

Abstract
Recurrent deletions have been associated with numerous diseases and genomic disorders. Few, however, have been resolved at the molecular level because their breakpoints often occur in highly copy-number-polymorphic duplicated sequences. We present an approach that uses a combination of somatic cell hybrids, array comparative genomic hybridization, and the specificity of next-generation sequencing to determine breakpoints that occur within segmental duplications. Applying our technique to the 17q21.31 microdeletion syndrome, we used genome sequencing to determine copy-number-variant breakpoints in three deletion-bearing individuals with molecular resolution. For two cases, we observed breakpoints consistent with nonallelic homologous recombination involving only H2 chromosomal haplotypes, as expected. Molecular resolution revealed that the breakpoints occurred at different locations within a 145 kbp segment of >99% identity and disrupt KANSL1 (previously known as KANSL1). In the remaining case, we found that unequal crossover occurred interchromosomally between the H1 and H2 haplotypes and that this event was mediated by a homologous sequence that was once again missing from the human reference. Interestingly, the breakpoints mapped preferentially to gaps in the current reference genome assembly, which we resolved in this study. Our method provides a strategy for the identification of breakpoints within complex regions of the genome harboring high-identity and copy-number-polymorphic segmental duplication. The approach should become particularly useful as high-quality alternate reference sequences become available and genome sequencing of individuals' DNA becomes more routine.
Introduction
Structural variation, including copy-number variation, accounts for a significant proportion of human genetic diversity.1–4 A notable feature of copy-number variation is the potential for recurrent events to occur at “hotspots” within the human genome as a resultof nonallelic homologous recombination (NAHR) between repetitive sequences. Most notable in this regard are segmental duplications (SDs)—contiguous regions (>1 kbp) with high sequence identity (>90%).5,6 Recurrent, de novo copy-number variants (CNVs) have been associated with a variety of phenotypes, including schizophrenia (MIM 181500),7 autism (MIM 209850),8 epilepsy (MIM 604827),9 intellectual disability,10 congenital anomalies (MIM 612474 and 187500),11,12 severe obesity (MIM 613444),13 and renal disease (MIM 137920).14
Although there have been significant advances in CNV discovery and genotyping, precise breakpoint delineation within SDs remains challenging. This information is, however, essential if we are to further our fundamental understanding of genome plasticity and processes underlying genomic rearrangements. Traditionally, breakpoint resolution of genomic rearrangements required a combination of pulse-field gel electrophoresis and Southern blot analysis to reveal an atypical hybridizing band that harbored the breakpoint of interest.15,16 Sequence-level breakpoint identification of the genome has advanced considerably with more modern molecular methods that leverage the high quality of the human reference genome.17 For unique regions, the procedure is relatively straightforward and typically includes array comparative genomic hybridization (array CGH) followed by long-range PCR,18 subcloning, and direct Sanger sequencing.19,20 More recently, next-generation methods have allowed researchers to rapidly capture breakpoints by using split-read21 and paired-end-read mapping approaches.19,20,22
In contrast, few breakpoints mapping to repetitive regions, particularly those with large and highly identical duplications (>10 kbp and >95%), have been cloned and sequenced.16,23 Unlike unique regions, breakpoints that map to repeated sequences are much more problematic. Array CGH is unable to localize CNV breakpoints within blocks of near-perfect sequence identity, which may span hundreds of kilobases, because of probe cross-hybridization. Long-range PCR is relatively ineffective over such large distances of high sequence identity. Similarly, paired-end-read or split-read approaches generally fail to identify the breakpoints because of short library inserts and short read lengths that cannot successfully traverse the distances needed to anchor PCR primers to unique identifiers on either side of the breakpoint. Breakpoint resolution is further complicated by both structural polymorphisms and gaps in the human genome reference sequence, which often occur precisely at the breakpoints of interest. Such differences make determination of the true breakpoint particularly difficult because both variation and sequences exist at these sites, which are not present in the human reference sequence.
Here, we present an approach for determining sequence-level breakpoints occurring within SDs by using a combination of somatic cell hybrids, array CGH, and high-throughput sequencing. We take advantage of the specificity of next-generation sequencing data and the fact that large duplicated sequences with near-perfect sequence identity will still carry hundreds of sequence variants that distinguish the copies. A singly unique nucleotide (SUN) identifier is defined as a paralogous sequence variant (PSV) that tags a specific sequence paralog by uniquely distinguishing it from all other paralogs in the human genome. Such variants allow for interrogation of individual paralogs that are otherwise difficult to distinguish. In practice, SUNs are identified from next-generation sequencing data with SUN k-mers (SUNKs), sequences that have length k and map to exactly one genomic location containing one or more SUNs. Previously, we developed a catalog of these variants, and here we apply them to define breakpoints24 in individuals. We examine recurrent microdeletions on 17q21.31, one of the most structurally complex regions of the genome, as a model locus. Structural variation at this locus has been extensively characterized, most notably in haplotype-specific sequence assemblies of the H1 and H2 haplotypes, making the locus ideal for further study.25,26
Material and Methods
H2 Reference Assembly
Analysis of the H1 and H2 haplotypes was based on previously reported haplotype-specific sequence assemblies.26
Generation of Somatic Cell Hybrids
Somatic cell hybrids were generated at Mayo Medical Laboratories. After electrofusion of Epstein-Barr Virus (EBV) cells with E2 cells, mouse-human hybrid colonies were observed at 18 days. Subsequently, 88 clones were selected for initial expansion and genotyping. Six A and six B chromosome 17 homologs were selected for additional subculture. At pass three, all 12 hybrid clones were tested for chromosome 17 by FISH. On the basis of the FISH results, two A and two B hybrid clones were selected for confirmatory genotyping, and all cases confirmed retention of the appropriate A or B genotype. This study was approved by the institutional review board of the University of Washington and Radboud University, and all subjects provided informed consent.
Sample Genotyping
As previously described,27 H1/H2 genotyping was determined via gel electrophoresis on the basis of a deletion in intron 9 of MAPT. After generation of somatic cell hybrids, initial confirmatory genotyping was performed at Mayo Medical Laboratories (AFMa061za9, AFM192yh2, AFMa154za9, and AFM044xg3). Additional markers, AFM298wg5, AFMb364yh9, AFM155xd12, and AFMa110wb5, were identified as being close to the 17q21.31 deletion on the basis of the Marshfield genetic map,28 and these were subsequently genotyped at the University of Washington with the primers specified in the UniSTS marker database. To examine microsatellites within SDs, we chose a subset of the reported markers and primers used in a previously reported BAC assembly of the H2 haplotype.25 After amplification, all microsatellite genotypes were determined with an ABI 3730 DNA analyzer. All primers used are listed in Table S1.
Haplotype-Specific Array CGH
By using hybrid cell line DNA, we performed array CGH to compare the H1, H2, and 17q21.31 deletion-bearing chromosomes to one another. Because the hybrid cell lines are haploid for human chromosome 17, unique regions of the human genome removed by deletion have an extremely low signal, corresponding to copy number 0. In contrast, deletions within SDs, regions of the genome for which there exist additional paralogs, display intermediate levels of signal loss proportional to the number of paralogous copies elsewhere in the genome. Although a mouse genome is present in hybrid cells, we expected minimal cross-hybridization because even single mismatches are known to affect probe hybridization,29–31 and at exons within 17q21.31, the average human-mouse identity is ∼85%, corresponding to nine mismatches on a 60 bp probe.32 Finally, we visualized array CGH data on the H2 haplotype by remapping probes.26
Array Design and Analysis
We designed a custom 244K Agilent array specifically to interrogate 17q21.31 contained within hybrid cell lines (Table S2; GEO accession code GSE34867). At the deletion locus and flanking sequence (NCBI build 36, chr17:40.25M–42.75M), probes were placed at high density at 1 probe per 100 bp. Sample labeling was achieved with Roche NimbleGen Dual-Color DNA Labeling kits according to the manufacturer's protocol, but half (500 ng) the input DNA was used, and the protocol was scaled appropriately. For array hybridization, 25 ng each of labeled test and reference DNA was then brought to a 158 μl volume. Subsequently, the labeled DNA was hybridized to a custom Agilent array according to the Agilent hybridization protocol. In brief, the recommended hybridization master mix for a 1× microarray was prepared and added to the labeled DNA, and hybridization at 65°C on a rotator rack (20 rpm) followed for 72 hr. Array wash and scanning proceeded according to the manufacturer's protocol. However, feature extraction was carried out with a normalization set consisting of probes on human chromosome 17 but outside of 17q21.31.
Array CGH oligonucleotide probes were remapped to the H2 assembly with BLAST (blastn parameters −e 1e−10 −m 8 −W 7).33 Partial BLAST hits were extended without gaps to encompass the entire probe sequence, and probes with no BLAST hits were aligned with JAligner (see Web Resources), an implementation of the Smith-Waterman algorithm (NUC.4.4 matrix; gap open and extension penalties were equal to 10). Finally, probes were mapped to a given location on the H2 assembly if and only if the global alignment mapped with a ≤1 bp mismatch and a ≤1 bp gap. Using these criteria, we mapped 11,967 distinct probes to 18,914 positions in the H2 assembly. To calculate the haploid copy number of probes mapping to the H2 assembly, we aligned each probe to the human genome (build 36), mouse genome (mm8), and the H2 assembly by using BLAST (with the same parameters as those used in probe mapping). To avoid double-counting between the human genome and the H2 assembly, we excluded human genome BLAST hits to the 17q21 deletion region (chr17: 40799295–42204344). To provide a ceiling on the copy number of a given probe, we defined a probe's copy number as the number of BLAST hits covering ≥90% of the probe with ≤3 mismatches and ≤1 bp gap. Consistent with a tendency to overestimate probe copy number, for the 3,231 probes that were within the H2 assembly between 700,000–1,000,000 bp, a region predicted to be almost entirely unique sequence in a haploid human genome, 99% (3,186/3,231) of probes were predicted to have a copy number of 1, and the remaining probes were predicted to have a copy number >1.
We determined copy-number loss at each probe given NAHR between a particular pair of paralogous sequences. The expected relative copy number for a given probe was defined as the copy number of a probe after the deletion divided by the estimated probe copy number in the H2 assembly. We compared expected changes in relative copy number to observed log2 ratios to determine the most likely pair of paralogous sequences mediating each deletion (Figure S1B).
Gap Closure
To close gap 2, we used the previously identified BAC RP11-84A7 (AC243906). To close gap 1, we screened for clones mapping to gap regions by using a method similar to that previously reported for placing fosmids in the genome.20 We locally aligned fosmid end sequences to the H1 assembly and H2 pseudo-assembly by using MegaBLAST.34 Clones under consideration were subsequently limited to those with an alignment either within the spacer sequence (represented in AC217768) or at the proximal end of AC139677. Local alignments were then extended into global alignments with needle, a Needleman-Wunsch algorithm implementation from the EMBOSS software suite.35 We scored global alignments for mismatches and gaps by only using bases with Q30 or higher quality. Paired end-sequence placements were then screened on the basis of concordant clone-end orientation and estimated insert size. Subsequently, clone-end orientation and size-concordant placements were assigned to the H1 haplotype, other paralogous sequence in the H2 haplotype, or sequence that mapped adjacent to or within the proximal gap; sequence identity was used as a tie-breaker. Importantly, for all clones chosen, end sequences were best assigned to sequence adjacent to the gap or inferred sequence within the gap and not at paralogous sequence elsewhere in the H1 or H2 assemblies. We selected three clones for sequencing: two clones extending proximally and distally from the spacer sequence on AC217768 (1134622_I19 and 50932900_K17; AC244164 and AC244161, respectively) and one clone (1013914_P2; AC244163) extending proximally from the proximal end of AC139677 (Figure S2). The three fosmids and the BAC clone used for closing gaps in the H2 haplotype were sequenced and assembled at The Genome Institute at Washington University. Consistent with our hypothesized structure for RP11-374-N3, distal portions of 50932900_K17 (AC244161) and proximal portions of 1013914_P2 (AC244163), which mapped to gap 1, were paralogous and in direct orientation to SDs on the H1 and H2 haplotypes proximal to unique deleted sequence (Figure S2, Figure S3, and Figure S4). Similarly, 1134622_I19 (AC244164) mapped entirely to finished sequence (all from AC217768; Figure S5) in the H2 assembly and contained sequence that was paralogous, but of inverted orientation (based on end-sequence placement), to SDs on the H1 and H2 haplotypes proximal to unique deleted sequence.
Next-Generation Sequencing, Complete Genome Sequencing, and Breakpoint Mapping with SUNs
Massively parallel sequence data were generated from three probands with both SOLiD and Illumina sequencing platforms. For members of family 2, long mate-paired libraries were generated from 100 μg of genomic DNA, which was isolated from peripheral blood samples via QIAamp mini columns (QIAGEN). Library preparation was essentially as described in the SOLiDv3.5 library preparation manual (Applied Biosystems). Of note, we performed DNA size selections directly after CAP adaptor ligation to select genomic fragments between 2 and 3 kbp and, moreover, to reduce the presence of concatamers. Additionally, we performed a size selection after library amplification. To assess the presence of adaptors and determine the average insert sizes, we cloned libraries and chose 384 clones per library for capillary sequencing. Initially, we sequenced two 50 bp mates for each library (F3 and R3 tags) on a SOLiD 3PLUS instrument and thereby used a single quadrant for the father and mother of the sequencing slide, but two quadrants for the proband. To obtain additional read depth for the mother and proband, we subsequently performed a 50-bp-fragment run on the same libraries by using a full sequencing slide for each on a SOLiD4 instrument.
For the family 1 proband (31928) and family 3 proband (31873), 3 μg of genomic DNA was sheared, end-repaired, an A-tail added, and adaptors were ligated to the fragments as described in Igartua et al.36 After ligation, the samples were run on a 6% pre-cast polyacrylamide gel (Invitrogen, catalog number EC6265BOX). The band at 400 bp was excised, diced, and incubated. Size-selected fragments were amplified with 0.5 μl of primers, 25 μl of 2× iProof, 0.25 μl of SYBR Green, and 8.25 μl of dH2O under the following conditions: 98°C for 30 s, 30 cycles of 98°C for 10 s, 60°C for 30 s, 72°C for 30 s, 72°C for 15 s, and 72°C for 2 min. Fluorescence was assessed between the 30 and 15 s 72°C step. Amplified, size-selected libraries were quantified with an Agilent 2100 Bioanalyzer and paired-end sequenced (101 bp reads) on an Illumina HiSeq 2000.
Using a pipeline similar to that previously described,24 we identified 36-mer SUNKs that uniquely distinguish paralogs potentially mediating 17q21.31 deletions in the H2 assembly. We identified PSVs by one of two methods: First, for sequence present in the current assembly, we used whole-genome assembly comparison (WGAC)-defined global alignments to identify single-base-pair differences between paralogs (Figure S6). Second, for sequence in the proximal gap, we identified and sequenced fosmids (AC244161 and AC244163) extending into either side of the gap. We subsequently identified PSVs from alignment of fosmid draft sequences against inferred regions of paralogy on the H1 and H2 haplotypes (H1:219,599–261,693 and H2:452,165–261,693, respectively) by using stretcher, a Needleman-Wunsch algorithm implementation from the EMBOSS software suite (Figure S6).35
For each identified PSV, we generated all possible 36-mers incorporating the variant. Subsequently, we passed the 36-mers through a series of filters. First, those containing repeat sequence as identified by RepeatMasker and TandemRepeatFinder37 or those within 36 bp of such sequence were excluded. Second, we used mrFAST38 to identify all possible mappings, including that to the H2 haplotype (GRCh37), of each 36-mer to the mouse (mm8) and human reference assembly, allowing for up to two mismatches, insertions, or deletions (edit distance ≤2). For PSVs outside gap 1, we identified SUNKs as those reads with one exact match in the human reference assembly or the H2 haplotype, no exact matches to the mouse genome, ≤10 mrFAST hits with edit distance ≤2 in the human genome, and ≤10 mrFAST hits with edit distance ≤2 in the mouse genome. SUNKs within gap 1 were defined similarly, but no matches to the current reference assembly or H2 haplotype were allowed.
Because of high sequence identity within AC217768 in the current H2 assembly, relatively few SUNs were identified in gap 1. However, because all sequence in AC217768 is lost in NAHR-mediated 17q21.31 deletions, gap 1 PSVs that are only present elsewhere in the genome within AC217768 are still breakpoint-informative for H2/H2 NAHR. Similarly, gap 1 PSVs that are only present on the H2 haplotype proximal to or within AC217768 are breakpoint-informative for H1/H2 NAHR. Using these criteria, we identified additional H1/H2 or H2/H2 breakpoint-informative PSVs.
Finally, we empirically validated the presence or absence of SUNs by using data from the 1000 Genomes Project.39 As a positive control, we identified candidate SUNKs in the combined sequence data from nine H1/H2 CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) individuals (mean coverage 3×), and H2-specific candidate SUNs without observed mapped reads were excluded. As a negative control, we identified candidate SUNKs in combined sequence data from a CEU trio (mean coverage 27.6×; NA12878, NA12891, and NA12892) and from an YRI trio (mean coverage 21x; NA19238, NA19239, and NA19240), all with H1/H1 genotypes. H2-specific candidate SUNs were discarded if observed at a read depth above the minimum H1-specific SUN read depth in two or more samples. A similar validation procedure was carried out for H1-specific SUNs. We used next-generation sequencing data from probands to refine the breakpoints of the rearrangement on the basis of the absence or presence of reads mapping to these unique identifiers.
Results
We briefly review the structural features of the 17q21.31 microdeletion locus. Within the current reference assembly (GRCh37), the locus is defined approximately by chr17:43.4–44.8 Mbp. The locus encompasses ∼600 kbp of unique sequence. This sequence contains several genes, including MAPT, CRHR1, and KANSL1 (previously known as KIAA1267), and is flanked by extensive SDs. The 17q21.31 locus has two major structural haplotypes spanning ∼1.5 Mbp: the H1 haplotype, which is most common, and the H2 haplotype, which is present at a frequency of 20% in Europeans.25,27,40 BAC-based, haplotype-specific sequence assemblies of the H1 and H2 haplotypes have previously been created from the BAC library RP11, which was derived from an H1/H2 individual.26 The reference assembly at 17q21.31 represents the H1 haplotype, and the H2 is presented as an alternate haplotype (chr17_ctg5_hap1). These two haplotypes are distinguished by the presence of an approximately 970 kbp inversion in addition to more than 300 kbp of differences in the copy number and content of SDs (Figure S7).25,26 Importantly, the H2 haplotype contains 95 kbp of SD in direct orientation flanking the unique region, whereas no such sequence is observed in the H1 haplotype. Recurrent deletions at this locus cause the 17q21.31 microdeletion syndrome (MIM 610433), in which deletions only arise in parents with one or more H2-bearing chromosomes.41–43 NAHR involving only this H2-specific duplication is hypothesized to underlie the H2 predisposition to microdeletion.26
Our goal was to localize the breakpoints of recurrent 17q21.31 deletions in six individuals of European descent. This set included three families wherein de novo microdeletions had been previously identified41 and for which transformed cell lines had been constructed from the proband and both parents, as well as three unrelated probands with the 17q21.31 deletion, for further analysis.9 To assess the accuracy of our experiments, we proceeded in a series of steps whereby we developed genomic resources to simplify and validate our findings as needed. To remove the potential confounding effects of large-scale differences on different structural haplotypes on chromosome 17, we initially isolated deletion-bearing chromosomes by using somatic cell hybrids (reviewed in Trask et al.44) from both the transmitting parent and the proband (Figure 1). This allowed us to design the ideal array CGH experiment, where duplicated sequences flanking the critical region could be compared in the isolated donor and deleted chromosomes (Figure 1B, Figure 2). Once we refined the location of the paralogous segments where breakpoints were likely to occur, we focused on obtaining sequence-level breakpoint resolution in the three probands with parental information. It then became necessary to discover and characterize sequence that mapped to gaps within the H2 haplotype; the additional sequence allowed us to attain sequence-level breakpoint delineation by using a combination of next-generation sequencing and SUN identifiers.24 This breakpoint delineation was consistent with results obtained by array CGH of somatic cell hybrids. These results give us confidence that genome sequencing of individuals in conjunction with SUN mapping will provide a robust method for routine breakpoint characterization in the future.
Figure 1.
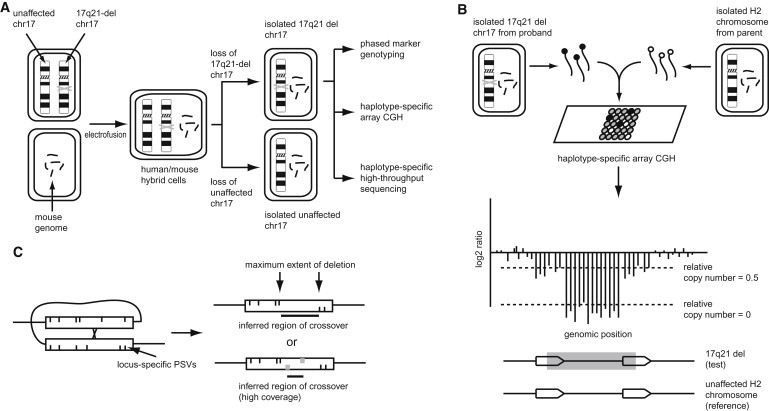
Schematic of SD-Breakpoint Detection Approach
(A) After the creation of human/mouse hybrid cells, clonal populations that carried only one of two chromosome 17 homologs were selected. The 17q21.31 deletion-bearing chromosome could then be studied in isolation from the unaffected chromosome 17.
(B) Hybrid cell lines permit haplotype-specific array CGH. NAHR-mediated deletions (bottom schematic, gray box) remove both unique sequence and SD (block arrows). Deletions in unique sequence are seen as extremely low signal representing relative copy number 0 (log2 ratio plot schematic). Copy-number loss in SD displays intermediate signal loss proportional to the number of remaining paralogous copies elsewhere in the genome (in the schematic, relative copy number = 0.5).
(C) For NAHR-mediated deletions, unequal crossover within SDs (rectangles) removes PSVs specific to the proximal and distal duplicons (vertical hashes in upper and lower rectangle halves, respectively), which can be used to infer the maximal extent of the deletion and the region of crossover. At low coverage, the absence of reads mapping to a PSV might reflect lack of sequence coverage. At sufficiently high coverage, however, the absence of reads mapping to a PSV (gray vertical hashes) implies the absence of the PSV in the sample and can further refine the crossover region.
Figure 2.
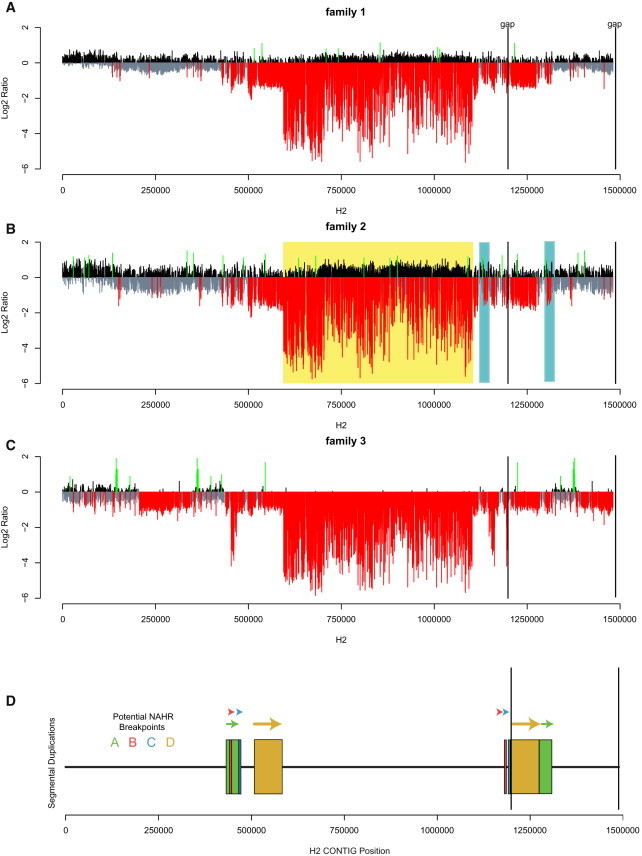
Haplotype-Specific Comparative Genomic Hybridization of Three 17q21.31 Deletion-Bearing Chromosomes versus an Unaffected H2 Chromosome 17
(A) Somatic cell hybrid DNA allowed for array CGH comparing specific 17q21 haplotypes. Relative gain (black), loss (gray) and gains and losses >3 standard deviations beyond the chromosome 17 mean (green and red, respectively) are plotted against genomic position on a previously described sequence assembly of the H2 haplotype.26
(B) Pairs of segmental duplications (SDs) in direct orientation as determined by sequence comparison6 are shown as pairs of colored blocks. If we assume that the deletions occurred due to NAHR, there are four pairs of directly oriented SDs that can mediate the rearrangement (breakpoints A–D). The percent identity between SDs is 98.6%, 99.2%, 99.3%, and 99.7% for breakpoints A, B, C, and D, respectively. Because chromosome 17 homologs are initially haploid within somatic cell hybrids, deletions within unique regions of the genome (family 2, yellow highlight) are seen as an extremely low signal corresponding to relative copy number 0. In contrast, deletions within SDs display intermediate levels of signal loss as a result of cross-hybridization from paralogous sequence elsewhere in the genome. The light blue highlights in family 2 (A) represent a deletion that occurred within SDs (not shown) and that resulted in a relative loss of signal at both locations, potentially confounding breakpoint analysis.
Somatic Cell Hybrid Characterization
We constructed 36 somatic cell hybrids derived from three parent-child trios in which the child harbored a de novo 17q21.31 deletion and from three unrelated 17q21.31-deletion-bearing probands for whom no parental DNA samples were available (Figure 1; Table S3). H1/H2 haplotype status was determined with a previously described 238 bp deletion marker within intron 9 of MAPT.27 In all three cases for which parental DNA samples were available, one parent was either homozygous or heterozygous for the H2 haplotype, and the other was homozygous for the H1 haplotype. For each of the six probands and the parents containing an H2 haplotype, we constructed at least two human-mouse somatic cell hybrid cell lines such that each of the chromosome 17 homologs (referred to as A and B; see Material and Methods) was isolated. The creation of somatic cell hybrids isolates the 17q21.31 deletion-bearing chromosome and the progenitor parental chromosome prior to deletion and thereby facilitates breakpoint detection (Figure 1).
We initially genotyped the somatic cell hybrids by using eight microsatellite markers (Figure S8 and Table S3) to assess the integrity of each chromosome 17 homolog and confirm that deletions originated from the parent carrying the H2 haplotype. In family 1, markers immediately flanking the deletion locus in the proband (31928) indicate that it probably arose as a result of interchromatidal NAHR (between sister chromatids), as expected. In family 2, the deletion occurred in the gamete of the mother (31918), who is homozygous for the H2 chromosomes and is also suggestive of interchromatidal NAHR. Finally, in family 3, crossover between the H1 and H2 haplotypes and the 17q21.31 deletion co-occur within a genetic distance of less than 0.54–1.32 cM, as determined by the Marshfield map and HapMap, respectively.28,45 Because of the short genetic distance separating the events, these preliminary results suggested the possibility that unequal crossover between the H1 and H2 haplotypes generated the deletion within this family. We tested an additional seven microsatellite markers flanking the deletion locus (Table S3). The results remained consistent with interchromosomal but not intrachromatidal NAHR for family 3 (Figures S8 and S9).
Haplotype-Specific Array CGH
We next performed haplotype-specific array CGH by using matched chromosome 17 hybrid cell lines (Figure 1B; Material and Methods; GEO accession code GSE34867). For each family, we hybridized DNA from a line containing the 17q21.31-deletion-bearing chromosome of the child against the corresponding H2-haplotype-bearing hybrid cell line from the parent. As expected, deletions within the unique portion of 17q21.31 were readily apparent (relative copy number 0; Figure 2). Deletions within the SDs were detectable but displayed intermediate levels of signal loss proportional to the number of paralogous copies elsewhere on chromosome 17. We observed similar patterns and log2 ratio signal intensity for both families 1 and 2, whereas the deletion in family 3 showed a different pattern by array CGH. We noted, for example, that some signal loss proximal to 340 kbp and distal to 1.38 Mbp was not observed in the other individuals (Figure 2; Figure S10).
We hypothesized that the array CGH signature observed in family 3 was a consequence of interchromosomal NAHR and sought to assess its relative frequency in 17q21.31-deletion-bearing probands. Further examination of the three additional unrelated 17q21.31-deletion-bearing probands by array CGH showed log2 ratios similar to those in families 1 and 2 (Figure S11). The breakpoints for these three additional individuals had been previously analyzed by array CGH of diploid DNA42 and provided a benchmark for comparison. We also surveyed 12 additional 17q21.31 spontaneous deletions by using a combination of a lower-resolution array CGH platform and marker segregation and noted only one further case, which was consistent with the H1/H2 recombination pattern identified in family 3. Thus, on the basis of our analysis with somatic cell hybrids (1/6) and examination of other data (1/12), H1/H2 deletions account for ∼10% of cases.
Under the assumption that the 17q21.31 deletions arose as a result of NAHR between high-identity SDs, we developed a breakpoint analysis method that compares the array CGH signal intensity to the expected changes in relative copy number of high-identity SDs bracketing the critical region (see Material and Methods). Analysis of the H2 assembly predicted four possible pairs of paralogous sequences (breakpoint regions A–D; Figure 2; Figures S7 and S10) under a model of H2 interchromatidal NAHR.
Examining SDs at the proximal deletion breakpoint, we observed a predicted region of copy number 0 (yellow highlight, Figures S10A and S10C) for breakpoints A–C. Although array CGH data from family 3 demonstrated a log2 signal consistent with a copy number of 0 in this region, the same degree of signal loss was not observed in either family 1 or family 2. This suggests that deletions for both family 1 and family 2 are mediated by sequences at breakpoint D. Similarly, the distal breakpoint, a region of predicted copy number 0 (yellow highlight, Figures S10B and S10D), for breakpoints A–C is inconsistent with the log2 ratios observed in families 1 and 2. Thus, the most likely sequences mediating NAHR for families 1 and 2 are those of breakpoint D, corresponding to a pair of directly oriented SDs with >99% identity and a length of ∼75 kbp in the current H2 assembly.
In contrast to that in families 1 and 2, relative copy-number loss proximal to 340 kbp and distal to 1.38 Mbp in family 3 (orange highlight, Figure S10) was not consistent with intrachromosomal NAHR involving any of the breakpoints A–D but was consistent with the previous microsatellite data suggesting that the family 3 deletion might be mediated by interchromosomal NAHR between the H1 and H2 haplotypes. This was paradoxical; it would require sequence proximal to the unique deleted sequence on the H1 haplotype to directly orient with paralogous sequence distal to the unique deleted sequence on the H2 haplotype. However, such sequences are not currently observed in the current H2 assembly (Figure S7 and Table S4).26 This suggested several possible hypotheses. If the H1/H2 crossover and the deletion were separate events, then the family 3 deletion could have occurred on an H2 haplotype with altered copy number within SDs or might not have been the result of NAHR. Alternatively, interchromosomal crossover between the H1 and H2 haplotypes might have occurred as a result of sequences not currently represented in the H2 assembly.
We performed array CGH between hybrid cell lines containing the H2 chromosome from the mother in family 3 and the mother in family 2 and observed no copy-number differences across the region (Figure S12). This suggested that the unusual log2 ratio observed for the deletion in family 3 was not the result of structural variation or polymorphism on the H2 haplotype.
Closing the Sequence Gaps in the H2 Assembly
We explored the possibility that crossover between H1 and H2 haplotypes is mediated by previously unrepresented sequence in the current haplotype assembly. There are two gaps within the current H2 assembly in GRCh37 (gap 1 and gap 2; Figure 3), both of which lie distal to the unique deleted sequence (Figure 3; Figure S7). Previously reported marker data suggested that gap 2 (spanned by RP11-84A7) does not contain sequence that can mediate 17q21.31 deletions by H1/H2 NAHR.25 In contrast, a draft sequence of RP11-374N3 (AC048388) contained sequence paralogous to SDs proximal to the unique deleted sequence on both the H1 and H2 haplotypes, in agreement with our hypothesis that H1/H2 NAHR might occur. This was additionally supported by the presence and orientation of microsatellites DG17S133 and DG17S435 in RP11-374N3 (Figure S9).25
Figure 3.
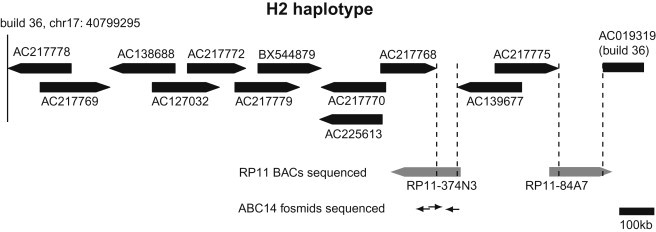
Completion of the H2 Contig with Clone-Based Resources
Two gaps exist in the H2 contig (dotted vertical lines). The distal gap (gap 2, ∼130 kbp) is spanned by the previously placed BAC RP11-84A7.25 So that the proximal gap (gap 1, ∼70 kbp) can be closed, assembly of RP11-374N3 will be completed with the assistance of additional clones from the fosmid library of an H1/H2 individual (ABC14, NA12156).
We noted that, to close gap 2 (∼130 kbp), Steffansson et al.25 had placed RP11-84A7, which was not used in the H2 sequence assembly,26 in a BAC assembly to connect the distal end of the H2 haplotype to the reference assembly. To reconfirm placement of RP11-84A7 (AC243906) on the H2 haplotype, we first end-sequenced the clone and noted that the T7 end maps to the distal portion of either the H1 or H2 assembly from Zody et al.26 and that the SP6 end maps to AC019319 in build 36. In order to distinguish placement of RP11-84A7 on the H2 haplotype versus the H1 haplotype, we compared microsatellites on RP11-84A7 with those on RP11-619A10 (AC217775), the last BAC in the H2 assembly, by using RP11-113E17, a clone assigned by Stefansson et al.25 to the H1 haplotype, as a negative control (Figure S13 and Table S5). Marker genotyping confirmed the predicted overlap between RP11-619A10 and RP11-84A7 and also demonstrated RP11-84A7 and RP11-113E17 to be on opposite haplotypes. Finally, the size of gap 2 was estimated as the average size of a BAC from RP11 minus its overlap, based on end-sequence placement, with sequence on either side of the gap (130 kbp = 180 kbp – 25 kbp – 25 kbp).
RP11-374N3 (AC048388) was previously determined to span gap 1 (∼70 kbp) in the H2 assembly but could not be assembled by shotgun sequencing alone.26 We hypothesized that this was due to the presence of two arms of oppositely oriented, highly identical sequence separated by a spacer sequence unique within the clone (Figure S2). Importantly, the hypothesized structure suggested that gap 1 contains sequence paralogous to SDs on the H1 and H2 haplotypes and that this sequence might mediate NAHR. If sequence in gap 1 largely corresponds to one of two highly identical arms of sequence in RP11-374N3 (Figure S2), then the other duplicated arm of sequence, entirely contained within the neighboring finished clone AC217768, provides a good approximation of the sequence in gap 1. On the basis of this hypothesized structure, we estimated that gap 1 contains 40 kbp and 70 kbp of sequence with ∼99% identity to the H1 and H2 haplotypes proximal to the unique deleted sequence, respectively.
We sequenced RP11-84A7 and additional clone-based resources to aid in the assembly of RP11-374N3 (Figure 3). A draft assembly of RP11-84A7 (spanning gap 2; AC243906) did not contain sequence that could mediate 17q21.31 deletions. Because RP11-374N3 (spanning gap 1) previously could not be assembled by shotgun sequencing alone,26 we identified three additional smaller clones of a fosmid clone library (ABC14) from an H1/H2 individual to effectively provide subassembly and resolve near-perfect local duplications of the larger BAC (Material and Methods and additional references19,46,47). As predicted, draft sequences from these clones (AC244161, AC244163, and AC244164) identified the presence of an additional ∼70 kbp of SD in direct orientation (∼99% estimated identity) between gap 1 and the H2 haplotype proximal to unique deleted sequence and an additional ∼40 kbp of SD in direct orientation (>99% estimated identity) between gap 1 and the H1 haplotype. This confirmed our hypothesized structure of RP11-374N3 and therefore that previously uncharacterized sequence in the H2 assembly could mediate NAHR between the H1 and H2 haplotypes in family 3. Additionally, it suggested that the length of breakpoint D, which probably mediated deletions in the remaining five probands, is nearly twice as large (∼145 kbp versus 75 kbp) as what is annotated in the human genome reference.
Identification of Breakpoint-Informative Paralogous Sequence Variants
To achieve sequence-level resolution, we identified SUNs, PSVs unique to specific loci in the genome (Figure 1C), as well as other breakpoint-informative PSVs within the SDs mediating the observed 17q21.31 deletions.24 We used two different techniques to identify breakpoint-informative PSVs (Figure S6). For sequences present in the current H2 assembly, we identified PSVs by using WGAC as described previously6 to generate alignments of paralogous sequence. To create SUNs, we then filtered PSVs by determining which PSVs could generate unique 36 bp reads with respect to the human and mouse genomes (Material and Methods). For sequences mapping to gaps in the current H2 assembly, PSVs were identified from the alignment of the fosmid draft sequences mapping to gap 1 with the expected regions of paralogous sequence on the H1 and H2 haplotypes (Material and Methods). This technique could be useful with other regions that have alternate structural haplotypes and where a haplotype-specific sequence assembly might not exist, yet where the haplotype of a given clone is known. Subsequent filtering of these PSVs revealed relatively few SUNs in gap 1 (Table 1). This was due to the near identity of sequence within gap 1 to sequence immediately proximal on AC217768 in the H2 assembly (Figure S2). This sequence, however, would be lost in the event of H1/H2 or H2/H2 NAHR. Therefore, gap 1 PSVs present elsewhere only within AC217768 would still be breakpoint informative in the event of H2/H2 NAHR and would thus effectively act as SUNs (Material and Methods). Similarly, gap 1 PSVs present elsewhere in the genome but exclusively on the H2 haplotype within or proximal to AC217768 would effectively act as SUNs in the event of H1/H2 NAHR.
Table 1.
Summary of Identified Breakpoint-Informative PSVs
| Name | H2 Proximal and Distal | H1/H2 Inferred Proximal | H1/H2 Inferred Distal | H2/H2 Inferred Proximal | H2/H2 Inferred Distal | H2/H2 Informative | H1/H2 Informative |
|---|---|---|---|---|---|---|---|
| Region(s) | H2:519,560–593,627 bp, H2:1,198,880–1,273,881 bp | H1:219,599–261,693 bp | H2, gap 1 | H2:452,165–519,559 bp | H2, gap 1 | NA | NA |
| Description | breakpoint D, proximal and distal paralogs | inferred H1 paralog to gap 1 | PSVs inferred from alignment to H1 | inferred H1 paralog to gap 1 | PSVs inferred from alignment to H2 | H2 proximal, H2 distal, and H2/H2 inferred proximal and distal | H2 proximal, H2 distal, and H1/H2 inferred proximal and distal |
| k-mers | 2,627 | 845 | 440 | 858 | 1,195 | 4,680 | 3,912 |
| PSVs (SUNs) | 86 (86) | 37 (37) | 19 (1) | 40 (40) | 61 (2) | 187 | 142 |
After quality control (Material and Methods), we identified 4,680 36-mers corresponding to 187 distinct PSVs that can be used to distinguish deletions due to H2 interchromatidal NAHR and 3,912 36-mers corresponding to 142 distinct PSVs that can be used to distinguish H1/H2 interchromosomal NAHR (Table S6).
Resolution of CNV Breakpoints within Paralogous Sequence
We leveraged the specificity of next-generation sequence data to achieve sequence-level breakpoint resolution in the three parent-child trios by mapping genome sequence data to this set of SUN identifiers. We initially compared sequence patterns between the proband and mother for family 2 by generating whole-genome sequence from both individuals. We generated ∼26 Gbp of sequence (∼9-fold coverage) for the family 2 mother, who was an H2-homozygote, by using the SOLIDv4 sequencing platform. As expected, reads aligned to breakpoint-informative PSVs across both the proximal and distal paralogs of the breakpoint D region: an ∼145 kbp region of near-perfect sequence identity including previously uncharacterized sequence mapping to the gap in the H2 assembly. This finding is consistent with the finding, from array CGH results from somatic cell hybrids, that the mother is diploid across the 17q21.31 microdeletion region (Figure 4). In stark contrast, when genome sequence (44 Gbp, ∼15-fold coverage) was generated from the proband in family 2 and mapped to these variants, we observed no aligned reads to PSVs on the proximal paralog of breakpoint D past the H2 position at 508,415 bp and no aligned reads to PSVs before the H2 position at 1,209,274 bp on the distal paralog. This localizes the crossover between the paralogs and refines the deletion breakpoints from a 145 kbp region based on array CGH to a ∼22 kbp window (H2:508,415–529,961 on the proximal paralog and Gap 1: 56,251 to H2:1,209,274 on the distal paralog; chr17_ctg5_hap1:567,056–588,595 on the proximal paralog and gap 1 to chr17_ctg5_hap1:1,317,189 in the GRCh37 genomic sequence). This breakpoint includes the 5′ UTR of KANSL1.
Figure 4.
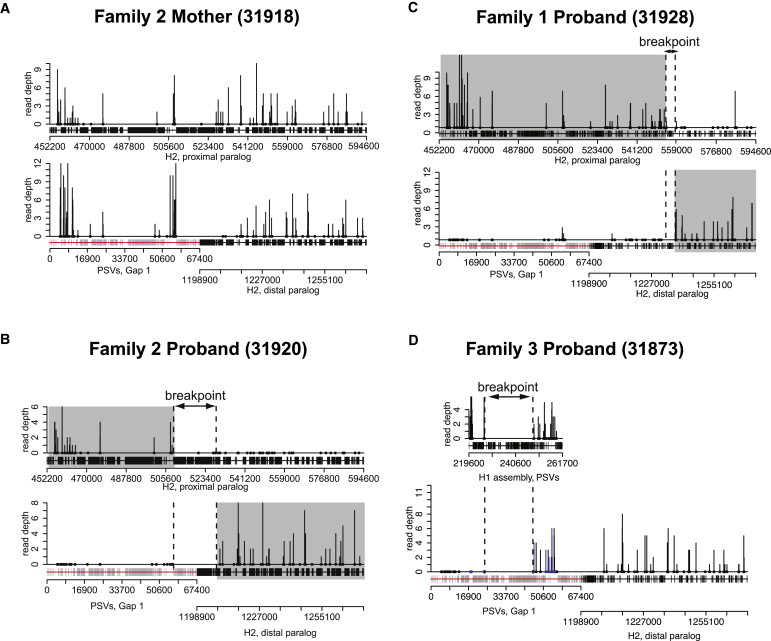
Breakpoint-Informative PSVs Identify 17q21.31 Deletion Breakpoints within SDs
Read depth (vertical lines) at breakpoint-informative PSVs (dots) has been plotted over an alignment of the proximal (top plot) and distal (bottom) paralogs of breakpoint D in two probands (B and C) with 17q21.31 deletions and the mother from family 2 (A), who is homozygous for the H2 haplotype. For the proband of family 3 (D), the paralogous H1 region (D, top plot) is plotted in approximate alignment with the inferred region of directly oriented paralogy in gap 1. The distribution of breakpoint-informative PSVs is determined, in part, by the relative density of repeat sequences in finished sequence (black blocks) or is inferred to be present in gap 1 (gray blocks). As expected in unaffected H2 chromosomes (A), breakpoint-informative PSVs can be observed along the entire length of the proximal and distal paralogs of breakpoint D. In contrast, sequence data from a 17q21.31 deletion in family 2 (B) demonstrates no PSVs past the H2 position at 508,415 bp on the proximal paralog and no PSVs proximal to the H2 position at 1,209,274 bp on the distal paralog of breakpoint D. These define the deletion breakpoints (dotted highlight) and the resulting chimeric SD product (gray highlight) of NAHR. A similar deletion pattern is observed in family 1 (C), although with a different breakpoint (H2 position at 554,425 bp and H2 position at 1,237,776 bp on the proximal and distal paralogs, respectively), reflecting the recurrent nature of the deletion. Finally, in family 3, H1-specific PSVs are uninformative because of the paternally inherited H1 chromosome (D), but H2-specific sequences demonstrate no PSVs from the proximal paralog of breakpoint D, consistent with H1/H2 NAHR.
We repeated this mapping strategy by focusing on the remaining two probands. We generated ∼42 Gbp of whole-genome sequence (∼14-fold coverage) for the proband from family 1 (31928) and ∼46 Gbp of sequence (∼15-fold coverage) for the proband from family 3 (31873) by using Illumina Hi-Seq2000 platform. In family 1, we narrowed the deletion breakpoints to a ∼4 kbp window (H2:554,425–558,503 and H2:1,233,725–1,237,776 on the proximal and distal paralogs, respectively; chr17_ctg5_hap1:613,066–617,144 and chr17_ctg5_hap1:1,341,640–1,345,691 in the GRCh37 genomic sequence) that includes the first coding exon of KANSL1. Although we observe a few sequence read alignments to PSVs outside of these breakpoint intervals, the hits are not collinear, and we attribute these to either polymorphisms between the H1 and H2 haplotypes or spurious PCR-induced mutations that arose during library prep. Finally, we observed no reads aligning to PSVs from the proximal segment of breakpoint D in family 3, but we did observe sequence alignments after the gap 1 position at 45,302 bp on the distal paralog, which aligns to the position at 248,866 bp on the H1 assembly. The first PSV observed on the H1 assembly proximal to this is at the H1 position at 224,601 bp. This places the breakpoint in a ∼24 kbp window (chr17:43,668,073–43,692,338 in the GRCh37 genomic sequence) upstream of CRHR1 on the H1 chromosome and completely within the gap 1 sequence of the H2 chromosome. This pattern is consistent with our previous hypothesis of H1/H2-mediated NAHR because such a crossover occurs within the expected region of directly oriented H1/H2 SDs and would remove the proximal paralog of breakpoint D in its entirety.
Discussion
We employed a combination of technologies and analyses that allow for breakpoint delineation within genomic regions previously refractory to analysis. We note three key components of our analysis. First, generation of somatic cell hybrids isolating chromosome 17 homologs greatly simplified microsatellite and array CGH analysis by providing haplotype-specific genetic data. Marker genotypes were phased and allowed inferences to be made on the basis of markers within SDs. Removal of the confounding effects of an alternate haplotype was of particular relevance for 17q21.31 so that copy-number polymorphisms of NSF on the H1 haplotype could be resolved.25 Although it is impractical to routinely design somatic cell hybrids for individuals, these reagents proved powerful in helping to interpret and validate our findings in this study. Final validation of our results would benefit from future technology that allows Mbp-scale sequencing of single molecules from proband DNA.
Second, when examining copy-number losses within SDs, we found that it was crucial to discern the degree of loss as a function of duplication copy number. Analysis of observed log2 ratios versus expected relative copy number bolstered initial evidence from marker genotyping that breakpoints in family 3 deletions, for example, were distinct from those of families 1 and 2. This underscores the utility of somatic cell hybrids in helping to provide a sensitive framework of copy-number loss for medically relevant regions of the genome. That is, if a particular SD is present in two copies and if it is of importance to discern whether zero, one, or both copies have been deleted, then with chromosome-specific array CGH, one would need to distinguish between relative copy number 1, 0.5, and 0, respectively. In contrast, for array CGH using genomic DNA, this would require distinguishing between relative copy numbers 1, 0.75, and 0.5, which is substantially more difficult. Moreover, modeling of expected versus observed copy-number losses allowed us to infer deficiencies in the current H2 assembly.
The final component of our analysis that permitted sequence-level breakpoint resolution is the discovery of phased, locus-specific paralogous sequence variation. For our model, locus-specific PSVs (SUNs) were known either by virtue of an accurate, haplotype-specific reference assembly or, for gaps in this assembly, sequencing of clone-based resources. It is perhaps not surprising that several (2/3) of the breakpoints map to the few remaining gaps in the duplicated regions given that these are the most highly identical, the most difficult to resolve, and the most likely to mediate NAHR.5,48 In some cases, we were able to refine the breakpoints to a small interval of 4 kbp, whereas in other cases the breakpoints are still quite large at 22 kbp. However, in large regions of perfect sequence identity, it will be impossible to refine the intervals any further unless discriminating SNPs specific to individual families can be discovered.
Our analysis also yielded biological insights regarding the 17q21.31 locus and its underlying rearrangements (summarized in Figure 5). We identified additional SDs critical to understanding the genetic basis for the unequal crossing over that mapped to the gap of the H2 assembly. First of all, we find that ∼90% of 17q21.31 rearrangement events (16/18 based on specific screening for the H1/H2 events) occurring as a result of interchromatidal NAHR are driven by European-specific SDs on the H2 haplotype. Second, all interchromatidal events were mediated by a single pair of SDs that were ∼145 kbp and had ∼99% identity, which accounts for 84% of the directly oriented SDs flanking the unique deleted sequence. In the two cases where we refined these breakpoints by using genome sequence data, the exact breakpoints differed but both localized to the same 99% identity segment. In both cases the rearrangements are predicted to disrupt KANSL1—for example, the family 2 breakpoints occur precisely in the first exon of this gene. It is noteworthy that the same duplications are highly stratified and have risen to high frequency in individuals of European descent.25
Figure 5.
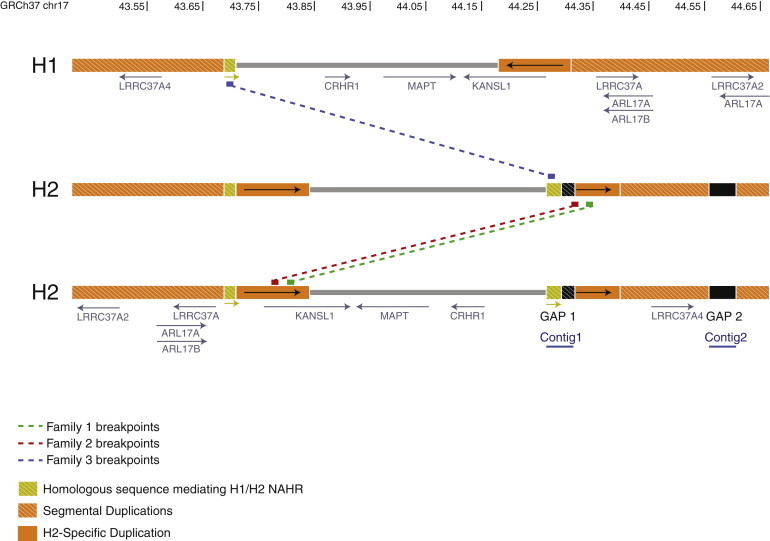
Summary of 17q21.31 Breakpoints on H1 and H2 Reference Assemblies
Sequence from breakpoint intervals was extracted from the H1 and H2 assemblies, aligned to the human reference sequence (GRCh37), and plotted on each haplotype. Coordinates represent the H1 haplotype on chromosome 17 (in Mbp), and hashed orange boxes represent segmental duplications. The H2-Specific Duplication, which contains sequence that mediates the NAHR event in family 1, is represented as solid orange blocks. Family 1 breakpoints were refined to a 4 kbp interval (green line) disrupting the first coding exon of KANSL1. The distal breakpoint of the microdeletion observed in family 2 (red line) falls in gap 1 (hashed black box) and has been refined to a 22 kbp interval within the 5′UTR of KANSL1. In addition, another segment of gap 1 sequence (hashed gold box) is homologous to H1 sequence that mediates the H1/H2 NAHR event leading to the microdeletion in family 3, which has been narrowed to a 24 kbp of perfect sequence identity. Gap 1 sequence has been resolved with fosmid clones, resulting in contig 1, and gap 2 sequence has been resolved with BAC clones, resulting in contig 2.
We also show that 17q21.31 deletions can occur as a result of interchromosomal NAHR between the H1 and H2 haplotypes. Our limited survey of 17q21.31 breakpoints indicates that interchromosomal NAHR is relatively uncommon. One case was previously identified,43 and we observed it independently twice in 18 probands, suggesting that such events account for ∼10% of 17q21.31 microdeletions. This is also compatible with previous population genetic data and theoretical predictions that crossovers between the H1 and H2 haplotypes are effectively suppressed. Interchromatidal deletions are probably more common than interchromosomal deletions for several reasons. First, sperm typing has shown NAHR due to interchromatidal deletions to be the predominant class of NAHR.49 Second, the interchromosomal paralogous segments mediating unequal crossover are smaller (40 kbp versus 145 kbp) and less numerous than those that can mediate interchromatidal NAHR. Finally, most crossover events between H1 and H2 in this region would be between allelic sequences in inverted orientation, creating the classic acentric and dicentric chromosomal products of a paracentric inversion, and are therefore inviable.
17q21.31 represents one of the most studied human genomic loci for which a complex alternate structural haplotype has been generated. Additional loci have either been implicated in pathogenic deletions or have been shown to have structural haplotypes predisposing an individual to such deletions.50,51 Unlike the 17q21.31 locus, none of these regions, to our knowledge, yet have haplotype-specific sequence assemblies. Although this presents a challenge, the methods we have developed provide a clear path forward to fine-mapping of breakpoints within segmental regions both in basic research and, ultimately, in a clinical setting. We propose the following strategy. In lieu of somatic cell hybrids, recently developed methods involving next-generation sequencing of flow-sorted chromosomes52 or pooled fosmids53 could be employed for the rapid generation of haplotype-specific sequence data, recovery of sequence information within the gaps, and discovery of large structural polymorphisms. Phased, locus-specific paralogous sequence variation could be generated through targeted sequencing of clone-based resources that now exist for more than 30 human genomes19,51,54 or through conventional46 or massively parallel sequencing53 methods. This would allow the establishment of high-quality alternate reference haplotypes of the human reference genome as is being pursued by the Genome Reference Consortium (Online Resources). These data could be used in the creation of a catalog of SUN identifiers so that breakpoints in deletion probands could be refined. Once such a catalog was established, it would be relatively trivial to routinely delineate the breakpoints of duplication and deletion probands with extraordinary precision by mapping complete genome sequencing to this catalog of sequence variants. This is important clinically for distinguishing breakpoints that are superficially similar (by array CGH) but that have different functional consequences with respect to breakpoints within duplicated genes or portions of genes (e.g., CHRNA7,55 SIRPB119 or KANSL1 [present study]). It is possible that these differences in breakpoints contribute to the variability of expressivity for genomic disorders and, as such, that it will be important to distinguish between them in the future.
Acknowledgments
We thank B. Coe, S. Ng and J. Hehir-Kwa for thoughtful discussion, T. Brown for assistance with manuscript preparation, A. Mackenzie, C. Igartua, C. Fields, S. Casadei, L. Vives, members of the Mayo Medical Laboratories, members of The Genome Institute at Washington University, and members of the Hubrecht Institute for assistance with data generation, and B. de Vries for clinical collection and evaluation of individuals with 17q microdeletions and their parents. K.M.S. was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) Fellowship (F32GM097807). This work was supported by National Institutes of Health grants HG002385 and HG004120 to E.E.E, and the Netherlands Organization for Health Research and Development (ZonMW 916.86.016 to L.E.L.M.V., and 917.66.363 to JAV). E.E.E. is an investigator of the Howard Hughes Medical Institute. E.E.E. is on the scientific advisory boards for Pacific Biosciences, Inc. and SynapDx Corp.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
The EMBOSS software suite, http://emboss.sourceforge.net/
Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
Genome Reference Consortium, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
JAligner Java implementation of the Smith-Waterman algorithm, http://jaligner.sourceforge.net/
Marshfield Genetic Maps, http://research.marshfieldclinic.org/genetics/
mrFAST, http://mrfast.sourceforge.net/
NCBI nucleotide database, http://www.ncbi.nlm.nih.gov/unists
NCBI BLAST and megaBLAST, http://blast.ncbi.nlm.nih.gov/
NCBI UniSTS database, http://www.ncbi.nlm.nih.gov/unists
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
RepeatMasker, http://www.repeatmasker.org/
Tandem Repeats Finder, http://tandem.bu.edu/trf/trf.html
UCSC Human Genome Browser (human reference genomes), http://genome.ucsc.edu
Accession Numbers
The NCBI nucleotide accession numbers for the four clone sequences reported in this paper are AC244161, AC244163, AC244164, and AC243906.
The GEO accession numbers for the nine microarray experiments in this paper are GSE34867.
References
- 1.Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 2.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vissers L.E., de Vries B.B., Veltman J.A. Genomic microarrays in mental retardation: From copy number variation to gene, from research to diagnosis. J. Med. Genet. 2010;47:289–297. doi: 10.1136/jmg.2009.072942. [DOI] [PubMed] [Google Scholar]
- 4.Girirajan S., Eichler E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010;19(R2):R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 6.Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 8.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharp A.J., Mefford H.C., Li K., Baker C., Skinner C., Stevenson R.E., Schroer R.J., Novara F., De Gregori M., Ciccone R. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries B.B., Pfundt R., Leisink M., Koolen D.A., Vissers L.E., Janssen I.M., Reijmersdal S., Nillesen W.M., Huys E.H., Leeuw N. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bochukova E.G., Huang N., Keogh J., Henning E., Purmann C., Blaszczyk K., Saeed S., Hamilton-Shield J., Clayton-Smith J., O'Rahilly S. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mefford H.C., Clauin S., Sharp A.J., Moller R.S., Ullmann R., Kapur R., Pinkel D., Cooper G.M., Ventura M., Ropers H.H. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 16.Chen K.S., Manian P., Koeuth T., Potocki L., Zhao Q., Chinault A.C., Lee C.C., Lupski J.R. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 17.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 18.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 19.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 21.Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pentao L., Wise C.A., Chinault A.C., Patel P.I., Lupski J.R. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- 24.Sudmant P.H., Kitzman J.O., Antonacci F., Alkan C., Malig M., Tsalenko A., Sampas N., Bruhn L., Shendure J., Eichler E.E., 1000 Genomes Project Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stefansson H., Helgason A., Thorleifsson G., Steinthorsdottir V., Masson G., Barnard J., Baker A., Jonasdottir A., Ingason A., Gudnadottir V.G. A common inversion under selection in Europeans. Nat. Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 26.Zody M.C., Jiang Z., Fung H.C., Antonacci F., Hillier L.W., Cardone M.F., Graves T.A., Kidd J.M., Cheng Z., Abouelleil A. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet. 2008;40:1076–1083. doi: 10.1038/ng.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker M., Litvan I., Houlden H., Adamson J., Dickson D., Perez-Tur J., Hardy J., Lynch T., Bigio E., Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 28.Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharp A.J., Itsara A., Cheng Z., Alkan C., Schwartz S., Eichler E.E. Optimal design of oligonucleotide microarrays for measurement of DNA copy-number. Hum. Mol. Genet. 2007;16:2770–2779. doi: 10.1093/hmg/ddm234. [DOI] [PubMed] [Google Scholar]
- 30.Benovoy D., Kwan T., Majewski J. Effect of polymorphisms within probe-target sequences on olignonucleotide microarray experiments. Nucleic Acids Res. 2008;36:4417–4423. doi: 10.1093/nar/gkn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee I., Dombkowski A.A., Athey B.D. Guidelines for incorporating non-perfectly matched oligonucleotides into target-specific hybridization probes for a DNA microarray. Nucleic Acids Res. 2004;32:681–690. doi: 10.1093/nar/gkh196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frazer K.A., Pachter L., Poliakov A., Rubin E.M., Dubchak I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z., Schwartz S., Wagner L., Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 35.Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 36.Igartua C., Turner E.H., Ng S.B., Hodges E., Hannon G.J., Bhattacharjee A., Rieder M.J., Nickerson D.A., Shendure J. Targeted enrichment of specific regions in the human genome by array hybridization. Curr. Prot. Hum. Genet. 2010 doi: 10.1002/0471142905.hg1803s66. Chapter 18, Unit 18.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alkan C., Kidd J.M., Marques-Bonet T., Aksay G., Antonacci F., Hormozdiari F., Kitzman J.O., Baker C., Malig M., Mutlu O. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conrad C., Andreadis A., Trojanowski J.Q., Dickson D.W., Kang D., Chen X., Wiederholt W., Hansen L., Masliah E., Thal L.J. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann. Neurol. 1997;41:277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- 41.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 42.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 43.Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 44.Trask B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat. Rev. Genet. 2002;3:769–778. doi: 10.1038/nrg905. [DOI] [PubMed] [Google Scholar]
- 45.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kidd J.M., Cheng Z., Graves T., Fulton B., Wilson R.K., Eichler E.E. Haplotype sorting using human fosmid clone end-sequence pairs. Genome Res. 2008;18:2016–2023. doi: 10.1101/gr.081786.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kidd J.M., Sampas N., Antonacci F., Graves T., Fulton R., Hayden H.S., Alkan C., Malig M., Ventura M., Giannuzzi G. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods. 2010;7:365–371. doi: 10.1038/nmeth.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper G.M., Nickerson D.A., Eichler E.E. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet. 2007;39(7, Suppl):S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 49.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharp A.J., Cheng Z., Eichler E.E. Structural variation of the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:407–442. doi: 10.1146/annurev.genom.7.080505.115618. [DOI] [PubMed] [Google Scholar]
- 51.Antonacci F., Kidd J.M., Marques-Bonet T., Teague B., Ventura M., Girirajan S., Alkan C., Campbell C.D., Vives L., Malig M. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat. Genet. 2010;42:745–750. doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan H.C., Wang J., Potanina A., Quake S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitzman J.O., Mackenzie A.P., Adey A., Hiatt J.B., Patwardhan R.P., Sudmant P.H., Ng S.B., Alkan C., Qiu R., Eichler E.E., Shendure J. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kidd J.M., Graves T., Newman T.L., Fulton R., Hayden H.S., Malig M., Kallicki J., Kaul R., Wilson R.K., Eichler E.E. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shinawi M., Schaaf C.P., Bhatt S.S., Xia Z., Patel A., Cheung S.W., Lanpher B., Nagl S., Herding H.S., Nevinny-Stickel C. A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat. Genet. 2009;41:1269–1271. doi: 10.1038/ng.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.