

Abstract
Syndromic diarrhea (or trichohepatoenteric syndrome) is a rare congenital bowel disorder characterized by intractable diarrhea and woolly hair, and it has recently been associated with mutations in TTC37. Although databases report TTC37 as being the human ortholog of Ski3p, one of the yeast Ski-complex cofactors, this lead was not investigated in initial studies. The Ski complex is a multiprotein complex required for exosome-mediated RNA surveillance, including the regulation of normal mRNA and the decay of nonfunctional mRNA. Considering the fact that TTC37 is homologous to Ski3p, we explored a gene encoding another Ski-complex cofactor, SKIV2L, in six individuals presenting with typical syndromic diarrhea without variation in TTC37. We identified mutations in all six individuals. Our results show that mutations in genes encoding cofactors of the human Ski complex cause syndromic diarrhea, establishing a link between defects of the human exosome complex and a Mendelian disease.
Main Text
Syndromic diarrhea (SD) is a rare and severe disease characterized by intractable diarrhea, facial dysmorphism, intrauterine growth retardation, immunodeficiency, and hair abnormalities.1 Trichohepatoenteric (THE [MIM 222470]) syndrome, described initially by Verloes et al. in 19972 as a different syndrome, has since been grouped with syndromic diarrhea because the main clinical features in both syndromes are identical3 (for simplicity, we now refer to both syndromes as a singular disorder, SD/THE syndrome). After a homozygosity-mapping analysis, we and others recently identified TTC37 mutations as being responsible for this syndrome in 21 individuals.4,5 The precise function of TTC37 (also called Thespin) was not elucidated even after the description of its involvement in SD/THE. This protein shares no sequence homology with other human proteins and shows no known functional domains except several tetratrico-peptide-repeat (TPR) domains that are structural motifs found in over 300 human proteins.6 In some databases, TTC37 is reported as being the ortholog of yeast SKI3, which encodes a key component of the Ski complex, a multiprotein complex required for exosome-mediated RNA surveillance.7 However, this lead was not explored in the initial studies. In our series, 6 out of 15 individuals did not carry a mutation in TTC37 but presented with typical characteristics of SD/THE syndrome. Considering the fact that TTC37 is homologous to Ski3p, we assumed that other genes encoding Ski-complex proteins might be responsible for SD/THE syndrome in these individuals; we confronted this hypothesis with the results of the linkage analysis of one of the consanguineous families.
Out of the six individuals with typical SD/THE syndrome and no mutation in TTC37, only one was previously reported.8 All procedures followed for clinical and genetic analyses in this study were in accordance with the ethical standards of the institutional and national committees on human experimentation, and proper informed consent was obtained from the parents of the affected children. All individuals presented with severe and intractable diarrhea that occurred between 1 and 12 weeks after birth, hair abnormalities (sparse, fragile, and uncombable hair and trichorrhexis nodosa), and facial dysmorphism characterized by hypertelorism, a broad flat nasal bridge, and a prominent forehead (Figure 1). All children received parenteral nutrition, but the amount of time varied between individuals and ranged from a few weeks to several years. Immunodeficiency was mostly due to low immunoglobulin levels and to the absence of an immune response to vaccines. One individual out of the three with immunodeficiency died from a measles infection. These six individuals harbor no mutation in TTC37, but their clinical presentation is undistinguishable from those who have TTC37 mutations (Table 1).
Figure 1.
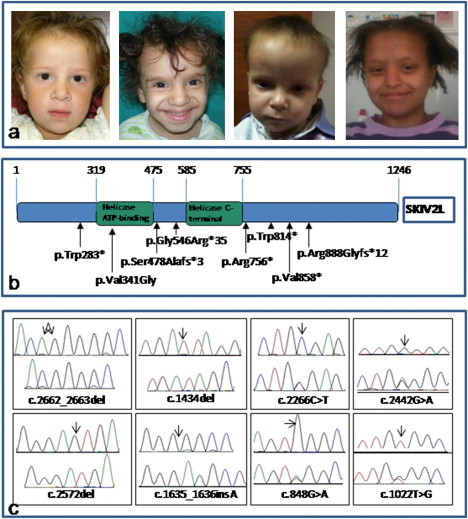
Clinical Presentation of Individuals with SD/THE Syndrome and SKIV2L Mutations
(A) Clinical presentation of four individuals with SD/THE syndrome. One individual has mutations in TTC37 (first picture on the left), and three others have mutations in SKIV2L (right panel).
(B) A schematic overview of the predicted SKIV2L structure with a helicase ATP-binding domain and a helicase C-terminal domain (UniProt). The location of the domains is annotated by amino acid position. The eight amino acid substitutions are indicated by arrows.
(C) Nucleotide sequences are shown for the controls (upper sequence) and for each SKIV2L variant (lower sequence). Arrows indicate mutation positions on the reference sequences.
Table 1.
Clinical Data of Individuals Affected by SD/THE Syndrome
| Individuals with Mutations in TTC37 (n = 18) | Individuals with Mutations in SKIV2L (n = 6) | |
|---|---|---|
| Premature birth (<37 weeks) | 9/17 | 2/5 |
| Intrauterine growth restriction | 14/17 | 4/6 |
| Birth weight (median and mean) in kg | 1.84 (0.78–3.58); 1.868 | 1.6 (1.01–2.00); 1.47 |
| Intractable diarrhea | 18/18 | 6/6 |
| Onset of diarrhea (median and mean) in weeks | 3.5 (1–32); 7.75 | 2.5 (1–12); 3.8 |
| Villous atrophy | 16/18 | 3/5 |
| Colitis | 5/6 | 3/3 |
| Facial dysmorphism | 18/18 | 6/6 |
| Hair abnormalities | 18/18 | 6/6 |
| Trichorrhexis nodosa | 17/18 | 5/5 |
| Immune deficiency | 17/18 | 3/6 |
| Liver disease | 9/16 | 3/6 |
| Siderosis | 3/14 | 1/3 |
| Cirrhosis | 7/15 | 2/3 |
| Skin abnormalities | 7/16 | 3/4 |
| Platelet abnormalities | 7/17 | 0/2 |
| Cardiac abnormalities | 4/16 | 2/4 |
| Outcome (deceased/alive) | 4/14 | 2/4 |
Searching for a candidate gene in this group, we investigated the possible homology (reported in the Ensembl database) between TTC37 and yeast SKI3. Interspecies sequence-alignment analysis (with the bioinformatics prediction software BLAST) revealed that TTC37 shares significant amino-acid-sequence similarity with yeast Ski3p. Between TTC37 residues 5 and 84, TTC37 and Ski3p were 35% identical and 58% similar, and between residues 488 and 1223, they were 20% identical and 38% similar, indicating that TTC37 is the human ortholog of SKI3. Because Ski3p is part of a multiprotein complex, this finding prompted us to test whether mutations in other human orthologs of the Ski proteins could be associated with SD/THE syndrome. For one consanguineous family, we performed a linkage analysis in which the TTC37 critical interval was excluded, and this analysis revealed that there was a region of homozygosity spanning nucleotides 9,138,488–40,453,008 and containing 856 genes in chromosomal region 6p21.2–6p24.3. When looking for functional candidate genes in this region, we noticed that SKIV2L (RefSeq accession number NM_006929.4), a gene described as being the human ortholog of the SKI2,9,10 was present in region 6p21.3. Thus, we performed direct sequencing of SKIV2L in our cohort of six individuals affected by typical SD/THE syndrome and found the presence of mutations predicted to be deleterious in all six. The identified mutations and their mode of inheritance are described in Table 2. All of the unaffected parents available for analysis were heterozygous for one of the mutations identified in their child. We identified eight different variations distributed throughout the protein (Figure 1). Seven of these variations correspond to nonsense or frameshift mutations that introduce a premature termination codon, and they are c.848G>A (p.Trp283∗), c.1434del (p.Ser479Alafs∗3), c.1635_1636insA (p.Gly546Argfs∗35), c.226C>T (p.Arg756∗), c.2442G>A (p.Trp814∗), c.2572del (p.Val858∗), and c.2662_2663del (p.Arg888Glyfs∗12). The mutation types suggest that the disease mechanism is loss of function. We identified one missense mutation, c.1022T>G (p.Val341Gly), that is located in the helicase ATP-binding domain (Figure 1) and concerns a highly conserved residue. PolyPhen predicted that this specific mutation is probably damaging (the prediction score was 2.8 [mutations predicted to be pathological have an index above 0.5]).
Table 2.
Mutations in SKIV2La, Geographical Origins, and Consanguinity in the Families of Individuals with SD/THE Syndrome
| Individual | Mutation 1 | Mutation 2 | Consanguinity | Geographical Origin |
|---|---|---|---|---|
| 1 | c.1635_1636insA (p.Gly546Argfs∗35) | c.1635_1636insA (p.Gly546Arg∗35) | yes | North Africa |
| 2 | c.2266C>T (p.Arg756∗) | c.2442G>A (p.Trp814∗) | no | France |
| 3 | c.848G>A (p.Trp283∗) | c.1022T>G (p.Val341Gly) | no | France |
| 4 | c.2572del (p.Val858∗) | c.2572del (p.Val858∗) | yes | Turkey |
| 5 | c.2662_2663del (p.Arg888Glyfs∗12) | c.2662_2663del (p.Arg888Glyfs∗12) | yes | Turkey |
| 6 | c.1434del (p.Ser479Alafs∗3) | c.1434del (p.Ser479Alafs∗3) | yes | Turkey |
RefSeq accession numbers NP_008860.4 and NM_006929.4.
It is now clear that the majority of genomic information is transcribed into RNA molecules; this process generates very abundant and complex pools of RNAs that the cells have to control. While performing numerous RNA-processing reactions, the cell must, at the same time, eradicate surplus and aberrant material.11 This task is, at least partially, performed by the RNA exosome multiprotein complex that contains both 3′-to-5′ exonuclease and 3′-to-5′ endonuclease activity and acts in both the nucleus and the cytoplasm.12 Initially discovered in yeast but also present in higher eukaryotes, including humans, this exosome complex is involved in the decay pathways of normal mRNA but is also required to maintain the fidelity of gene expression through RNA surveillance processes, such as nonsense-mediated decay, nonstop decay, and no-go decay.13–16 Genetic screens in yeast have identified three proteins (Ski2p, Ski3p, and Ski8p) that form the Ski (Superkiller) complex and act as specific cofactors required for all the functions of the cytoplasmic exosome but not the nuclear exosome.17 The putative DExH-box RNA helicase activity of Ski2 suggests that it is the only Ski protein with a catalytic function, whereas Ski3p and Ski8p contain repeated domains thought to be needed for protein-protein interactions.18 In humans, the cytoplasmic exosome is involved in various mRNA decay pathways and is required for normal cell growth.19
Here, we show that in six families, molecular defects in the cytoplasmic-exosome cofactor SKIV2L cause SD/THE syndrome, a severe autosomal-recessive condition mainly characterized by intractable diarrhea and woolly hair. This study establishes a formal link between a constitutional congenital disease and defects of human cytoplasmic-exosome cofactors. Although SD/THE syndrome is genetically heterogeneous and is associated with at least two different genes, it is extremely homogenous at the clinical level (Table 1), suggesting that a defect in Ski-complex function or structure is a key mechanism responsible for the main clinical features. Consequently, WDR61, the human ortholog of the third cofactor SKI8, will be a relevant candidate gene to be tested in persons that are affected by THE syndrome and that have no mutation in TTC37 or SKIV2L sequences (a situation not encountered in our series).
In human pathology, the exosome complex has previously been involved in autoimmune diseases, in which components of the nuclear or cytoplasmic exosome are the target of autoimmune response, or in cancer.20 Here, we point out that exosome dysfunction must be considered a cause of Mendelian disorders. The association between mutations in exosome-cofactor-encoding genes and human diseases provides a valuable model for investigating the role of this structure in human pathology but also in normal cellular function. The mechanism by which mRNA-surveillance defects lead to various clinical symptoms, such as severe diarrhea, hair abnormalities, or immunodeficiency, will need to be investigated in further studies.
Acknowledgments
We are extremely grateful to the individuals with trichohepatoenteric syndrome and the family members for their participation in the study. We also want to acknowledge Sylvain Baulande and Pascal Soularue (from Partnerchip), who kindly performed bioinformatics analysis after the linkage studies, Julies Salomon, who provided DNA samples, and Laurent Villard for helpful discussion. DNA extraction and storage were performed in the Biobank of the Department of Genetics of La Timone Hospital. This work was financially supported by the Assistance Publique-Hôpitaux de Marseille (AORC 2010). A.F. is supported by a scholarship from the Fondation de l'Université de la Méditerranée.
Web Resources
The URLs for the data presented herein are as follows:
Ensembl, http://www.ensembl.org
Homozygositymapper, http://www.homozygositymapper.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Polyphen, http://genetics.bwh.harvard.edu/pph/
UniProt, http://www.uniprot.org
References
- 1.Girault D., Goulet O., Le Deist F., Brousse N., Colomb V., Césarini J.P., de Potter S., Canioni D., Griscelli C., Fischer A. Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency. J. Pediatr. 1994;125:36–42. doi: 10.1016/s0022-3476(94)70118-0. [DOI] [PubMed] [Google Scholar]
- 2.Verloes A., Lombet J., Lambert Y., Hubert A.F., Deprez M., Fridman V., Gosseye S., Rigo J., Sokal E. Tricho-hepato-enteric syndrome: Further delineation of a distinct syndrome with neonatal hemochromatosis phenotype, intractable diarrhea, and hair anomalies. Am. J. Med. Genet. 1997;68:391–395. doi: 10.1002/(sici)1096-8628(19970211)68:4<391::aid-ajmg3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 3.Fabre A., André N., Breton A., Broué P., Badens C., Roquelaure B. Intractable diarrhea with “phenotypic anomalies” and tricho-hepato-enteric syndrome: Two names for the same disorder. Am. J. Med. Genet. A. 2007;143:584–588. doi: 10.1002/ajmg.a.31634. [DOI] [PubMed] [Google Scholar]
- 4.Hartley J.L., Zachos N.C., Dawood B., Donowitz M., Forman J., Pollitt R.J., Morgan N.V., Tee L., Gissen P., Kahr W.H. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy) Gastroenterology. 2010;138:2388–2398. doi: 10.1053/j.gastro.2010.02.010. 2398, e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabre A., Martinez-Vinson C., Roquelaure B., Missirian C., André N., Breton A., Lachaux A., Odul E., Colomb V., Lemale J. Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome. Hum. Mutat. 2011;32:277–281. doi: 10.1002/humu.21420. [DOI] [PubMed] [Google Scholar]
- 6.Blatch G.L., Lässle M. The tetratricopeptide repeat: A structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 7.Brown J.T., Bai X., Johnson A.W. The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA. 2000;6:449–457. doi: 10.1017/s1355838200991787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egritas O., Dalgic B., Onder M. Tricho-hepato-enteric syndrome presenting with mild colitis. Eur. J. Pediatr. 2009;168:933–935. doi: 10.1007/s00431-008-0861-4. [DOI] [PubMed] [Google Scholar]
- 9.Lee S.-G., Lee I., Park S.H., Kang C., Song K. Identification and characterization of a human cDNA homologous to yeast SKI2. Genomics. 1995;25:660–666. doi: 10.1016/0888-7543(95)80008-a. [DOI] [PubMed] [Google Scholar]
- 10.Dangel A.W., Shen L., Mendoza A.R., Wu L.-C., Yu C.Y. Human helicase gene SKI2W in the HLA class III region exhibits striking structural similarities to the yeast antiviral gene SKI2 and to the human gene KIAA0052: Emergence of a new gene family. Nucleic Acids Res. 1995;23:2120–2126. doi: 10.1093/nar/23.12.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 12.Houseley J., Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Schmid M., Jensen T.H. The exosome: A multipurpose RNA-decay machine. Trends Biochem. Sci. 2008;33:501–510. doi: 10.1016/j.tibs.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Parker R., Song H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- 15.Zhu B., Mandal S.S., Pham A.D., Zheng Y., Erdjument-Bromage H., Batra S.K., Tempst P., Reinberg D. The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev. 2005;19:1668–1673. doi: 10.1101/gad.1292105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toh-E A., Wickner R.B. “Superkiller” mutations suppress chromosomal mutations affecting double-stranded RNA killer plasmid replication in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1980;77:527–530. doi: 10.1073/pnas.77.1.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaeffer D., Clark A., Klauer A.A., Tsanova B., van Hoof A. Functions of the cytoplasmic exosome. Adv. Exp. Med. Biol. 2010;702:79–90. [PubMed] [Google Scholar]
- 18.Wang L., Lewis M.S., Johnson A.W. Domain interactions within the Ski2/3/8 complex and between the Ski complex and Ski7p. RNA. 2005;11:1291–1302. doi: 10.1261/rna.2060405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dijk E.L., Schilders G., Pruijn G.J. Human cell growth requires a functional cytoplasmic exosome, which is involved in various mRNA decay pathways. RNA. 2007;13:1027–1035. doi: 10.1261/rna.575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staals R.H., Pruijn G.J. The human exosome and disease. Adv. Exp. Med. Biol. 2010;702:132–142. [PubMed] [Google Scholar]