

Abstract
Virus infection initiates a number of cellular stress responses that modulate gene regulation and compartmentalization of RNA. Viruses must control host gene expression and the localization of viral RNAs to be successful parasites. RNA granules such as stress granules and processing bodies (PBs) contain translationally silenced messenger ribonucleoproteins (mRNPs) and serve as extensions of translation regulation in cells, storing transiently repressed mRNAs. New reports show a growing number of virus families modulate RNA granule function to maximize replication efficiency. This review summarizes recent advances in understanding the relationship between viruses and mRNA stress granules in animal cells and will discuss important questions that remain in this emerging field.
Keywords: stress granule, antiviral response, translation silencing, G3BP, TIA-1, eIF4G, PKR.
Stress granule formation and composition
Eukaryotic cells can contain multiple types of cytoplasmic mRNA-containing bodies, including processing bodies (PBs, also known as GW bodies) [1], exosome bodies 2, 3, neuronal bodies 4, 5 and stress granules (SGs) 6, 7. PBs and exosome granules are foci that are constitutively present in cells and contain components involved in mRNA decay 3, 8. Neuronal granules are also constitutively present in neurons but are instead associated with the concentration and transport of translationally silenced messenger ribonucleoproteins (mRNPs) moving along the axons to dendrites [5]. SGs are not constitutively present in cells, but similar to neuronal granules, SGs are concentrations of stable, translationally silent mRNA [9] that are thought to be sites of mRNA storage and triage [10]. SGs and PBs are found in the widest number of cells types. Although PBs are known to be modulated by some viruses, this review will focus on the many more publications describing viral modulation of stress granules.
Based on immunofluorescent microscopic analysis of SG constituents, SGs are defined as macromolecular aggregates of stalled 48S initiation complexes that form in response to stress conditions [11]. The best described pathway of SG formation initiates with phosphorylation of eukaryotic translation initiation factor (eIF) 2α (eIF2α) by the eIF2 kinases PKR, PERK, GCN2 or HRI 12, 13, 14, although alternative pathways exist such as inhibition of eIF4A RNA helicase 15, 16, 17 or viral infection [15]. PKR, a component of the interferon response, is commonly activated by RNA viruses producing double-stranded RNA as replication intermediates and PERK is activated by endoplasmic reticulum (ER) stress associated with a smaller group of viruses, many that express membrane glycoproteins (e.g. herpes viruses and others). HRI, activated by heme deprivation and oxidative stress, and GCN2, which is activated by nutrient starvation, are not commonly linked to virus infection, although GCN2 binding to Sindbis virus RNA induces its activation [18]. SG are foci of concentrated 48S translation preinitiation complexes, thus SGs are defined by the presence of translation initiation machinery including 40S ribosome subunits, eIF2, eIF3, eIF4A, eIF4B, eIF4E, eIF4G and eIF5 13, 15, 19, 20. SGs are also defined by certain key marker RNA binding proteins (RBPs) such as T-cell restricted intracellular antigen 1 (TIA-1), TIA-1-related protein (TIAR) and RasGAP SH3-domain binding protein 1 (G3BP1) 14, 21, however, SGs contain many other RBPs (Figure 1 ). Because SGs contain stable inert mRNA, they represent an intermediate step in the equilibrium between active translation that occurs on free polysomes and mRNA decay, which takes place in PBs. As such, they dynamically release contents for active translation 22, 23, 24, 25 as well as interact with PBs in a process that is thought to result in the exchange of mRNA ‘cargos’ [23]. The movement of RBPs between compartments is rapid, with a full replacement of some SG contents occurring in well under a minute 22, 23. Other evidence suggests that association of mRNA with the ER renders the mRNA resistant to inclusion in SGs [26]. Frequent interaction of SGs with PBs is observed in cells that are actively forming SGs and live cell imaging shows that this process is dynamic and transient [23]. Little is known about the mechanism or purpose of this interaction other than the proposed mRNP cargo exchanges (Figure 1), but the overexpression of tristetraprolin (TTP) and related protein BRF1 is known to promote and stabilize the association of SGs and PBs [23].
Figure 1.
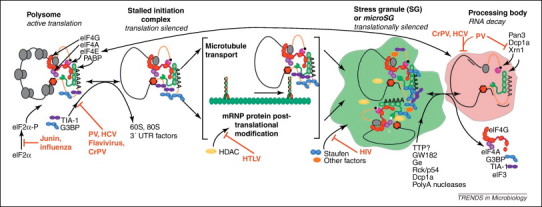
Stress granules (SGs) are intermediate compartments in mRNA metabolism. Inhibition of translation initiation leads to the disassembly of polysomes and the formation of stalled 48S initiation complexes. These messenger ribonucleoprotein (mRNP) complexes are recognized via an unknown mechanism and are remodeled, marking them for inclusion in SGs despite continued association with pro-translation initiation factors. SG components such as RasGAP SH3-domain binding protein 1 (G3BP1), Fragile X mental retardation protein (FMRP) and others are post-translationally modified, and small dispersed aggregates of remodeled mRNP complexes are transported by microtubule-associated motor proteins into larger SGs. The brackets around this central step indicate that it is not currently known which process is initially undertaken. SGs are thought to be sites of storage of stabilized mRNA, although it is known that mRNA can be released for translation or transported to processing bodies (PBs) for active decay by an unknown mechanism. Multiple virus systems (in red) have been found to interfere with the process of SG and PB formation and the points of interaction with the process are indicated. Stress granules also dock with PBs where mRNP modification and cargo exchange takes place. Initiation factors are lost except eukaryotic translation initiation factor (eIF4E) and deadenylase complexes (Pan2/3, Caf1/Ccr4) decapping complexes (Dcp1a/2) and exonucleases (Xrn1) become associated. Some viruses inhibit PB formation as indicated and poliovirus (PV) antagonizes specific PB components 43, 45, 71.
The molecular mechanism(s) [27] by which SGs form is undefined, but appears complex and involves several steps that include the self-oligomerization of certain constituent RNA-binding proteins, post-translational modifications of proteins and mRNP transport on microtubules (Table 1 , Figure 1). Theoretically, viral inhibition of any of these important steps may block or modulate SG formation in cells. Self-oligomerization of TIA-1 or TIAR and G3BP may play a crucial early role in the SG aggregation process and overexpression of these proteins induces spontaneous SG formation 21, 28. Expression of the C-terminal glutamine-rich prion related domain (PRD) of TIA-1 inhibits the formation of SGs and overexpression of TIA-1 lacking the PRD does not spontaneously induce SGs [28]. Additionally, murine embryonic fibroblasts (MEFs) that are null for TIA-1 or TIAR display deficient SG formation in response to various stressors [28]. G3BP can self-oligomerize in a phosphorylation-dependent manner and overexpression of the central domain of G3BP containing the arginine-rich and PxxP domains inhibits SG formation [21]. As is the case with TIA-1, cells with G3BP repressed by siRNA treatment [29] and G3BP–/– MEFs are also deficient in the formation of SGs [21]. Additionally, G3BP sequestration by the inactive kinase MK-STYX inhibits the formation of SGs in response to arsenite and G3BP overexpression [30], further confirming the importance of G3BP in SG formation. The ability of G3BP to mediate the formation of SGs is regulated by the phosphorylation of serine 149 by an unknown kinase. Overexpression of a phosphomimetic mutant, G3BPS149E, inhibits the formation of SGs whereas an exogenously expressed non-phosphorylatable mutant, G3BPS149A, localizes to SGs similarly to wild type [21]. Viral interference with G3BP and TIA-1 will be discussed below.
Table 1.
Stress granule mechanistic processes that viruses can potentially modulatea
| Process | How it can be modulated | Refs. |
|---|---|---|
| Cell Insult | ||
| Inhibit translation ternary complex formation | Phosphorylate eIF2α via activation of PKR, HRI, GCN2 and PERK | [14] |
| Block eIF4F function (scanning) | Hippuristanol inhibition of eIF4A, viral cleavage of eIF4G | [15] |
| Assembly/mRNP infiltration of key proteins | ||
| G3BP | RNA-binding protein can self-oligomerize, may sequester mRNA in SG, overexpression of mutants blocks SG formation | [21] |
| TIA-1, TIAR | RNA-binding protein can self-oligomerize, may sequester mRNA in SG, overexpression of mutants blocks SG formation | 14, 22, 28 |
| HDAC6 | Deacetylase function and SG infiltration associated with SG assembly | [36] |
| Movement and post-translational modifications of proteins | ||
| Microtubule transport | Required for assembly but not maintenance | 31, 32, 33, 34, 35, 36 |
| O-GlcNAc on ribosomal proteins | Required for SG assembly, multiple proteins modified | [72] |
| Acetylation | HDAC6 function associated with SG assembly | [36] |
| Methylation | Methylation is recruitment tag that controls SG assembly TDRD3 tudor domain interacts with methylated proteins Cold-inducible RNA binding protein (CIRBP) methylation controls nuclear translocation and SG entry FMRP methylation for RNA binding and SG localization |
[73] [74] [75] [76] |
| Phosphorylation/dephosphorylation | G3BP dephosphorylation required for SG assembly Grb7 non-phosphorylatable double mutant stabilizes SGs, focal adhesion kinase mutant stabilizes SGs |
[21] [77] |
| Ubiquitination | SGs contain ubiquitinated proteins, Ub-binding domain of HDAC6 required for localization to SGs | [36] |
| Interruption of SG disassembly | ||
| OGFOD | Interacts with G3BP, HRI, regulates eIF2α phosphorylation | [78] |
Abbreviated list only, particularly in terms of factors that infiltrate SGs.
In addition to the steps described above, SG formation involves post-translational modifications of several other proteins that regulate SG dynamics in complex, possibly hierarchical stages. These modifications include O-linked N-acetylglucosamine (O-Glc-Nac) modification, methylation, acetylation and phosphorylation and are summarized in Table 1. Some of these steps are less well characterized and have not yet been investigated by virologists in their systems, however, are potential targets for viral manipulation. Finally, multiple reports indicate that SG formation is mediated by microtubules (MTs) and its associated motor proteins dynein/dynactin and kinesin 31, 32, 33, 34, 35, 36. Disassembly of microtubules by pharmacological treatment abolishes the formation and dissolution of SGs, resulting in the formation of small, dispersed SGs at the onset of stress and prolonging their presence in cells recovering from stress 32, 33. However, MTs do not affect their maintenance once formed 31, 32. Inhibition of the motor proteins dynein/dynactin and kinesin similarly resulted in small dispersed puncta and extended SGs maintenance during stress recovery 34, 35. Taken together, the mechanism of SG formation appears to be multifactorial and may involve multiple types of protein modifications on key targets and RBP interactions with the cellular cytoskeleton. Many of these steps may be modulated by viruses.
Viral interactions with stress granules
The basic role of SGs and PBs in translation suppression and RNA decay suggest these processes will impact virus replication and force viral adaptation. Virus infection will induce stress responses on multiple levels as host processes are interrupted or co-opted. Indeed, numerous types of RNA viruses are now reported to manipulate SGs, reflecting the fact that SGs are involved in RNA silencing and storage, however DNA viruses also modulate SG responses. Viral interactions with the SG pathway produce varying phenotypes (Table 2 ). In general, most viruses appear to antagonize SG formation during infection, although some induce and may exploit portions of SG responses as part of the infectious cycle. For broad understanding of readers, we have provisionally categorized these virus systems discussed below into three classes according to the phenotype of virus interaction with the SG machinery. However, because there are conflicting data in some cases and overall the interactions are not yet probed in depth in most virus systems, these groupings may require revision with time.
Table 2.
Phenotypes of virus–SG interactions
| Virus | Phenotype | Refs |
|---|---|---|
| Virus induces then inhibits SGs | ||
| Mammalian orthoreovirus | SG formation induced by viral binding | [37] |
| Semliki Forest Virus | SG formation corresponds with host shutoff | [40] |
| Hepatitis C virus | SG components localize to HCV core-containing structures | [43] |
| Poliovirus | SGs induced early, correlated with translation shutoff | [15] |
| Poliovirus | SG inhibition due to G3BP1 cleavage | [29] |
| Poliovirus | Virus-induced vs. stress-induced SGs contain unique components | [41] |
| Poliovirus | G3BP cleavage unlinks TIA-1 aggregates from stalled initiation complexes | [42] |
| Virus inhibits SGs | ||
| Junin virus | Inhibition of eIF2α phosphorylation and SG by nucleoprotein N and GPC | [49] |
| Rotavirus | RV infection induces extended eIF2α phosphorylation; SGs not induced | [46] |
| Cardiovirus | Inhibition due to expression of the L protein | [48] |
| West Nile and dengue virus | TIA-1 interaction with viral genome inhibits SG formation | [51] |
| Cricket paralysis virus | Poly(A) mRNA form foci with PABP that lack other protein markers | [45] |
| Herpes simplex 1 | TIA-1 localizes to cytoplasm but not SGs; Vhs mutant virus forms SGs | [57] |
| Herpes simplex 1 | Vhs mutant viruses induce SGs in cell type-dependent manner | [56] |
| HIV-1 | Staufen–Gag interaction block SGs in favor of encapsidation | [54] |
| HTLV-1 | Tax protein sequesters HDAC6 from required SG functions | [55] |
| Influenza A virus | NS1 protein blocks eIF2α phosphorylation and SG formation | [50] |
| Virus tolerates or exploits SG responses | ||
| Respiratory syncytial virus | SG induction associated with increased viral replication | [27] |
| Respiratory syncytial virus | SG induction is PKR-dependent | [59] |
| Respiratory syncytial virus | No SGs in wild-type infection, only in trailer-deficient mutant viruses | [60] |
| Mouse hepatitis coronavirus | SG formation corresponds to translation inhibition | [47] |
| Transmissible gastroenteritis virus | PTB localization to SGs corresponds to increased replication | [58] |
| Mammalian orthoreovirus | SG induction is strain-specific and correlates with host shutoff | [39] |
| Vaccinia virus | SG-like bodies form in proximity to replication factories promoting translation | [62] |
| Vaccinia virus | Antiviral SG bodies form with ΔE3L mutant virus | [61] |
Viral inhibition of SGs in the mid-phase of infection
Mammalian orthoreoviruses (MRV) induce the formation of SGs via an eIF2α phosphorylation-dependent mechanism in a strain and cell-type-dependent manner, a phenotype due to the ability of strains to differentially enter cells. SGs are triggered by viral entry because infection with UV-inactivated particles or intermediate subvirion particles (ISVPs) induces SGs in a dose-dependent manner [37]. Further, viral gene expression was not required for SG formation but instead led to the inhibition of SGs as infection progressed [37]. Several strains of MRV inhibited SG formation in response to arsenite and other treatments despite producing high levels of eIF2α phosphorylation that should trigger SG formation, thus indicating MRV inhibits the formation of SGs downstream of eIF2 [38]. The ability of MRV to translate under stress conditions created by exogenous stressors correlated with the absence of SGs. Both cellular and viral translation was inhibited early when SG form, but viral translation was not blocked late when SGs were inhibited.
By contrast, another group analyzed different reovirus strains, two that inhibit host cell translation [clone 8 (c8) and c87] and the Dearing strain that does not, and found that SGs persist throughout infection. Additionally, they found that the ability of the reovirus strains to induce SG formation correlated with the strength of eIF2α phosphorylation and host cell translation inhibition, with the Dearing strain inducing the least eIF2α phosphorylation and a lower level induction of SG formation [39]. Together, these results indicate different virus strain–cell combinations may score differently in controlling SG responses, providing experimental tools to probe the most important governing mechanisms.
The alphavirus Semliki Forest virus (SFV) also induces SGs early during infection in an eIF2α phosphorylation-dependent manner, concomitant with the inhibition of host protein synthesis [40]. Infection of TIA-1-null MEFs resulted in delayed kinetics of host translation inhibition, suggesting that SFV permits some SG formation to aid translation shutoff. Like MRV, SFV prevented the formation of SG by exogenous stressors at late times post-infection. Contrary to MRV, SFV induction of SGs required viral replication, and there was a correlation between the levels of viral RNA (vRNA) staining and SGs. Interestingly, in cells with low vRNA content, SGs were still present in cytoplasmic areas that were not in close proximity to vRNA, suggesting SG formation was inhibited by a process closely linked to viral replication [40].
Poliovirus (PV) induces the formation of SGs in some cells early during infection in an eIF2-independent manner, unlike MRV and SFV. Formation of SGs containing G3BP and eIF4GI peaked between 2 and 3 hours post-infection (hpi) 15, 29 and then declined as infection matured [29] by a mechanism that requires viral replication. Similar to MRV and SFV, PV inhibits the formation of canonical SGs in response to exogenous stressors such as arsenite. In a report thought to be contrary to previously described PV-induced inhibition of SG, TIA-1-containing foci were observed late in virus infection and correlated with stable SGs [41]. However, it was later shown that TIA-1-positive foci persisting in PV-infected cells are actually devoid of other SG defining components such as initiation factors and most mRNA and thus do not represent canonical SGs [42]. Thus, PV gene products are able to unlink the process of TIA-1 aggregation from sequestration of stalled translation initiation complexes, thereby releasing sequestered translation apparatus to support viral translation. PV inhibition of SG is primarily mediated by the viral 3Cpro-mediated cleavage of the SG component G3BP, which separates the G3BP RNA-binding and protein-interaction domains (Figure 1, Figure 2 ). Rescue of formation of bonafide SG containing initiation factors and mRNA via the expression of cleavage-resistant G3BPQ326E led to an approximately sevenfold decrease in viral replication, indicating a potential antiviral role for SGs 29, 42.
Figure 2.
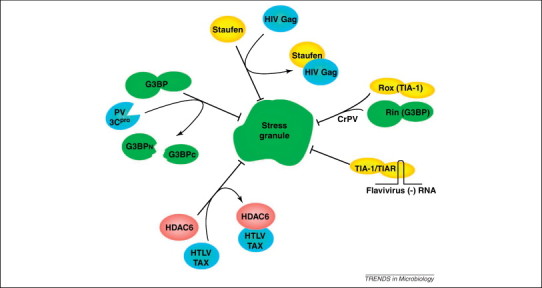
Inhibition of stress granule (SG) formation by several viral systems has been characterized in more detail and two types of mechanisms are illustrated: cleavage or sequestration of SG components. Cleavage of RasGAP SH3-domain binding protein 1 (G3BP1) by poliovirus (PV) 3C proteinase inhibits the inclusion of translation initiation factors, mRNA binding proteins and mRNA in SGs without modulating the ability of T-cell restricted intracellular antigen 1 (TIA-1) and TIA-1-related protein (TIAR) to aggregate. Human T-cell leukemia virus type-1 (HTLV-1) Tax protein interacts with histone deacetylase 6 (HDAC6) to inhibit SG formation, whereas the HIV Gag protein binds to Staufen1. The 3′ stem loop present in flavivirus negative sense RNA binds and sequesters TIA-1 and TIAR, leading to the inhibition of SGs. Through unknown mechanisms, cricket paralysis virus (CrPV) prevents accumulation of Drosophila homologs of TIA1 (Rox8) and G3BP (Rin) in SGs.
Hepatitis C virus (HCV) is also reported to induce SGs in response to infection, but inhibits their formation in response to exogenous stress stimuli as infection proceeds [43]. Unlike MRV, SFV and PV, HCV specifically recruits components of SGs to the viral replication factories (RFs) and several SG markers continue to co-localize with the HCV core protein even in the presence of stress [43]. This is likely due to the fact that G3BP1, and possibly other factors, interact with the viral NS5B protein and the 5′ terminus of the (–) strand RNA during HCV infection to mediate efficient replication of the viral genomic RNA [44]. Interestingly, SG constituents G3BP and ataxin2, plus PB component DDX6 and HCV core protein can be recruited to ring-like structures surrounding lipid droplets in cells [43].
Finally, cricket paralysis virus (CrPV), a member of the Dicistroviridae family, also modulates SG formation during infection by preventing the inclusion of Rox8 and Rin (Figure 1), the Drosophila homologues of TIA-1 and G3BP, respectively, but not polyadenylated mRNA or poly(A)-binding protein (PABP) [45]. Even after treatment with various stressors, CrPV-infected cells still maintain a diffuse distribution of Rox8 and Rin while forming polyA- and PABP-positive granules, indicating that modifications mediated by CrPV leads to selective inhibition of distinct SG markers [45]. Although there are differences, these results show that a basic phenotype of virus induction and repression of SGs is highly conserved between insect and animal viruses.
Viral inhibition of SGs throughout infection
In many viral systems, SGs are not readily observed during infection with wild-type virus and infection inhibits the formation of SGs in response to eIF2α phosphorylation and treatment with exogenous stressors. In some cases a viral mediator of SG inhibition has been implicated, but detailed mechanisms are lacking.
Rotavirus (RV) infection activates phosphorylation of eIF2α but not SG formation and infected cells do not form SGs in response to arsenite treatment [46]. Interestingly, eIF2α phosphorylation was not required for efficient RV replication but could be used to inhibit host cell protein synthesis at the expense of viral efficiency 46, 47 because infected MEFs expressing phosphorylation-null mutant eIF2αS51A translate more efficiently.
Cardioviruses, specifically Theiler's murine encephylomyelitis virus (TMEV), were similar and inhibited SG formation both in response to infection as well as to exogenous stress [48]. The TMEV leader protein (L) was linked to SG inhibition because viruses with mutant L proteins induce SG formation throughout infection. Ectopic expression of the L protein alone inhibited SGs in response to exogenous stress, and L proteins from other cardioviruses (Mengovirus and Saffold virus) also blocked SG formation [48].
Acute Junin virus infection blocks phosphorylation of eIF2α in response to arsenite treatment by an undefined mechanism that was dependent on the expression of the nucleoprotein (N) or glycoprotein precursor (GPC) [49]. However, persistently infected Vero cells expressing truncated N and low levels of GPC displayed SG formation phenotypes similar to uninfected cells [49].
Influenza A virus infection fails to induce SGs unless viruses with NS1 mutations are used. In this case, SGs form readily in a PKR-dependent fashion. Influenza virus NS1 protein inhibits PKR activation. Formation of SG was linked to repression of virus replication, but this could not be unlinked from negative effects of eIF2a phosphorylation on virus translation [50].
West Nile virus (WNV) and dengue virus (DV), both members of the Flaviviridae family, inhibit SG formation in response to exogenous stress by sequestering TIA-1 and TIAR through specific binding of either protein to the minus strand 3′-terminal stem loop structure (3′(–)SL) [51], an interaction that is required for viral replication [52] (Figure 2). DV 3′ UTR and 5′ UTR pulls down SG proteins G3BP1, caprin1 and USP10 as well as PB marker protein DDX6 (RCK/p54), and all these proteins were found to weakly colocalize with dsRNA that marked viral replication sites. However, SG marker proteins were not found aggregated in SG-like foci. Functional roles for the other SG proteins were not described but DDX6 interaction with the 3′UTR was required for replication [53].
The effects of two retroviruses on SG formation have been investigated and both have been found to be inhibitory. Human immunodeficiency virus 1 (HIV-1) inhibits the formation of SGs in response to arsenite treatment despite activating elevated levels of phosphorylated eIF2α but, paradoxically, not in response to puromycin treatment [54]. This indicates that the inhibition of SGs occurs at a step downstream of eIF2α phosphorylation. Another SG marker protein, Staufen1, interacts with the viral Gag protein, forming stable HIV-1 ribonucleoproteins (RNPs) destined for encapsidation instead of SG translation silencing (Figure 2) [54].
Human T-cell leukemia virus type-1 (HTLV-1) also inhibits SG formation through expression of the viral Tax protein [55]. Inhibition of SGs is dependent on the cellular localization of Tax, as cells displaying a nuclear Tax signal contain SGs even in the absence of stress whereas cells displaying a cytoplasmic Tax signal do not contain SGs. Legros et al. determined the inhibition of SGs is due to the interaction of Tax with histone deacetylase 6 (HDAC6), a protein crucial to the formation and maintenance of SGs (Figure 1, Figure 2) [36].
Finally, DNA viruses such as Herpes simplex virus 1 (HSV1) also induce SGs or SG-like structures, but only with mutant viruses. Infection with wild type HSV1 results in cytoplasmic focal localization of TIA-1 and TIAR but not overt SG formation. However, infection with HSV1 ΔUL41, a mutant strain lacking the virion host shutoff (Vhs) protein, results in SG formation in a cell type-specific manner 56, 57 that did not correlate with eIF2α phosphorylation. These initial data suggest HSV, which controls host translation eIF2α phosphorylation, strongly limits SG formation. Interestingly, these results also stress that eIF2α phosphorylation is not the only route to initiate SG formation.
Viral tolerance or exploitation of SG responses?
Viruses are master manipulators of cellular processes so it is not surprising that some SG components such as G3BP and TIA-1 may be utilized in virus replication as described for HCV and flaviviruses above. However, some viruses may co-opt steps of SG induction to aid virus replication. This may be suspected where virus infection induces but does not block aggregation of bonafide SGs or key SG components. In some cases, there may be unlinkage of eIF2α phosphorylation and aggregation of some SG components that are used for viral functions.
Two RNA viruses, respiratory syncytial virus (RSV) [27] and coronaviruses 47, 58, may benefit from inducing SG formation as part of the mechanism by which they inhibit host cell protein synthesis. In the case of RSV, infection induces SGs in ∼30% of infected cells at 24 hpi, and this may promote RSV replication because cells with SGs contained larger viral inclusion bodies than cells not forming SGs. Further, the inhibition of SG formation via the stable knockdown of G3BP resulted in a 10-fold decrease in replication [27]. A followup report showed SG induction by RSV was mediated by PKR-dependent eIF2α phosphorylation and a PKR knockout had a decreased ability to induce SGs in response to RSV infection. However, unlike the initial findings, the lack of SG formation was not correlated with a decrease in viral replication [59]. By contrast, Hanley et al. used the same cell line and RSV strain and found that only RSV viruses containing mutations or truncations in the 5′-trailer region induce SGs during infection [60]. The trailer is a region of extragenic RNA that is required for genomic RNA transcription. The cause of these contrasting results is yet to be determined and owing to the opposing nature of these results, the question of the role of SGs in the RSV lifecycle is unanswered.
Two different coronaviruses, mouse hepatitis coronavirus (MHV) [47] and transmissible gastroenteritis coronavirus (TGEV) [58] induce SGs that are present during phases of active virus gene expression, and may persist throughout much of infection. MHV induces the formation of SGs and PBs at 6 hpi; this is coincident with partial eIF2α phosphorylation, shutoff of host translation, but robust virus translation. However, although SG coexist with high virus translation rate, it was not reported if MHV antagonized or inhibited SG at later timepoints in infection. In contrast to RSV, MHV replication was enhanced in cells deficient in eIF2α phosphorylation or SG formation [47], indicating that although the virus triggers eIF2α phosphorylation to inhibit host translation, its own translation is also susceptible to this inhibition but to a lesser degree. TGEV induces SGs that increase and persist for at least 16 h, through much of the infection cycle. These SGs contained TIA-1, TIAR and the nuclear protein PTB and it was shown that formation of these structures correlated with inhibition of viral RNA accumulation. PTB was shown to bind virus RNA by mass spectroscopy and viral guide RNA (gRNA) and subgenomic mRNA (sgmRNA) was included in precipitable complexes with TIA-1 and PTB, but SG structures with PTB were distinct from other foci containing (virus) dsRNA or replication complexes. In light of this, the authors suggest that SG induction and manipulation of PTB localization may play a role in regulating viral RNA replication, translation or packaging [58]. The effects of SG inhibition on TGEV mRNA expression were not analyzed and an antiviral role of the PTB-positive SGs cannot be ruled out.
Vaccinia virus (VV) is a DNA virus that may exploit aspects of SG responses by subverting SG components into novel aggregates that share properties with SG, but crucially differ by not containing translationally silenced mRNAs. Even though VV can suppress eIF2a phosphorylation, during replication SG-like structures form within and adjacent to its cytoplasmic replication factories (RFs) 61, 62. These aggregates contained colocalized G3BP and cytoplasmic activation/proliferation-associated protein-1 (p137 or Caprin1) plus initiation factors eIF4G, eIF4E and VV RNA. G3BP is an integral protein to the SG formation process [21] and G3BP:Caprin-1 heterodimer localizes to SGs [63]. G3BP and translation initiation factors become highly concentrated within aggregates, and are deficient elsewhere in the cytoplasm, a situation which occurs in uninfected cells only when SGs form. G3BP or Caprin1, may stimulate VV intermediate gene expression either individually or as a heterodimer through unknown mechanisms [64]. It was proposed that these sites were centers of viral translation [62], which is contrary to typical SGs containing the same proteins that are sites of stable, non-translating mRNA. Walsh et al. showed that viral RFs contained aggregates of eIF4E and eIF4G but not PABP or TIA-1, indicating an incomplete SG structure assembly [65]. Part of the VV mechanism of subversion of normal SG response may result from degradation of host mRNA, freeing aggregating SG components for alternate tasks.
Infection with a mutant VV lacking the dsRNA binding protein E3L (VVΔE3L) does not block PKR activation and eIF2α phosphorylation. These infections formed SG-like bodies that both surrounded and interlaced RFs and contained hallmark proteins G3BP, TIA-1, USP10, initiation factors and required eIF2α phosphorylation for their formation [61]. Because the formation of these SG were linked to PKR activation and reduced viral replication, the authors termed them antiviral granules (AVGs) [61]. It is likely that PKR activation triggers more complete activation of functional SGs. Together, the results suggest VV may steer G3BP and initiation factors into new roles, but does not silence translation function within SG due to anti-eIF2α phosphorylation function of E3L and VV-induced host mRNA degradation, but instead concentrating and supporting virus translation in or near RFs.
SGs as an antiviral response
Because many virus families antagonize SGs, they can be viewed as manifestations of an overt and integrated cellular stress response that has distinct antiviral aspects. SGs are potentially antiviral on several functional levels as they sequester and bind cell components that are vital for virus replication. For instance, SGs sequester TIA-1 and TIAR, which are required for flavivirus RNA replication by binding a 3′ stemloop that is complementary to minus strand RNA [51]. G3BP is also concentrated and utilized by HCV near replication complexes [43]. Translation initiation factors are required for any virus to replicate efficiently, so sequestration of 40S subunits and eIF4G, eIF4A, eIF4B and eIF3 can have negative consequences for virus replication. Further, internal ribosome entry site (IRES) transactivating factors such as PTB, PCBP2 and UNR that stimulate picornavirus translation are sequestered in SGs [42]. Thus, the act of SG-mediated sequestration of factors away from general cytoplasmic pools can be viewed as generally antiviral.
Although there is evidence that inclusion of viral RNAs into SGs can have inhibitory affects on viral replication under certain conditions [66], there is not yet evidence in most virus systems for significant inclusion of viral RNA into SGs under normal conditions. Therefore, antiviral activity due to inclusion of required factors is likely limited to cellular factors. Possible exceptions involve antiviral APOBEC3G and APOBEC3F proteins that bind mRNP complexes and shuttle into both SG and PB 67, 68. APOBEC3G binds HIV RNA, which also may be shunted into SG and PBs [69]. Other viral genomes at risk of sequestration into SGs are those that bind SG marker proteins. Examples are TIA-1/TIAR binding to flavivirus RNA and recent reports that indicate both HCV and DV RNA bind G3BP 44, 53.
Finally, although SGs can be considered a cell stress response that is largely antiviral, they may serve as an inhibitor of apoptosis, which is also a stress-induced antiviral response of greater negative consequence for viruses. SG can block apoptosis by negatively regulating the JNK/SAPK pathway via sequestration of RACK1 and other apoptosis-promoting factors into SG 7, 70. Thus, viral manipulation of SG may require fine tuning to sequester enough pro-apoptotic factors while not excessively depleting pro-viral factors and translation apparatus.
Concluding remarks
Much work remains to be done to fully understand the interactions between viruses and SGs, a field that is in its infancy and pressing questions are summarized in Box 1 . Major questions that currently remain to be answered center around mechanisms of interference with SGs. Viral sequestration or cleavage of the SG components G3BP, TIA-1, Staufen and HDAC6 provide initial clues, but the function of all these proteins in SG formation and regulation is poorly defined. Other viruses such as cardioviruses and Junin virus have identified viral products that mediate SG inhibition, but the targets and molecular details remain incomplete. Similarly, in most cases where SGs are inhibited during infection, the question remains open whether SG loss is due to inhibition of formation of SGs or stimulation of SG disassembly. Stimulation of SG dissolution as opposed to inhibition of SG formation would constitute a novel mechanism of SG modulation during infection and its investigation would provide crucial details into a largely unresearched stage of the SG process. Still other steps in SG assembly have not been evaluated during viral infection, notably microtubule transport or post-translational modifications. Viral interference in any of these processes could potentially restrict SG formation and function. Finally, PV, CrPV and HCV infection also disrupts PBs along with SGs 43, 45, 71. It will be important to determine which viruses block both types of RNA granules and whether integrated mechanisms are involved.
Box 1. Outstanding questions.
-
•
What additional virus systems manipulate SG responses?
-
•
What are the precise mechanisms by which viruses inhibit SG formation?
-
•
Are there cases of enhanced SG dissolution instead of inhibited assembly?
-
•
Are other steps in SG formation, such as microtubule transport, a target for viruses?
-
•
Is sequestration of factors such as G3BP or TIA-1 a result of a selective pressure to inhibit SGs or a by-product of their requirement for RNA replication?
-
•
What are the mechanisms of SG-mediated antiviral effects?
-
•
Do viruses alter the content of SG-sequestered mRNAs, e.g. exclude certain mRNAs?
-
•
What downstream effects does SG inhibition have on mRNA metabolism or PB formation?
-
•
Do SGs and PBs play a role in the innate immune response?
Further work is also necessary to determine the nature of antiviral effects mediated by SGs and the nature of the balancing act between virus induction of SG versus apoptosis. Although sequestration of cellular factors into SG is suggested for some systems, there has been no definitive test of this concept and it is clearly not antiviral in some cases where SG induction increases viral replication. An attractive hypothesis is that SG formation signals downstream stress signals that activate innate antiviral mechanisms as part of an integrated stress response. Mechanistic details of SG inhibition in other viral systems would potentially allow for suppression of inhibition, allowing for further characterization of the antiviral effects of SGs. Viruses are excellent probes of cellular function. As further investigation uncovers the mechanisms behind viral inhibition of SG formation, we will learn a great deal about the cell biology and biochemistry that supports RNA granule function.
References
- 1.Franks T.M., Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol. Cell. 2008;32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zurla C. Characterizing mRNA interactions with RNA granules during translation initiation inhibition. PLoS ONE. 2011;6:e19727. doi: 10.1371/journal.pone.0019727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin W.J. Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J. Biol. Chem. 2007;282:19958–19968. doi: 10.1074/jbc.M702281200. [DOI] [PubMed] [Google Scholar]
- 4.Krichevsky A.M., Kosik K.S. Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron. 2001;32:683–696. doi: 10.1016/s0896-6273(01)00508-6. [DOI] [PubMed] [Google Scholar]
- 5.Knowles R.B. Translocation of RNA granules in living neurons. J. Neurosci. 1996;16:7812–7820. doi: 10.1523/JNEUROSCI.16-24-07812.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas M.G. RNA granules: the good, the bad and the ugly. Cell Signal. 2011;23:324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchan J.R., Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheth U., Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stohr N. ZBP1 regulates mRNA stability during cellular stress. J. Cell Biol. 2006;175:527–534. doi: 10.1083/jcb.200608071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kedersha N., Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- 11.Kimball S.R. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am. J. Physiol. Cell Physiol. 2003;284:C273–C284. doi: 10.1152/ajpcell.00314.2002. [DOI] [PubMed] [Google Scholar]
- 12.McEwen E. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005;280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- 13.Kedersha N. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell. 2002;13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kedersha N.L. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999;147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazroui R. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell. 2006;17:4212–4219. doi: 10.1091/mbc.E06-04-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim W.J. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J. 2007;26:5020–5032. doi: 10.1038/sj.emboj.7601920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang Y. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J. Biol. Chem. 2006;281:32870–32878. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]
- 18.Berlanga J.J. Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 2006;25:1730–1740. doi: 10.1038/sj.emboj.7601073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchan J.R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 2008;183:441–455. doi: 10.1083/jcb.200807043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolster D. Immediate response of mammalian target of rapamycin (mTOR)-mediated signalling following acute resistance exercise in rat skeletal muscle. J. Physiol. 2003;553:213–220. doi: 10.1113/jphysiol.2003.047019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tourriere H. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003;160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Kedersha N. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kedersha N. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mollet S. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell. 2008;19:4469–4479. doi: 10.1091/mbc.E08-05-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang W.L., Tarn W.Y. A role for transportin in deposition of TTP to cytoplasmic RNA granules and mRNA decay. Nucleic Acids Res. 2009;37:6600–6612. doi: 10.1093/nar/gkp717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unsworth H. mRNA escape from stress granule sequestration is dictated by localization to the endoplasmic reticulum. Faseb. J. 2010;24:3370–3380. doi: 10.1096/fj.09-151142. [DOI] [PubMed] [Google Scholar]
- 27.Lindquist M.E. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J. Virol. 2010;84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilks N. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White J.P. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe. 2007;2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Hinton S.D. The pseudophosphatase MK-STYX interacts with G3BP and decreases stress granule formation. Biochem. J. 2010;427:349–357. doi: 10.1042/BJ20091383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov P.A. Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp. Cell Res. 2003;290:227–233. doi: 10.1016/s0014-4827(03)00290-8. [DOI] [PubMed] [Google Scholar]
- 32.Chernov K.G. Role of microtubules in stress granule assembly: microtubule dynamical instability favors the formation of micrometric stress granules in cells. J. Biol. Chem. 2009;284:36569–36580. doi: 10.1074/jbc.M109.042879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolobova E. Microtubule-dependent association of AKAP350A and CCAR1 with RNA stress granules. Exp. Cell Res. 2009;315:542–555. doi: 10.1016/j.yexcr.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loschi M. Dynein and kinesin regulate stress-granule and P-body dynamics. J. Cell Sci. 2009;122:3973–3982. doi: 10.1242/jcs.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai N.P. Dynein motor contributes to stress granule dynamics in primary neurons. Neuroscience. 2009;159:647–656. doi: 10.1016/j.neuroscience.2008.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon S. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 2007;21:3381–3394. doi: 10.1101/gad.461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin Q. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 2009;83:11090–11101. doi: 10.1128/JVI.01239-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin Q. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2{alpha} phosphorylation and PKR. J. Virol. 2011;85:8798–8810. doi: 10.1128/JVI.01831-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith J.A. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 2006;80:2019–2033. doi: 10.1128/JVI.80.4.2019-2033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McInerney G.M. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell. 2005;16:3753–3763. doi: 10.1091/mbc.E05-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piotrowska J. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 2010;84:3654–3665. doi: 10.1128/JVI.01320-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White J.P., Lloyd R.E. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J. Virol. 2011;85:12442–12454. doi: 10.1128/JVI.05888-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ariumi Y. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 2011;85:6882–6892. doi: 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J. Virol. 2011;85:6996–7004. doi: 10.1128/JVI.00013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khong A., Jan E. Modulation of stress granules and P bodies during dicistrovirus infection. J. Virol. 2011;85:1439–1451. doi: 10.1128/JVI.02220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montero H. Rotavirus infection induces the phosphorylation of eIF2alpha but prevents the formation of stress granules. J. Virol. 2008;82:1496–1504. doi: 10.1128/JVI.01779-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raaben M. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol. 2007;9:2218–2229. doi: 10.1111/j.1462-5822.2007.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borghese F., Michiels T. The leader protein of Cardioviruses inhibits stress granule assembly. J. Virol. 2011;85:9614–9622. doi: 10.1128/JVI.00480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linero F.N. Junin virus infection impairs stress granule formation in Vero cells treated with arsenite via inhibition of eIF2{alpha} phosphorylation. J. Gen. Virol. 2011;92:2889–2899. doi: 10.1099/vir.0.033407-0. [DOI] [PubMed] [Google Scholar]
- 50.Khaperskyy D.A. Influenza A virus inhibits cytoplasmic stress granule formation. Faseb. J. 2011 doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 51.Emara M., Brinton M. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9041–9046. doi: 10.1073/pnas.0703348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002;76:11989–12000. doi: 10.1128/JVI.76.23.11989-12000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward A.M. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011;8:1173–1186. doi: 10.4161/rna.8.6.17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abrahamyan L.G. Novel Staufen1 ribonucleoproteins prevent formation of stress granules but favour encapsidation of HIV-1 genomic RNA. J. Cell Sci. 2010;123:369–383. doi: 10.1242/jcs.055897. [DOI] [PubMed] [Google Scholar]
- 55.Legros S. The HTLV-1 Tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene. 2011;30:4050–4062. doi: 10.1038/onc.2011.120. [DOI] [PubMed] [Google Scholar]
- 56.Dauber B. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 2011;85:5363–5373. doi: 10.1128/JVI.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esclatine A. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc. Natl. Acad. Sci. U.S.A. 2004;101:18165–18170. doi: 10.1073/pnas.0408272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sola I. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 2011;85:5136–5149. doi: 10.1128/JVI.00195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindquist M.E. Activation of protein kinase R is required for induction of stress granules by respiratory syncytial virus but dispensable for viral replication. Virology. 2011;413:103–110. doi: 10.1016/j.virol.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanley L.L. Roles of the respiratory syncytial virus trailer region: effects of mutations on genome production and stress granule formation. Virology. 2010;406:241–252. doi: 10.1016/j.virol.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simpson-Holley M. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J. Virol. 2011;85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katsafanas G.C., Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe. 2007;2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Solomon S. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell Biol. 2007;27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katsafanas G.C., Moss B. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J. Biol. Chem. 2004;279:52210–52217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- 65.Walsh D. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in Poxvirus-infected cells. Mol. Cell. Biol. 2008;2008:2648–2658. doi: 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Henao-Mejia J. Suppression of HIV-1 Nef translation by Sam68 mutant-induced stress granules and nef mRNA sequestration. Mol. Cell. 2009;33:87–96. doi: 10.1016/j.molcel.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gallois-Montbrun S. Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J. Virol. 2008;82:5636–5642. doi: 10.1128/JVI.00287-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gallois-Montbrun S. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 2007;81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kozak S.L. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 2006;281:29105–29119. doi: 10.1074/jbc.M601901200. [DOI] [PubMed] [Google Scholar]
- 70.Arimoto K. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008;10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 71.Dougherty J.D. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 2011;85:64–75. doi: 10.1128/JVI.01657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohn T. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008;10:1224–1231. doi: 10.1038/ncb1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie W., Denman R.B. Protein methylation and stress granules: posttranslational remodeler or innocent bystander? Mol. Biol. Int. 2011;2011:137459. doi: 10.4061/2011/137459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goulet I. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules. Human Mol. Genet. 2008;17:3055–3074. doi: 10.1093/hmg/ddn203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Leeuw F. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp. Cell Res. 2007;313:4130–4144. doi: 10.1016/j.yexcr.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 76.Dolzhanskaya N. Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J. Cell Sci. 2006;119:1933–1946. doi: 10.1242/jcs.02882. [DOI] [PubMed] [Google Scholar]
- 77.Tsai N.P. Regulation of stress granule dynamics by Grb7 and FAK signalling pathway. EMBO J. 2008;27:715–726. doi: 10.1038/emboj.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wehner K.A. OGFOD1: a novel modulator of eIF2{alpha} phosphorylation and the cellular response to stress. Mol. Cell Biol. 2010;30:2006–2016. doi: 10.1128/MCB.01350-09. [DOI] [PMC free article] [PubMed] [Google Scholar]