast, sequencing of the Drosophila rogdi homolog, ROGDI (FLJ22386) (RefSeq accession number NM_024589.1), yielded a homozygous nonsense mutation (c.469C>T in exon 7) causing a premature stop codon, p.Arg157∗ (Figure 1B). This nonsense mutation cosegregated with KTS in all five families; all affected individuals were homozygous for the mutation, all parents were heterozygous, and unaffected siblings were either heterozygous or homozygous for the wild-type allele. Of 100 unaffected adults from the same Druze community, ten were heterozygous carriers of the mutation; none were homozygous for the mutation. The mutation was not observed in 100 Druze individuals from other parts of Israel. Primer sequences and PCR conditions are available upon request. The c.469C>T mutation was not reported in the Exome Variant Server. No clinical links were reported for SNPs in the gene.
ROGDI (FLJ22386) is the human homolog of Drosophila rogdi. The human gene encodes a 287 amino acid leucine zipper protein of unknown function. Drosophila rogdi encodes 343 and 268 amino acid isoforms. The stop at residue 157 of the human homolog corresponds to residue 156 of the shorter isoform and residue 231 of the longer isoform in Drosophila. The most conserved domain of the protein is the C terminus (residues 253–281 of the human protein), shared by the human protein and both Drosophila isoforms and truncated by the stop mutation in the KTS-affected families. Meta-analysis of genome-wide expression studies indicates that in both humans and mice, ROGDI is expressed more in the hippocampus than in other tissues.10 Our RT-PCR analysis indicates that ROGDI is widely expressed and has higher levels in the adult brain, spinal cord, peripheral blood, heart, and bone marrow but lower (and still detectable) levels in many other tissues, including the fetal brain (Figure S2).
Examination of the primary sequence of ROGDI does not provide any clues about the subcellular localization of the protein. To address this issue, we generated an expression plasmid encoding wild-type ROGDI (pROGDI) after amplification of the full-length ROGDI cDNA by PCR with cDNA from an adult human brain as a template (Clontech). Cellular localization of ROGDI was thereafter evaluated in three experiments. Human embryonic kidney (HEK) 293 cells were transfected with pROGDI and were examined by indirect immunofluorescence micoroscopy with a rabbit anti-ROGDI polyclonal antibody (1 μg/ml rabbit anti-ROGDI polyclonal antibody, Protein Tech Group, Chicago, IL, USA; Catalog No. 17047-1-AP) generated against the entire protein and a 1:1,000 dilution of secondary Alexa Fluor-488 (green) goat anti-rabbit antibody (Molecular Probes); the HEK 293 cells revealed a strong nuclear labeling of multiple bright spots and virtually no cytoplasmic staining (Figures 2A–2F). Blood mononuclear cells—treated with the same antibodies and a 1:1,000 dilution of a mouse monoclonal LAMIN A antibody (Abcam, Cambridge, UK) and conjugated with a secondary Alexa Fluor-594 (red) goat anti-mouse antibody (Molecular Probes)—were counterstained with DAPI and examined by confocal microscopy (Nikon D-Eclipse C1 with EZ-C1 3.91 software) (Figures 2G–2R). Native ROGDI colocalized with the nuclear envelope marker LAMIN A (Figures 2J, 2N, and 2R), suggesting again that ROGDI might belong to the nuclear envelope. The same procedure was undertaken for the labeling of dermal fibroblasts cultured from individual II-1 and the control, except that in this case, the ROGDI and LAMIN A antibodies were conjugated with secondary Alexa Fluor-594 (red) and Alexa Fluor-488 (green) antibodies (Molecular Probes), respectively. As shown in Figures 3A–3C, this experiment revealed that in dermal fibroblasts, ROGDI localizes to the nucleus, and a strong labeling of the nuclear envelope is associated with faint spots within the nucleus. Most importantly, ROGDI was not detected in the fibroblasts from affected individual II-1 (Figures 2D–3F). Consistent with these data, the protein was also not detected by immunoblotting (Figure 3G). These latter results confirm the specificity of the labeling obtained with the ROGDI antibody used in these experiments and are consistent with the loss-of-function mutation (p.Arg157∗) identified in individuals with KTS.
Figure 2.
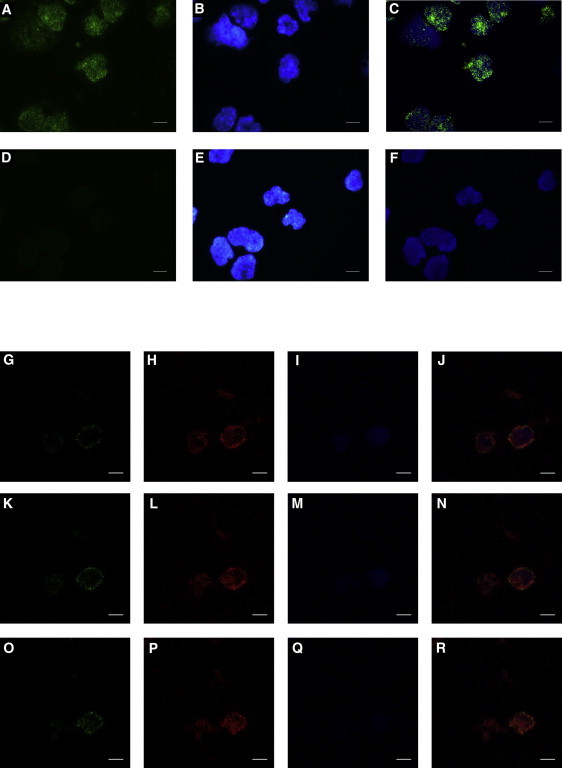
Subcellular Localization of ROGDI in Transfected HEK 293 Cells and Blood Mononuclear Cells
(A–F) The immunostaining of ROGDI-transfected HEK 293 cells was performed with a ROGDI polyclonal antibody incubated in the presence of a permeabilizing reagent (0.2% saponin). ROGDI labeling (A) was revealed with an Alexa Fluor-488 goat anti-rabbit secondary antibody (green). Nuclei (B) were stained with DAPI (blue). The merged picture (C) shows the ROGDI-antibody staining (green) together with nuclei staining (blue). As a control, the same experiment was performed with the secondary antibody in the absence of ROGDI antibody (D, E, and F).
(G–R) The same ROGDI antibody was used for immunolocalization of native ROGDI in blood mononuclear cells (green signal in G, K, and O). Colabeling was performed with a LAMIN A monoclonal antibody (red signal in H, L, and P). Nuclei (I, M, and Q) were stained with DAPI (blue). The partial colocalization of ROGDI with LAMIN A is shown in (J), (N), and (R). Cells were observed by confocal microscopy. Three cell sections from the middle to the top of cells are shown (G–J, K–N, and O–R, respectively). White scale bars represent 10μm.
Figure 3.
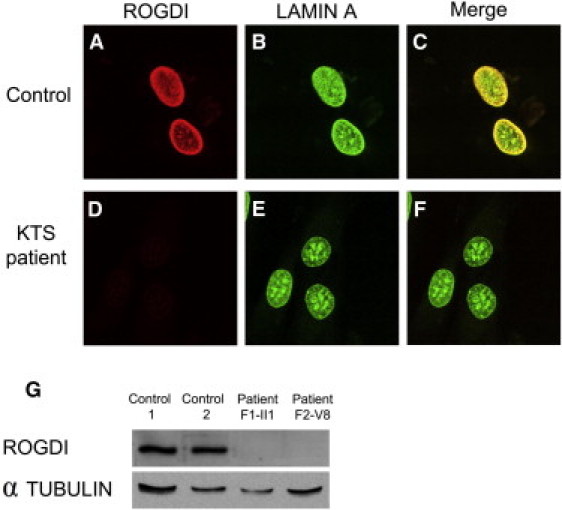
Immunostaining in Dermal Fibroblasts and Immunoblot Analysis from a KTS-Affected Individual and a Control
(A–F) Double immunostaining of LAMIN A and ROGDI in control (A–C) and KTS (D–F) fibroblasts (II-1 in family 1). LAMIN A (green) and ROGDI (red) were labeled with specific antibodies. Merged images are shown in (C) and (F).
(G) Immunoblot shows absence of ROGDI in Epstein-Barr virus (EBV)-transformed lymphoblasts from affected individuals compared to controls. Lanes 1 and 2 show EBV-transformed lymphoblasts of controls. Lanes 3 and 4 display EBV-transformed lymphoblasts from affected individuals (II-1 in family 1; V-8 in family 4). The upper bands indicate a molecular weight of ∼32 kDa. Tubulin was used as a loading control.
ROGDI emerges as new player in neurogenesis. The expression pattern of the gene, showing strong expression in the adult brain and spinal cord, is in line with the disease characteristics relevant to cortical dysfunction and spasticity. However, its detectable expression in many other sites that do not seem to be affected by the disease raises the question of its physiological role in these tissues. ROGDI (FLJ22386) has been reported to interact with a protein called disrupted in schizophrenia 1 (DISC1) (MIM 605210) in yeast two-hybrid screens.11 DISC1 is deemed necessary for neuronal proliferation, the migration of cortical interneurons, and their proper differentiation in the cerebral cortex.12 A plausible interaction between ROGDI and DISC1, if confirmed by a proper experimental set such as coimmunoprecipitation studies in native cells, could offer a clue regarding the role of ROGDI in the pathophysiology of KTS.
In summary, 14 KTS-affected individuals from a consanguineous Druze community share homozygosity for a nonsense mutation in the human homolog of Drosophila rogdi, which encodes a leucine zipper protein of unknown function. The nonsense mutation would truncate a highly conserved C-terminal domain. Mammalian ROGDI is highly expressed in the hippocampus, and the corresponding protein localizes to the nuclear envelope. Age of onset and the severity of the degenerative KTS phenotype vary considerably among individuals who are homozygous for the same disease allele and who are from the same small community.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneCards, http://www.genecards.org/
Mutalyzer, http://www.mutalyzer.nl/2.0
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PRIMER3, http://frodo.wi.mit.edu/primer3/
SNP Database, http://www.ncbi.nlm.nih.gov/snp/
SuperLink, http://bioinfo.cs.technion.ac.il/superlink-online/
Tandem Repeat Finder, http://tandem.bu.edu/trf/trf.html
UCSC, http://genome.ucsc.edu
References
- 1.Kohlschütter A., Chappuis D., Meier C., Tönz O., Vassella F., Herschkowitz N. Familial epilepsy and yellow teeth—a disease of the CNS associated with enamel hypoplasia. Helv. Paediatr. Acta. 1974;29:283–294. [PubMed] [Google Scholar]
- 2.Witkop C.J., Jr., Sauk J.J., Jr. Heritable defects of enamel. In: Stewart R.E., Prescott G.H., editors. Oral Facial Genetics. C.V. Mosby; St. Louis: 1976. pp. 200–202. [Google Scholar]
- 3.Christodoulou J., Hall R.K., Menahem S., Hopkins I.J., Rogers J.G. A syndrome of epilepsy, dementia, and amelogenesis imperfecta: Genetic and clinical features. J. Med. Genet. 1988;25:827–830. doi: 10.1136/jmg.25.12.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zlotogora J., Fuks A., Borochowitz Z., Tal Y. Kohlschütter-Tönz syndrome: epilepsy, dementia, and amelogenesis imperfecta. Am. J. Med. Genet. 1993;46:453–454. doi: 10.1002/ajmg.1320460422. [DOI] [PubMed] [Google Scholar]
- 5.Petermöller M., Kunze J., Gross-Selbeck G. Kohlschütter syndrome: Syndrome of epilepsy—dementia—amelogenesis imperfecta. Neuropediatrics. 1993;24:337–338. doi: 10.1055/s-2008-1071567. [DOI] [PubMed] [Google Scholar]
- 6.Musumeci S.A., Elia M., Ferri R., Romano C., Scuderi C., Del Gracco S. A further family with epilepsy, dementia and yellow teeth: The Kohlschütter syndrome. Brain Dev. 1995;17:133–138. doi: 10.1016/0387-7604(95)00013-2. discussion 142–143. [DOI] [PubMed] [Google Scholar]
- 7.Wygold T., Kurlemann G., Schuierer G. Kohlschütter syndrome—an example of a rare progressive neuroectodermal disease. Case report and review of the literature. Klin. Padiatr. 1996;208:271–275. doi: 10.1055/s-2008-1046481. [DOI] [PubMed] [Google Scholar]
- 8.Donnai D., Tomlin P.I., Winter R.M. Kohlschutter syndrome in siblings. Clin. Dysmorphol. 2005;14:123–126. [PubMed] [Google Scholar]
- 9.Haberlandt E., Svejda C., Felber S., Baumgartner S., Günther B., Utermann G., Kotzot D. Yellow teeth, seizures, and mental retardation: A less severe case of Kohlschütter-Tönz syndrome. Am. J. Med. Genet. A. 2006;140:281–283. doi: 10.1002/ajmg.a.31071. [DOI] [PubMed] [Google Scholar]
- 10.Kapushesky M., Adamusiak T., Burdett T., Culhane A., Farne A., Filippov A., Holloway E., Klebanov A., Kryvych N., Kurbatova N. Gene Expression Atlas update—a value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012;40(Database issue):D1077–D1081. doi: 10.1093/nar/gkr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camargo L.M., Collura V., Rain J.C., Mizuguchi K., Hermjakob H., Kerrien S., Bonnert T.P., Whiting P.J., Brandon N.J. Disrupted in Schizophrenia 1 Interactome: Evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- 12.Kamiya A., Kubo K.I., Tomoda T., Takaki M., Youn R., Ozeki Y., Sawamura N., Park U., Kudo C., Okawa M. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.