

Abstract
Mutational events along the human mtDNA phylogeny are traditionally identified relative to the revised Cambridge Reference Sequence, a contemporary European sequence published in 1981. This historical choice is a continuous source of inconsistencies, misinterpretations, and errors in medical, forensic, and population genetic studies. Here, after having refined the human mtDNA phylogeny to an unprecedented level by adding information from 8,216 modern mitogenomes, we propose switching the reference to a Reconstructed Sapiens Reference Sequence, which was identified by considering all available mitogenomes from Homo neanderthalensis. This “Copernican” reassessment of the human mtDNA tree from its deepest root should resolve previous problems and will have a substantial practical and educational influence on the scientific and public perception of human evolution by clarifying the core principles of common ancestry for extant descendants.
Introduction
Nested hierarchy of species, resulting from the descent with modification process,1 is fundamental to our understanding of the evolution of biological diversity and life in general. In molecular genealogy, the sequential accumulation of mutations since the time of the most recent common ancestor (MRCA) is reflected within the ever-evolving phylogeny of any genetic locus. Accordingly, the reconstructed ancestral sequence of a locus should optimally serve as the reference point for its derived alleles.2 The human mtDNA phylogeny3–7 is an almost perfect molecular prototype for a nonrecombining locus, and knowledge on its variation has been and is extensively used in medical, genealogical, forensic, and population genetic studies.8–11 Boosted by rapid advances in sequencing and genotyping technology, its mode of inheritance, high mutation rate, lack of recombination, and high cellular copy number have proved critical in making this locus the primary choice in the field of archaeogenetics and ancient DNA.12–14 Although its early synthesis was based on restriction-fragment-length polymorphisms,15–18 control-region variation,19,20 or a combination of both,21 the human mtDNA phylogeny is now reconstructed from complete mtDNA sequences,4,6,7,22 thus stretching the phylogenetic resolution to its maximum. mtDNA also became the main target of ancient-DNA studies because it is much more abundant than nuclear DNA.13 The recently published Homo neanderthalensis mitogenomes23,24 represent the best available outgroup source for rooting the human mtDNA phylogeny known to lay inside the contemporary African variation.22,25,26 Despite these major advances, the extinct human mtDNA complete root sequence was never precisely determined, and mtDNA nomenclature remains cumbersome because it refers to the first completely sequenced mtDNA,27,28 labeled rCRS, which is now known to belong to the recently coalescing European haplogroup H2a2a1.7 The use of the rCRS as a reference resulted in a number of practical problems such as (1) the misidentification of derived versus ancestral states of alleles and (2) the count of nonsynonymous mutations that map to the path between the rCRS and the case sequences.29 For instance, clinical and functional studies frequently include among the putative nonsynonymous candidate mutations the haplogroup-HV-defining transition at position 14766 (CYTB) simply because the revised Cambridge Reference Sequence (rCRS) belongs to its derived haplogroup H.30
In this study, to definitively address these issues, we propose a “Copernican” reassessment of the human mtDNA phylogeny by switching to a Reconstructed Sapiens Reference Sequence (RSRS) as the phylogenetically valid reference point. To this end, the previously suggested root7,22,25 was updated to most parsimoniously incorporate the available mitogenomes from H. neanderthalensis.23,24 Moreover, we further refined the human mtDNA phylogeny to an unprecedented level by adding information from 8,216 mitogenomes and evaluated the ranges of nucleotide substitutions from the root RSRS rather than the rCRS28 as a reference point (Figure 1 and Figure S1, available online).
Figure 1.
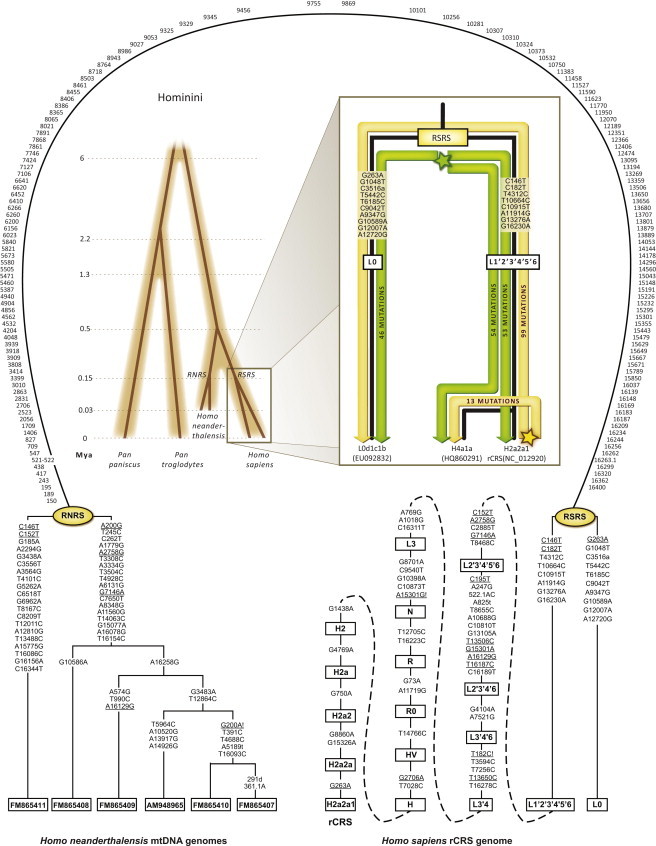
Schematic Representation of the Human mtDNA Phylogeny within Hominini
(Left) Hominini phylogeny illustrating approximate divergence times of the studied species. The positions of the RSRS and the putative Reconstructed Neanderthal Reference Sequence (RNRS) are shown.
(Right) Magnification of the human mtDNA phylogeny. Mutated nucleotide positions separating the nodes of the two basal human haplogroups L0 and L1′2′3′4′5′6 and their derived states as compared to the RSRS are shown. The positions of the rCRS and the RSRS are indicated by golden and a green five-pointed stars, respectively. Accordingly, the number of mutations counted from the rCRS (NC_012920) or the RSRS (Sequence S1) to the L0d1c1b (EU092832) and H4a1a (HQ860291) haplotypes retrieved from a San and a German, respectively, are marked on the golden and green branches. The principle of equidistant star-like radiation from the common ancestor of all contemporary haplotypes is highlighted when the RSRS is preferred over the rCRS as the reference sequence.
Subjects and Methods
Updating the Human mtDNA Phylogeny and Inference of the Ancestral Root Haplotype
MtDNA Genomes Comprising the Phylogeny
A total of 18,843 complete mtDNA sequences were used to refine the human mtDNA phylogeny of which 10,627 were previously reported and used for the mtDNA tree Build 13 (28 Dec 2011) as posted by PhyloTree.7 The remaining 8,216 sequences are mainly from the large complete mtDNA database available at FamilyTreeDNA and in part from data sets maintained by the authors. The large database available at FamilyTreeDNA was privately obtained by the sample donors, usually for genealogical purposes. Most donors were of western Eurasian ancestry, but donors with matrilineal ancestry from other geographical regions have also contributed. Once the mtDNA sequences were obtained, donors had several options: keep them confidential, share them with peer genealogists, submit them to the National Center for Biotechnology Information (NCBI) GenBank, and/or consent to contribute them anonymously to a research database maintained by FamilyTreeDNA to improve the mtDNA phylogeny. In turn, this contribution rewards and enriches the genealogical experience as well as benefits the scientific community. All the procedures followed in this study were in accordance with the ethical standards of the responsible committee on human experimentation of the participating research centers.
Likewise, it is important to clarify that because the complete sequences were obtained privately, some donors have independently uploaded their sequence to NCBI. Currently (as of February 28, 2012), a total of 1,220 complete mtDNA sequences that were generated at FamilyTreeDNA were privately deposited in NCBI GenBank. Most of these sequences were already considered in the previous PhyloTree Builds.7 Because we have no way to know which of the sequences were autonomously uploaded to NCBI, all duplicate sequences that matched precisely between NCBI and our database were excluded from our analysis. Therefore, even if multiple samples were excluded, no topological information was lost. Accordingly, out of the 8,216 sequences used to verify the phylogeny, a total of 4,265 sequences are released and deposited in NCBI GenBank under accession numbers JQ701803–JQ706067. The complete mtDNA sequences of the Neanderthals were retrieved from the literature.23,24
Complete mtDNA Sequencing
DNA was extracted from buccal swabs. MtDNA was amplified with 18 primers to yield nine overlapping fragments as previously reported.22 PCR products were cleaned with magnetic-particle technology (BioSprint 96; QIAGEN). After purification, the nine fragments were sequenced by means of 92 internal primers to obtain the complete mtDNA genome. Sequencing was performed on a 3730xl DNA Analyzer (Applied Biosystems), and the resulting sequences were analyzed with the Sequencher software (Gene Codes Corporation). Mutations were scored relative to the rCRS and the suggested RSRS. Sample quality control was assured as follows:
-
(1)
After the PCR amplification of the nine fragments, DNA handling and distribution to the 96 sequencing reactions was aided by the Beckman Coulter Biomek FX liquid handler to minimize the chance for human pipetting errors.
-
(2)
All 96 sequencing reactions of each sample were performed simultaneously in the same sequencing run. Most observed mutations were determined by at least two sequence reads. However, in a minority of the cases only one sequence read was available because of various technical reasons, usually related to the amount and quality of the DNA available.
-
(3)
Any fragment that failed the first sequencing attempt or any ambiguous base call was tested by additional and independent PCR and sequencing reactions. In these cases, the first hypervariable segment (HVS-I) of the control region was resequenced too to assure that the correct sample was retrieved.
-
(4)
Genotyping history for each sample was recorded to help in the search for DNA handling errors and artificial recombination events.
-
(5)
All sequences were aligned with the software Sequencher (Gene Codes Corporation), and all positions with a Phred score less than 30 were manually evaluated by an operator. Two independent operators read each sequence. All positions that differed from the reference sequences were recorded electronically to minimize typographic errors.
-
(6)
Any sequence that did not comfortably fit within the established human mtDNA phylogeny was highlighted and resequenced to exclude potential lab errors.
-
(7)
Any comments and remarks raised by external investigators after release of the data will be addressed by reassessing the original sequences for accuracy. After that, any unresolved result will be further examined by resequencing and, if necessary, immediately corrected.
Tree Reconstruction and Notation of Mutations
The phylogeny was reconstructed by evaluating both all previously available published and the herein released complete mtDNA sequences aiming at the most parsimonious solution and aided by the software mtPhyl. Polymorphic positions are shown on the branches and reticulations were resolved by considering the degree of mutability of individual positions as counted by their number of occurrences in the overall phylogeny. Both the ancestral and derived base status for each mutation appearing in the phylogeny according to the International Union Of Pure And Applied Chemistry (IUPAC) nucleotide code are reported. We use capital letters for transitions (e.g., G73A) and lowercase letters for transversions (e.g., A73t). Although heteroplasmies are not noted in the phylogeny, we recommend labeling them by using IUPAC code and capital letters (e.g., G73R). Throughout the phylogeny indels are given with respect to the RSRS and maintain the traditional nucleotide position numbering as in the rCRS. Sequencing alignment prefers 3′ placement for indels, except in cases where the phylogeny suggests otherwise.31 Deletions are indicated by a “d” after the deleted nucleotide position (e.g., T15944d). Insertions are indicated by a dot followed by the position number and type of inserted nucleotide(s) (e.g., 5899.1C for a C insertion at the first inserted nucleotide position after position 5899 and 5899.2C for a subsequent C insertion, and these are abbreviated as 5899.1CC when occurring on the same branch). We label polynucleotide stretches of unknown length as follows: 573.XC. In cases where an insertion occurred at an ancestral branch but a reversion of this insertion (= deletion) took place at a descendant branch, we noted the latter as follows: 5899.1Cd. An exclamation mark (!) at the end of a labeled position denotes a reversion to the ancestral state. The number of exclamation marks stands for the number of sequential reversions in the given position from the RSRS (e.g., C152T, T152C!, and C152T!!). Some indel positions have been a source of confusion because multiple alignment solutions enable alternative scoring. Notably, the dinucleotide repeat in hypervariable segment II (HVS-II) of the control region can be viewed either as a CA repeat starting at position 514 or as an AC repeat starting at position 515, leading to two different notations being in use for a repeat loss: 522–523d versus 523–524d. We adhered to the guidelines for consistent treatment of mtDNA-length variants that were established by the forensic genetic community31 and favor the AC interpretation. As the RSRS has one AC unit less compared to the rCRS, we filled positions 523 and 524 of the RSRS with “NN,” thereby preserving the historical genome annotation numbering. Consequently, an AC insertion compared to the RSRS is scored as 522.1AC, whereas an AC deletion is scored as 521–522d. Table S2 presents all common indel positions throughout the complete mtDNA sequence and the way we labeled them. Transitions at the hypervariable position 16519, insertions of one or two Cs at positions 309, 315, and 16193, A to C transversions at 16182 and 16183, as well as length variation of the AC dinucleotide repeat spanning 515–522, were excluded from the phylogeny.
Haplogroup labels were re-evaluated and the following suggestions were made:
-
(1)
Monophyletic clades that are composed of two or more previously named haplogroups are labeled by concatenating their names and separating them by apostrophe (e.g., L0a'b). This is not applied in the case of capital-letter-only labeled haplogroups (e.g., JT);
-
(2)
We suggest labeling an extant sample that matches a haplogroup root with the superscript case letter n for “nodal” (e.g., Hn);
-
(3)
We note that when complete mtDNA sequences are considered, the inability to differentiate a nodal haplotype from an unresolved paraphyletic clade is eliminated. Accordingly, the haplogroup label of each observed complete mtDNA sequences can: (1) mark it in a nodal position; (2) affiliate it with a previously labeled haplogroup; (3) suggest a, so far, unlabeled haplogroup; or (4) in the absence of two additional samples to justify the labeling of a, so far, unidentified haplogroup, affiliate it with the ancestral haplogroup. So, the label of a given sample as “H” means that it is an unlabeled descendent of haplogroup H that cannot be affiliated to any known H haplogroup clade at the time of report and based on complete mtDNA sequence. We suggest restricting the use of label “H∗” to cases where the haplogroup labeling is based on partial mtDNA sequence;
-
(4)
To aid the nonexpert in understanding the mtDNA haplogroup nomenclature system, we summarize in Table S3 the cases where haplogroup labels do not logically follow from the hierarchy and hence could lead to confusion. Changing these haplogroup labels to make them more logical is undesirable at this stage because they are already used extensively in the literature and therefore changing them would probably cause even more confusion. In addition, we note that for the most basal nodes of the phylogeny, historically the following shorthand names have been in use: L1′5 = L1′2′3′4′5′6; L2′5 = L2′3′4′5′6; L2′6 = L2′3′4′6; and L4′6 = L3′4′6, which we will herein refer to by their full name. One shorthand haplogroup name, M4”67, is maintained because writing it in full (M4′18′30′37′38′43′45′63′64′65′66′67) seems impractical.
It is important to note that the aim of this study is to publish the most up-to-date human mtDNA phylogeny, and it cannot be regarded by any means as a population-level survey exploring the frequencies and distributions of the various haplogroups. Therefore, although all sequences were used to establish the tree topology, the subset of sequences actually presented in the phylogeny is lower because for each branch up to two representative example sequences are provided. In most cases, we labeled haplogroups only when supported by at least three distinct haplotypes to maximize the accuracy of the haplogroup defining array of mutations and to avoid the establishment of haplogroups resulting from sequencing errors. Exceptions included previously established haplogroups or haplogroups supported by a particularly long array of mutations. Accordingly, the tips of the herein released phylogeny are in fact internal haplogroup nodes, thus private mutations (if any) of individual haplotypes were not included.
Evaluation of the mtDNA Clock and Age Estimates
Substitution Counts and Molecular Clock
To calculate the substitution counts from the RSRS to every extant mitogenome (which is a tip in the mtDNA phylogeny), we summed up the number of mutations on the path leading to each noted haplogroup in the phylogeny and added to this the number of positions that differed between the tip and the root of the haplogroup. Thus, we are guaranteed to correctly count all parallel and back mutations, except for the case where two mutations affecting the same position occurred on a branch in the tree (in which case we either count zero instead of two, if the second is a back mutation, or one instead of two, if the second mutation is not back to the initial state). As has been argued in the past, such repeated mutations within a single branch in the highly resolved human mtDNA tree are highly unlikely,32 and are even more so if the fastest mutating sites (16519 and the A to C transversions and poly-C insertions around the HVS-I position 16189) are eliminated, as was done in our analysis.
To test the validity of molecular clock assumption on human mtDNA substitutions, we used PAML 4.4 with the HKY85 substitution model to generate maximum likelihood estimates of branch lengths with and without the molecular clock assumption. We chose to sample around 200–300 sequences and analyze their coalescent tree (a subtree of the complete tree) in each PAML run, to accommodate PAML's computational limitations, and also to sample mostly deep branches (such as M44), rather than the recent and very short branches (such as D4a1b1) of the oversampled haplogroups such as H and D. Thus, we preferentially sampled haplogroups whose coalescence with other samples in the tree was more ancient. This ensured that even in such a sample, the deeper clades such as the basal M clades would be represented with high probability, whereas more recently coalescing haplogroups such as the ones of haplogroup D would be rarely sampled.
The generalized likelihood ratio (GLR) test for validity of the clock assumption then uses the test statistic 2 × (log-likelihood of non-clock model − log-likelihood of clock model), which, under the null hypothesis of molecular clock, has a χ2 distribution with degrees of freedom equal to the number of parameters under no clock (= number of branches in the tree) minus number of parameters under clock (= number of internal nodes in the tree).
We performed the analyses on two sets of the mtDNA sequences: once by using the coding region alone and once on the entire molecule. This was done as another sanity check for the validity and generality of our results. All obtained p values are presented in Table S4.
Age Calculations Assuming a Molecular Clock
In spite of the discovered clock violations, we were still interested in applying the best available tools for estimating the ages of ancestral nodes in the tree assuming a molecular clock. We adopted the calculation approach and mutation rate estimate of,32 who suggest to estimate ages in substitutions and then transform them to years in a nonlinear manner accounting for the selection effect on nonsynonymous mutations. We used PAML 4.433 with the HKY85 substitution model to generate maximum likelihood estimates of internal node ages under a molecular clock assumption. Because PAML is computationally limited in the size of trees it can analyze, we performed estimation for the whole tree in several separate runs. We divided the tree into seven collections of haplogroups:
-
•
All L haplogroups (i.e., the entire phylogeny excluding M and N)
-
•
All of M excluding D
-
•
D and JT
-
•
H excluding H1 and H5
-
•
B4'5 and HV excluding H but including H1 and H5
-
•
U
-
•
N excluding HV, U, JT and B4'5
For each PAML run, we selected all sequences belonging to one of these sets, and added a small random sample of other samples from the rest of the phylogeny to maintain “calibration.” Putting together the estimates from all seven runs provided us with age estimates for all nodes in our tree. Estimates are given in Table S5.
Data Transition
We are aware that the suggested change can raise difficulties and even antagonism from the scientific community. On the other hand, a scenario in which a reference sequence of a genetic locus does not represent its ancestral sequence should, indisputably, be corrected. The realization of the superiority of complete mtDNA sequence analysis compared to other approaches, combined with the emergence of deep sequencing technologies, will possibly shift the entire field into the use of only complete mtDNA sequences in the near future.34–36 Therefore, the sooner the change is made the less “painful” it will be. As the common practice for reporting complete mtDNA sequences is by posting the sequences as FASTA files to NCBI, rather than reporting the substitutions with respect to a reference sequence (as in the case of many data sets restricted to control-region variation), no major change is needed. When a FASTA file is available or created, the only change needed is to switch the reference sequence to the RSRS. For control-region-based data sets, the conversion might be more problematic as the common practice to report the sequences in literature did not involve FASTA files but recorded mutations as compared to the rCRS. Table S6 compares the classic diagnostic mutations for the major haplogroups relative to the rCRS or the RSRS.
To facilitate data transition we release the tools “FASTmtDNA,” which allows transformation of Excel list-type reports of mtDNA haplotypes into FASTA files, and “mtDNAble,” which labels haplogroups, performs a phylogeny-based quality check and identifies private substitutions. These noted features are fully supported in a web interface or as standalone versions, which can be freely downloaded from the website including their manual and example files. In addition, the web interface allows the benefit of comparing private substitutions between submitted and previously stored mitogenomes to suggest the labeling of additional haplogroups. Following quality check and consent, the web interface enables the storing of complete mtDNA sequences by members of the mtDNA community to enrich a growing database. This in turn is expected to strengthen the data set used by the website to label haplogroups, perform quality control and refine the phylogeny. Additional tools will be periodically added and updated.
Results
The RSRS
Since the sub-Saharan haplogroup L0 was defined,37 it became clear that the root of the extant variation of human mitochondrial genomes is allocated between haplogroups L0 and L1′2′3′4′5′6, which are separated from each other by 14 coding and four control-region mutations22 (Figure 1). Until now, our understanding of the root of the human mtDNA tree was incomplete because of the absence of reliable closely related outgroup mitogenomes, and the exact placement of the 18 mutations separating the L0 and L1′2′3′4′5′6 nodes remained vague. In principle, ancient mtDNA from early human fossils might be informative but unreachable because of considerable technical problems inherent to the analysis process.13 However, as the split between H. sapiens and H. neanderthalensis certainly predates the appearance of the RSRS,38 a resolution of the deepest node might be achieved by rooting the human phylogeny with H. neanderthalensis complete mtDNA sequences23,24 (Figure 1). Table S1 shows all substitutions separating haplogroup L0 from L1′2′3′4′5′6, their status in the six H. neanderthalensis mitogenomes and their most parsimonious allocation around the human root. Accordingly, the ancestral mtDNA sequence of extant humans should correspond to the bifurcation of L0 and L1′2′3′4′5′6. Although it cannot be excluded that further sampling of the African mtDNA variation might reveal yet another more basal clade of the human mtDNA tree, it is at least equally valid to indicate that, in spite of the many thousands of reported complete mtDNA sequences,7 such a clade has not been found so far. Operating under this assumption we established the reference point, RSRS, which is made available as Sequence S1.
We present the most resolved human mtDNA phylogeny by compiling the information from 18,843 mitochondrial genomes of which 10,627 were previously summarized in PhyloTree Build 13 (28 Dec 2011).7 We followed the established cladistic notation for haplogroup labeling adjusted for complete mtDNA genomes.7,39 Yet, in contrast with the previously reported phylogeny, all mutational changes noted on the branches of the tree indicate the actual descendant nucleotide state relative to the state in the RSRS. Although this has no effect on the tree topology per se, it is critical to emphasize its major consequences in the way of reporting the list of mutations denoting an mtDNA haplotype. Accordingly, although the HVS-I haplotype of a nodal haplogroup H2a2a1 mitogenome will show no differences when compared to the rCRS, its differentiation relative to the RSRS is now documented by the transitions A16129G, T16187C, C16189T, T16223C, G16230A, T16278C and C16311T. This common practice of expressing haplotypes as a string of differences from the rCRS (Figure 1) led, for instance, many inexperienced readers to incorrectly hold the “fact” that African haplogroup L mitogenomes have more substitutions separating them from the rCRS as compared to western Eurasian haplogroup H mitogenomes as a “proof” of an African origin for all contemporary humans.
Indications for Violation of the Molecular Clock
The accepted notion of a molecular clock means that contemporary mtDNA haplotypes should show statistically insignificant differences in the number of accumulated mutations from the RSRS.40 Triggered by the suggested change in the reference sequence that facilitates substitution counts from the ancestral root, we further evaluated this hypothesis. The range of substitution counts separating contemporary mitogenomes belonging to major haplogroups from the RSRS is shown in Figure S2. The mean distance is 57.1 substitutions, the median is 56 and the empirical standard deviation is 5.9. Widely different distances ranging from 41 substitutions in some L0d1a1 mitogenomes to 77 in some L2b1a mitogenomes are observed. Interestingly, the ranges of substitution counts within haplogroups M and N, which are hallmarks of the relatively recent out-of-Africa exodus of humans, are also very large. For example, within M there are two mitogenomes with 43 substitutions (in M30a and M44) and two mitogenomes with as many as 71 substitutions (in M2b1b and M7b3a). This is especially striking because the path from the RSRS to the root of M already contains 39 substitutions. Hence, the difference between the M root and its M44 descendant is only four substitutions (two in the coding region and two in the control region) as compared to 32 substitutions in the M2b1b and M7b3a mitogenomes. These observations raise the possibility that the tree in general, and haplogroup M in particular, might not adhere uniformly to the assumed molecular clock, under which substitutions occur at a fixed rate on all branches of the tree over time. We evaluated this scenario by performing generalized likelihood ratio tests of the molecular clock by using PAML33 on subsets of samples from the entire tree, on haplogroup L2 (following past evidence of clock violations in this haplogroup40) and on the sister haplogroups M and N. Our results demonstrate violations of the molecular clock in M (0.00015 ≤ p value ≤ 0.0003 for χ2 GLR test in three different analyses) and give mixed results for the entire tree (p = 0.005 and p = 0.018 for two analyses, which might be sensitive to the parts of the tree randomly sampled) and L2 (GLR χ2 p value = 5 × 10−5 and p value = 0.033 for two analyses) and borderline results in N (GLR χ2 p value = 0.049 and p value = 0.054 in two analyses). We are currently unable to offer well-founded explanations for these findings, which remain the scope of future studies.
As the clock violation was observed only in a restricted number of specified cases, we applied the best available tools for estimating the ages of ancestral nodes. We adopted a conventional calculation approach and mutation rate32 and used PAML 4.4 to generate maximum likelihood estimates for internal node ages under a molecular clock assumption.33 Figure 2 displays the phylogeny and density of extant haplogroups as a function of both the number of substitutions occurring since the RSRS and the estimated coalescence times.
Figure 2.
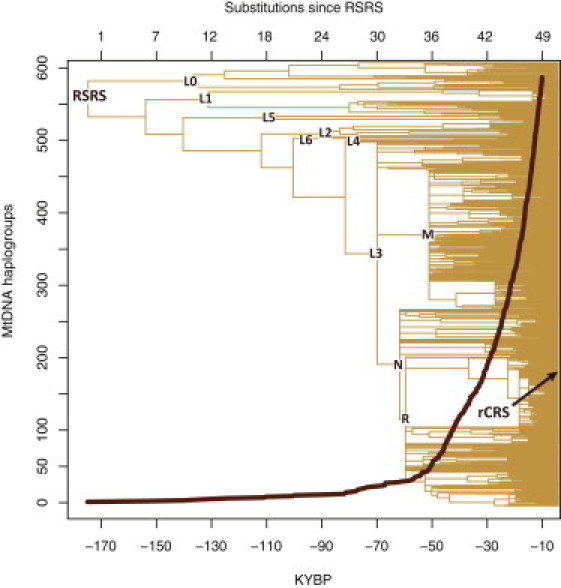
Human mtDNA Phylogeny
A schematic representation of the most parsimonious human mtDNA phylogeny inferred from 18,843 complete mtDNA sequences with the structure shown explicitly for bifurcations that occurred 40,000 years before present (YBP) or earlier, and a graph showing the explosion of haplogroups since then. The y axis indicates the approximate number of haplogroups from each time layer that have survived to nowadays. The upper and lower x axes of the rooted tree are scaled according to the number of accumulated mutations since the RSRS and the corresponding coalescence ages, respectively.
Approaching a Perfect Phylogeny
The mitochondrial genomes released herein almost double the number of sequences that were previously available. Despite the fact that the sequences released in this study are not equally representative of all human populations but are mainly from donors of western Eurasian matrilineal ancestry, a few additional advantages arise from this combined data. First, an almost final level of resolution for a number of western Eurasian clades was achieved, and the nodes of ancestral and derived haplogroups are often differentiated by a single mutation. For example, Figure 3 compares the resolution of haplogroup H4 as first41 and as currently resolved. This comprehensive level of resolution minimizes the chance of additional nomenclature issues arising in future studies. Second, the highly resolved phylogeny is a powerful tool for quality assessment.29,42–44 Mapping any additional complete mtDNA haplotype to such highly resolved phylogeny will highlight potential sequencing errors and problems such as sample mix-up, contamination, and typographical errors. Third, the phylogeny itself is a useful resource for future evolutionary, clinical, and forensic studies.45–51
Figure 3.
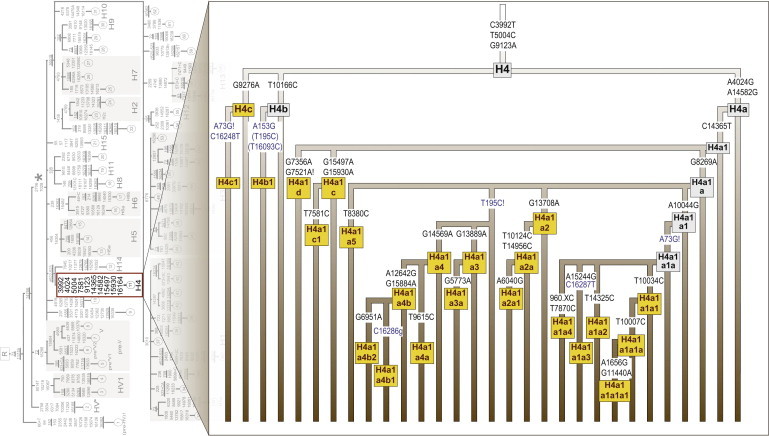
Haplogroup H4 internal cladistic structure
(Left) Haplogroup H4 as first reported.41 Mutations in bold were considered diagnostic for the haplogroup.
(Right) Haplogroup H4 as currently resolved with a total of 236 H4 mitogenomes. An almost perfect resolution of the nested hierarchy is achieved. Additional haplogroups suggested herein are shown in yellow. Control-region mutations are noted in blue.
Discussion
Thirty-one years ago, Anderson and colleagues27 published the first complete sequence of human mtDNA. This became the reference sequence in multidisciplinary studies that revolutionized human genetics, leading, for instance, to the concept of “late-out-of-Africa” (“African Eve”) peopling of the world by modern humans,17,18 the identification of a wide range of pathological mtDNA mutations,52,53 and the possibility of reconstructing the origins and the relationships of modern as well as ancient populations.12,14,54 The publication of globally selected complete mtDNA genomes about 10 years ago marked the beginning of the genomic era in this field.4 Since then, progress has been impressive. Most admirable is the penetration of the principles applied in the field of archaeogenetics to hundreds of thousands of people around the world who became interested in their matrilineal descent. In fact, in this paper we add information from more than 8,000 complete mtDNA sequences resulting largely from the curiosity and enthusiasm of lay people to the ∼10,000 publicly available complete mtDNA sequences. However, as discussed above, the entire field faces a problem: the traditional manner of reporting variation observed in human mitochondrial genome sequences is, to be blunt, conceptually incorrect.
Supported by a consensus of many colleagues and after a few years of hesitation, we have reached the conclusion that on the verge of the deep-sequencing revolution,47,55 when perhaps tens of thousands of additional complete mtDNA sequences are expected to be generated over the next few years, the principal change we suggest cannot be postponed any longer: an ancestral rather than a “phylogenetically peripheral” and modern mitogenome from Europe should serve as the epicenter of the human mtDNA reference system. Inevitably, the proposed change could raise some temporary inconveniences. For this reason, we provide tables and software to aid data transition.
What we propose is much more than a mere clerical change. We use the Ptolemaian geocentric versus Copernican heliocentric systems as a metaphor. And the metaphor extends further: as the acceptance of the heliocentric system circumvented epicycles in the orbits of planets, switching the mtDNA reference to an ancestral RSRS will end an academically inadmissible conjuncture where virtually all mitochondrial genome sequences are scored in part from derived-to-ancestral states and in part from ancestral-to-derived states. We aim to trigger the radical but necessary change in the way mtDNA mutations are reported relative to their ancestral versus derived status, thus establishing an intellectual cohesiveness with the current consensus of shared common ancestry of all contemporary human mitochondrial genomes.
Note that the problem is not restricted to mtDNA. Indeed, in the much larger perspective of complete nuclear genomes in which comparisons are often currently made relative to modern human reference sequences, often of European origin, it seems worthwhile to begin considering, as valuable alternatives, public reference sequences of ancestral alleles (common in all primates) whereby derived alleles (common to some human populations) would be distinguished.
Acknowledgments
We thank the genealogical community for donating their privately obtained complete mtDNA sequences for scientific studies and FamilyTreeDNA for compiling the data. We thank FamilyTreeDNA for supporting the establishment of the herein released website. We thank Eileen Krauss-Murphy of FamilyTreeDNA for help with assembly of the database. We thank Rebekah Canada and William R. Hurst for help with the assembly of haplogroup H and K samples, respectively. R.V. and D.M.B. thank the European Commission, Directorate-General for Research for FP7 Ecogene grant 205419. D.M.B. is a shareholder of FamilyTreeDNA and a member of its scientific advisory board. R.V. and M.M. thank the European Union, Regional Development Fund for a Centre of Excellence in Genomics grant, and R.V. thanks the Swedish Collegium for Advanced Studies for support during the initial stage of this study. M.M. thanks Estonian Science Foundation for grant 8973. A.T. received support from Fondazione Alma Mater Ticinensis and the Italian Ministry of Education, University and Research: Progetti Ricerca Interesse Nazionale 2009. S.R. thanks the Israeli Science Foundation for grant 1227/09 and IBM for an Open Collaborative Research grant. FCT, the Portuguese Foundation for Science and Technology, partially supported this work through the personal grant N.M.S. (SFRH/BD/69119/2010). Instituto de Patologia e Imunologia Molecular da Universidade do Porto is an Associate Laboratory of the Portuguese Ministry of Science, Technology and Higher Education and is partially supported by the Portuguese Foundation for Science and Technology.
Contributor Information
Doron M. Behar, Email: behardm@usernet.com.
Mannis van Oven, Email: m.vanoven@erasmusmc.nl.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FASTmtDNA, http://www.mtdnacommunity.org
mtDNAble, http://www.mtdnacommunity.org
mtPhyl, http://eltsov.org/mtphyl.aspx
PhyloTree, http://www.phylotree.org
Accession Numbers
The 4,265 complete mtDNA sequences reported herein have been submitted to GenBank (accession numbers JQ701803–JQ706067).
References
- 1.Darwin C. John Murray; London: 1859. Natural Selection. On the Origin of Species by Means of Natural Selection, or, The Preservation of Favoured Races in the Struggle for Life, Chapter 4. [Google Scholar]
- 2.Delsuc F., Brinkmann H., Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005;6:361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- 3.Kivisild T., Metspalu E., Bandelt H.J., Richards M., Villems R. The world mtDNA phylogeny. In: Bandelt H.J., Macaulay V., Richards M., editors. Human mitochondrial DNA and the evolution of Homo sapiens. Springer-Verlag; Berlin: 2006. pp. 149–179. [Google Scholar]
- 4.Ingman M., Kaessmann H., Pääbo S., Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408:708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 5.Richards M., Macaulay V. The mitochondrial gene tree comes of age. Am. J. Hum. Genet. 2001;68:1315–1320. doi: 10.1086/320615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torroni A., Achilli A., Macaulay V., Richards M., Bandelt H.J. Harvesting the fruit of the human mtDNA tree. Trends Genet. 2006;22:339–345. doi: 10.1016/j.tig.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 7.van Oven M., Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 8.Underhill P.A., Kivisild T. Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu. Rev. Genet. 2007;41:539–564. doi: 10.1146/annurev.genet.41.110306.130407. [DOI] [PubMed] [Google Scholar]
- 9.Salas A., Bandelt H.J., Macaulay V., Richards M.B. Phylogeographic investigations: The role of trees in forensic genetics. Forensic Sci. Int. 2007;168:1–13. doi: 10.1016/j.forsciint.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 10.Shriver M.D., Kittles R.A. Genetic ancestry and the search for personalized genetic histories. Nat. Rev. Genet. 2004;5:611–618. doi: 10.1038/nrg1405. [DOI] [PubMed] [Google Scholar]
- 11.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert M.T., Kivisild T., Grønnow B., Andersen P.K., Metspalu E., Reidla M., Tamm E., Axelsson E., Götherström A., Campos P.F. Paleo-Eskimo mtDNA genome reveals matrilineal discontinuity in Greenland. Science. 2008;320:1787–1789. doi: 10.1126/science.1159750. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert M.T., Hansen A.J., Willerslev E., Rudbeck L., Barnes I., Lynnerup N., Cooper A. Characterization of genetic miscoding lesions caused by postmortem damage. Am. J. Hum. Genet. 2003;72:48–61. doi: 10.1086/345379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haak W., Forster P., Bramanti B., Matsumura S., Brandt G., Tänzer M., Villems R., Renfrew C., Gronenborn D., Alt K.W., Burger J. Ancient DNA from the first European farmers in 7500-year-old Neolithic sites. Science. 2005;310:1016–1018. doi: 10.1126/science.1118725. [DOI] [PubMed] [Google Scholar]
- 15.Denaro M., Blanc H., Johnson M.J., Chen K.H., Wilmsen E., Cavalli-Sforza L.L., Wallace D.C. Ethnic variation in Hpa 1 endonuclease cleavage patterns of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 1981;78:5768–5772. doi: 10.1073/pnas.78.9.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown W.M. Polymorphism in mitochondrial DNA of humans as revealed by restriction endonuclease analysis. Proc. Natl. Acad. Sci. USA. 1980;77:3605–3609. doi: 10.1073/pnas.77.6.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cann R.L., Stoneking M., Wilson A.C. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 18.Vigilant L., Stoneking M., Harpending H., Hawkes K., Wilson A.C. African populations and the evolution of human mitochondrial DNA. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 19.Richards M., Côrte-Real H., Forster P., Macaulay V., Wilkinson-Herbots H., Demaine A., Papiha S., Hedges R., Bandelt H.J., Sykes B. Paleolithic and neolithic lineages in the European mitochondrial gene pool. Am. J. Hum. Genet. 1996;59:185–203. [PMC free article] [PubMed] [Google Scholar]
- 20.Torroni A., Bandelt H.J., D'Urbano L., Lahermo P., Moral P., Sellitto D., Rengo C., Forster P., Savontaus M.L., Bonné-Tamir B., Scozzari R. mtDNA analysis reveals a major late Paleolithic population expansion from southwestern to northeastern Europe. Am. J. Hum. Genet. 1998;62:1137–1152. doi: 10.1086/301822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torroni A., Schurr T.G., Cabell M.F., Brown M.D., Neel J.V., Larsen M., Smith D.G., Vullo C.M., Wallace D.C. Asian affinities and continental radiation of the four founding Native American mtDNAs. Am. J. Hum. Genet. 1993;53:563–590. [PMC free article] [PubMed] [Google Scholar]
- 22.Behar D.M., Villems R., Soodyall H., Blue-Smith J., Pereira L., Metspalu E., Scozzari R., Makkan H., Tzur S., Comas D., Genographic Consortium The dawn of human matrilineal diversity. Am. J. Hum. Genet. 2008;82:1130–1140. doi: 10.1016/j.ajhg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briggs A.W., Good J.M., Green R.E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajkovic D., Kucan Z. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 24.Green R.E., Malaspinas A.S., Krause J., Briggs A.W., Johnson P.L., Uhler C., Meyer M., Good J.M., Maricic T., Stenzel U. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kivisild T., Shen P., Wall D.P., Do B., Sung R., Davis K., Passarino G., Underhill P.A., Scharfe C., Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kivisild T., Reidla M., Metspalu E., Rosa A., Brehm A., Pennarun E., Parik J., Geberhiwot T., Usanga E., Villems R. Ethiopian mitochondrial DNA heritage: Tracking gene flow across and around the gate of tears. Am. J. Hum. Genet. 2004;75:752–770. doi: 10.1086/425161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 28.Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 29.Yao Y.G., Salas A., Bravi C.M., Bandelt H.J. A reappraisal of complete mtDNA variation in East Asian families with hearing impairment. Hum. Genet. 2006;119:505–515. doi: 10.1007/s00439-006-0154-9. [DOI] [PubMed] [Google Scholar]
- 30.Pello R., Martín M.A., Carelli V., Nijtmans L.G., Achilli A., Pala M., Torroni A., Gómez-Durán A., Ruiz-Pesini E., Martinuzzi A. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum. Mol. Genet. 2008;17:4001–4011. doi: 10.1093/hmg/ddn303. [DOI] [PubMed] [Google Scholar]
- 31.Bandelt H.J., Parson W. Consistent treatment of length variants in the human mtDNA control region: A reappraisal. Int. J. Legal Med. 2008;122:11–21. doi: 10.1007/s00414-006-0151-5. [DOI] [PubMed] [Google Scholar]
- 32.Soares P., Ermini L., Thomson N., Mormina M., Rito T., Röhl A., Salas A., Oppenheimer S., Macaulay V., Richards M.B. Correcting for purifying selection: An improved human mitochondrial molecular clock. Am. J. Hum. Genet. 2009;84:740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 34.Tang S., Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;48:287–296. doi: 10.2144/000113389. [DOI] [PubMed] [Google Scholar]
- 35.Li M., Schönberg A., Schaefer M., Schroeder R., Nasidze I., Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010;87:237–249. doi: 10.1016/j.ajhg.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaragoza M.V., Fass J., Diegoli M., Lin D., Arbustini E. Mitochondrial DNA variant discovery and evaluation in human Cardiomyopathies through next-generation sequencing. PLoS ONE. 2010;5:e12295. doi: 10.1371/journal.pone.0012295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishmar D., Ruiz-Pesini E., Golik P., Macaulay V., Clark A.G., Hosseini S., Brandon M., Easley K., Chen E., Brown M.D. Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. USA. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards M.B., Macaulay V.A., Bandelt H.J., Sykes B.C. Phylogeography of mitochondrial DNA in western Europe. Ann. Hum. Genet. 1998;62:241–260. doi: 10.1046/j.1469-1809.1998.6230241.x. [DOI] [PubMed] [Google Scholar]
- 40.Torroni A., Rengo C., Guida V., Cruciani F., Sellitto D., Coppa A., Calderon F.L., Simionati B., Valle G., Richards M. Do the four clades of the mtDNA haplogroup L2 evolve at different rates? Am. J. Hum. Genet. 2001;69:1348–1356. doi: 10.1086/324511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Achilli A., Rengo C., Magri C., Battaglia V., Olivieri A., Scozzari R., Cruciani F., Zeviani M., Briem E., Carelli V. The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am. J. Hum. Genet. 2004;75:910–918. doi: 10.1086/425590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parson W., Bandelt H.J. Extended guidelines for mtDNA typing of population data in forensic science. Forensic Sci. Int. Genet. 2007;1:13–19. doi: 10.1016/j.fsigen.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Salas A., Carracedo A., Macaulay V., Richards M., Bandelt H.J. A practical guide to mitochondrial DNA error prevention in clinical, forensic, and population genetics. Biochem. Biophys. Res. Commun. 2005;335:891–899. doi: 10.1016/j.bbrc.2005.07.161. [DOI] [PubMed] [Google Scholar]
- 44.Bandelt H.J., Lahermo P., Richards M., Macaulay V. Detecting errors in mtDNA data by phylogenetic analysis. Int. J. Legal Med. 2001;115:64–69. doi: 10.1007/s004140100228. [DOI] [PubMed] [Google Scholar]
- 45.Ballantyne K.N., van Oven M., Ralf A., Stoneking M., Mitchell R.J., van Oorschot R.A., Kayser M. MtDNA SNP multiplexes for efficient inference of matrilineal genetic ancestry within Oceania. Forensic Sci. Int. Genet. 2011 doi: 10.1016/j.fsigen.2011.08.010. in press. Published online September 20, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Pereira L., Soares P., Radivojac P., Li B., Samuels D.C. Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am. J. Hum. Genet. 2011;88:433–439. doi: 10.1016/j.ajhg.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Behar D.M., Harmant C., Manry J., van Oven M., Haak W., Martinez-Cruz B., Salaberria J., Oyharçabal B., Bauduer F., Comas D., Quintana-Murci L., Consortium. TG The Basque paradigm: Genetic evidence of a maternal continuity in the Franco-Cantabrian Region since pre-Neolithic times. Am. J. Hum. Genet. 2012;90:486–493. doi: 10.1016/j.ajhg.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeviani M., Carelli V. Mitochondrial disorders. Curr. Opin. Neurol. 2007;20:564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 49.Gunnarsdóttir E.D., Nandineni M.R., Li M., Myles S., Gil D., Pakendorf B., Stoneking M. Larger mitochondrial DNA than Y-chromosome differences between matrilocal and patrilocal groups from Sumatra. Nat. Commun. 2011;2:228. doi: 10.1038/ncomms1235. [DOI] [PubMed] [Google Scholar]
- 50.Baum D.A., Smith S.D., Donovan S.S. Evolution. The tree-thinking challenge. Science. 2005;310:979–980. doi: 10.1126/science.1117727. [DOI] [PubMed] [Google Scholar]
- 51.Behar D.M., Metspalu E., Kivisild T., Rosset S., Tzur S., Hadid Y., Yudkovsky G., Rosengarten D., Pereira L., Amorim A. Counting the founders: The matrilineal genetic ancestry of the Jewish Diaspora. PLoS ONE. 2008;3:e2062. doi: 10.1371/journal.pone.0002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J., 2nd, Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 53.MITOMAP. (2011) A Human Mitochondrial Genome Database. http://www.mitomap.org.
- 54.Quintana-Murci L., Harmant C., Quach H., Balanovsky O., Zaporozhchenko V., Bormans C., van Helden P.D., Hoal E.G., Behar D.M. Strong maternal Khoisan contribution to the South African coloured population: A case of gender-biased admixture. Am. J. Hum. Genet. 2010;86:611–620. doi: 10.1016/j.ajhg.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schönberg A., Theunert C., Li M., Stoneking M., Nasidze I. High-throughput sequencing of complete human mtDNA genomes from the Caucasus and West Asia: High diversity and demographic inferences. Eur. J. Hum. Genet. 2011;19:988–994. doi: 10.1038/ejhg.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.