

Abstract
Mechanisms mediating closure of the dorsal vertebrae are not clear. Previously, we showed that deletion of TGFβ type II receptor (Tgfbr2) in sclerotome in mice results in failure in the formation of the spinous process, mimicking spina bifida occulta, a common malformation in humans. In this study, we aimed to determine whether missing dorsal structures in Tgfbr2 mutant mice were due to defects in mesenchymal migration and to clarify mechanism of TGFβ-mediated migration. First, we showed that gross alterations in dorsal vertebrae were apparent by E16.5 days in Tgfbr2 mutants. In addition, histological staining showed that the mesenchyme adjacent to the developing cartilage was thin compared to controls likely due to reduced proliferation and migration of these cells. Next, we used a chemotaxis migration assay to show that TGFβ promotes migration in mixed cultures of embryonic sclerotome and associated mesenchyme. TGFβ stimulated expression of PDGF ligands and receptors in the cultures and intact PDGF signaling was required for TGFβ-mediated migration. Since PDGF ligands are expressed in the sclerotome-derived cartilage where Tgfbr2 is deleted and the receptors are predominantly expressed in the adjacent mesenchyme, we propose that TGFβ acts on the sclerotome to regulate expression of PDGF ligands, which then act on the associated mesenchyme in a paracrine fashion to mediate proliferation, migration and subsequent differentiation of the adjacent sclerotome.
Keywords: vertebrae, sclerotome, axial skeleton, TGFβ type II receptor, PDGF, migration
Introduction
In vertebrates, the axial skeleton is composed of cartilage and bone produced by paraxial mesoderm. The vertebral column is the central part of the axial skeleton, and it includes the vertebral body, the neural arch, and the intervertebral discs between adjacent vertebral bodies. During development, the paraxial mesoderm gives rise to the segmental units of the somite. The somites then subdivide into the dorsal dermomyotome and the ventral sclerotome under the induction of signals from the notochord and the floor plate of the neural tube (Pourquie et al., 1993). Sclerotome, a population of undifferentiated mesenchymal cells, differentiate to form three subcompartments: ventral, lateral, and dorsal, with each giving rise to distinct parts of the vertebrae and ribs (Christ et al., 2000). Following a process of patterned anterior-posterior fusion of consecutive sclerotomes, the vertebrae forms as a cartilagenous template that is later converted into bone by endochondral ossification.
Spina bifida (SB), or failure of the neural tube and vertebrae to close at the midline, is one of the most common (1–2 cases per 1000 births) congenital malformations in humans leading to infant mortality or severe disability. This condition is divided into four categories with spina bifida occulta (SBO) as the mildest form that only involves the vertebrae. As with many other problems, SB appears to result from a combination of genetic and environmental risk factors, but the major cause of SB is believed to be failure of neural tube closure during the first 4 weeks of embryogenesis. However, accumulating evidence suggests that SB can result from defects in the somatic mesoderm surrounding the neural tube (Furumoto et al., 1999; Payne et al., 1997; Pickett et al., 2008; Stottmann et al., 2006).
The transforming growth factor beta (TGFβ) superfamily contains signaling molecules that regulate many aspects of cell physiology including skeletal development. TGFβ signals through a dual receptor system of type I and type II transmembrane serine/threonine kinases (Janssens et al., 2005). Upon TGFβ binding to type II receptor (TGFbR2), which is a constitutively active kinase, type I receptor (TGFbR1) can be recruited and becomes phosphorylated and activated. The canonical Tgfbr2 signaling pathway is mediated by a group of transcription factors called Smads. Smads directly regulate the transcription of target genes (Janssens et al., 2005). The role of members of the TGFβ superfamily in specific aspects of skeletal development and pathology is most clearly illustrated in mice and humans with mutations or targeted deletions in their respective genes (Kingsley, 1994; Serra and Chang, 2003). Targeted deletion of the mouse Tgfb2 gene results in several skeletal abnormalities including failure in the dorsal closure of the neural arches suggesting an important role for the TGFβ2 ligand in vertebrae development (Sanford et al., 1997). Little is known about the mechanisms of neural arch closure or of TGFβ action in this process.
Platelet-derived growth factor (PDGF) is a potent mitogenic and chemotactic factor for cells of mesenchymal origin, including chondrocytes and mesenchymal stem cells. The PDGF signaling family consists of four different polypeptide chains, designated A, B, C and D, as well as two receptors α (PDGFRα) and β (PDGFR β). The four PDGF chains assemble into five different dimeric isoforms: PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC and PDGF-DD. PDGF-BB, AB and CC can bind and activate both PDGFRα and β, whereas PDGF-AA binds only PDGFRα (Bornfeldt et al., 1995). Upon binding to the receptor, PDGF ligands transduce signals to regulate many biological functions of cells, including proliferation, migration, differentiation, and apoptosis (Heldin and Westermark, 1999). The PDGF ligand-receptor system has also been implicated to play roles in wound healing (Barrientos et al., 2008), tendon and cartilage repair (Molloy et al., 2003; Schmidt et al., 2006), atherosclerosis (Ross, 1993), and tumorigenesis (Sun et al., 2005). A recent study showed that the PDGFRα pathway plays a central role in regulating formation of the spinous process (Pickett et al., 2008). Disruption of PDGFRα signaling in the mesenchyme surrounding sclerotome-derived cartilage led to SBO whereas disruption in the sclerotome itself had no effect on vertebral development (Pickett et al., 2008). Since PDGF ligands were shown to be expressed in sclerotome derived tissues and the receptors were expressed in the surrounding mesenchyme, it was suggested that paracrine signaling between the sclerotome and surrounding mesenchyme is required for closure of the neural arches and formation of the spinous process (Pickett et al., 2008). Deletion of Pdgfc in mice also resulted in SBO and it was suggested that PDGF-C acts through PDGFRα to mediate development of the vertebrae (Ding et al., 2004). TGFβ induces the expression of PDGF ligands in many cell types, such as mesangial cells (Haberstroh et al., 1993) and endothelial cells (Taylor and Khachigian, 2000), suggesting that TGFβ might mediate physiological and pathological processes by regulating PDGF signaling. In fact, various studies have implicated PDGF acting in concert with TGFβ in development of organ fibrosis, including pulmonary and hepatic fibrosis (Trojanowska, 2008).
Our previous results showed that deletion of Tgfbr2 in Col2a expressing tissue in mice resulted in alterations in the formation of the vertebrae and intervertebral disc (IVD) (Baffi et al., 2004). These mice demonstrated small vertebrae and the spinous process failed to form, mimicking SBO in humans. These results suggested that TGFβ plays an important role in regulating embryonic development of the axial skeleton; however, the mechanism of this action has not been clarified. In this study, we aimed to determine whether the missing dorsal structures in Tgfbr2 mutant mice were due to defects in proliferation and migration, and to further clarify the underlying mechanism of migration mediated by TGFβ. We first show that TGFβ signaling is required for dorsal closure of vertebrae and formation of the spinous process, and that gross disruption to development is evident by E16.5 days. We then used a chemotaxis migration assay to show that TGFβ signaling promotes the migration of sclerotome-associated mesenchyme, and that the downstream effects of PDGF are required. We propose that TGFβ acts through PDGF in a paracrine manner to regulate migration of the mesenchyme adjacent to the sclerotome and subsequent formation of the spinous process.
Material and methods
Mice
All experiments were carried out with the approval of the UAB institutional animal care committee. The generation of Tgfbr2fl/fl mice were previously described (Chytil et al., 2002). Col2a-Cre mice were obtained from Jackson laboratories (Ovchinnikov et al., 2000). The Rosa26/+ reporter strain was obtained from Jackson labs (Soriano. 1999). Col2a-Cre mice were crossed to Tgfbr2fl/fl mice to obtain Col2a-Cre+;Tgfbr2fl/wt mice, which were subsequently crossed to Rosa26/Rosa26 mice to get the mice with a genotyping of Col2aCre+;ROSA26/+;Tgfbr2fl/wt. These mice were then used to backcross with Tgfbr2fl/fl to get Col2aCre+;ROSA26/+;Tgfbr2fl/wt as controls and Col2aCre+;ROSA26/+;Tgfbr2fl/fl as the experimental group.
Whole mount X-gal staining
Embryos were collected with the day of the vaginal plug designated as E0.5. Whole-mount X-gal staining was performed on the staged embryos as described (Chai et al., 2000). Between five and ten embryos of each genotype were generated. Briefly, embryos were taken and skin was removed before the fixation in 4% paraformaldehyde for 2 hrs at room temperature. The fixed embryos were soaked in the permeablization solution for 1 hr, followed by the staining solution overnight at room temperature. Stained embryos were post-fixed in 4% paraformaldehyde overnight at 4°C and then cleared with glycerol for photography. Alterations in the dorsal most edge of the vertebra were measured as the length of the gap between the edges of the neural arches at the level of the second lumbar vertebrae in E15.5, E16.5 and E17.5 day embryos. Measurements were taken from 5 mice of each genotype. Two embryos from each group were used for sectioning. Frozen sections (10 μm) were prepared by embedding the stained embryos in OCT (Tissue-Tek).
Histology, immunostaining, and cell proliferation
At least two E15.5, 16.5, and 17.5 day embryos from each group were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned at 6 μm. For histological analysis, sections were stained with either hematoxylin and eosin or alcian blue. For immunofluorescence and immunohistochemical staining, sections were de-waxed and hydrated, followed by antigen retrieval by boiling the sections in citrate acid buffer (pH 6.0) for 20 mins. After blocking in 5% BSA for 1 hr, the sections were incubated overnight at 4°C with a 1:100 dilution of primary antibodies. Antibodies used for this experiment include Ki67 (NeoMarkers) and anti-mouse PDGF-BB (Abcam). Biotinylated goat-anti-rabbit IgG or biotinylated rabbit-anti-goat IgG (1:200, Vector lab) was added as the secondary antibody. Fluorescent signal was detected with avidin-conjugated Cy3, with DAPI as counterstain. Color was developed with DAB (Vector laboratories), with hematoxylin as counterstain.
Proliferation of sclerotome and associated mesenchymal cells was detected by immunostaing for Ki67 on deparaffinised sections as described above. Ki67-positive cells and DAPI-stained total cells were counted from five randomly selected boxed areas in the region of neural arch. Percentage of proliferation was obtained by dividing the number of Ki67-positive cells by the total number of cells. Data were presented by multiplying 100. Significance was determined by T-test.
Micromass culture of sclerotome and associated mesenchyme
Sclerotome and associated mesenchyme ventral to the neural tube was isolated from E11.5 day mouse embryos. The micromass culture was set up using a method described previously (Sohn et al., 2010). Briefly, mesenchymal cells were dissociated into a single cell suspension with incubation in 1 mg/ml collagenase D at 37°C for 30 minutes and reconstituted at a density of 1 × 107 cells/ml. Twenty microliters of cell suspension was dropped into each well of a 24 well plate. After a pre-incubation time of 1 hr at 37°C to allow cells to attach, the cultures were then flooded with F-12:DMEM (1:1) containing 10% FBS, 50 μg/ml ascorbic acid, 10 mM β-glycerolphosphate, 2 mM glutamine, antibiotics with or without different concentrations of TGFβ1. For the cells infected with adenovirus, virus was added to the cell drops with 2 μl per 20 μl drop (100 MOI, Multiplicity of Infection).
Quantitative real-time RT-PCR
mRNA samples were collected from micromass cultures in which cells were treated with either TGFβ1 (2.5, 5 ng/ml) for 8 hours or Cre virus (5, 10, 20 MOI) using the standard Trizol method (Paaske et al., 1987). RNA concentration was determined by Nanodrop Spectrophotometer. QuantiFast SYBR Green RT-PCR Kit (Qiagen) was used for quantitative real-time RT-PCR analysis with specific primers (Table 1). Data analysis was performed with REST software (Pfaffl et al., 2002).
Table 1.
Primers used in real-time PCR
| Gene Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| Pdgfa | TGTCAAGGTGGCCAAAGTGGAGTA | TGCACACTCCAGGTGTTCCTCTAA |
| Pdgfb | TGAAATGCTGAGCGACCACTCCAT | AGTGTGCTCGGGTCATGTTCAAGT |
| Pdgfc | AAGTTGAGGAGCCCAGTGATGGAA | AAGGTACTGAAGGCAGTCACAGCA |
| β2-microglobulin | GCCGTGTGAACCATGTGACTTT | CCAAATGCGGCATCTTCAAA |
Chemotaxis cell migration assay
Cell migration was performed using 96-well chemotaxis chamber as described (Pickett et al., 2008). Briefly, sclerotome and associated mesenchymal cells were isolated as described above. Cells in single cell suspension were stained with CellTracker Green CFDA or Red CMPTX (5 μM) following the manufacture’s protocol (Invitrogen). Stained cells were then plated in triplicate at a density of 4 × 104 cells per well in a 96-well chemotaxis chamber (NeuroProbe), on a filter (5 μm pore size, PVP-free) pre-treated for 1 hr with 10 μg/ml rat tail collagen (Sigma, St Louis, MO). Recombination proteins were added to the bottom wells at the indicated concentrations. TGFβ1, PDGF-AA and BB ligands were obtained from R&D Systems. 10% FBS was used as a positive control. Almost all the cells migrated in response to FBS. AG1296 (25 μM, Sigma) treatment was 30 mins before plating cells in the chemotaxis chamber. The migration chamber was incubated at 37°C for 6 hrs. At the end of incubation, cells on top of the filter were gently wiped off with a cotton swab. Cells on the under-side of the filter were collected to the bottom of the microplate well by centrifuging the microplate assembly for 10 mins at 500 g. Cell migration was measured by reading the fluorescence in each well using a microplate reader (Synergy HT, BioTek). Each experiment was performed at least twice, and each condition was assayed in triplicate.
To determine which cells were migrating in the mixed sclerotome and sclerotome- associated cell cultures, cells isolated from Col2aCre+;ROSA26/+mice were used in the migration assay with TGFβ in the bottom of the well. The sclerotome from each embryo was individually held in eppendorf tubes and placed on ice until the genotyping was finished. Sclerotome from mice with the genotyping of Col2aCre+;ROSA26/+ were pooled and single cell suspension was obtained after digestion with Collagenase D (1 mg/ml, Roche) for 1 hour at 37°C. Some of the cells were used to perform the migration assay as described above with TGFβ, and some cells were directly plated on the bottom wells of the chemotaxis chamber at the density of 4 × 104 cells per well to use as a control for staining in the total cell population. At the end of the migration assay, cells on the under-side of the filter were collected to the bottom of the microplate well by centrifuging the microplate assembly for 10 mins at 500×g. Cells (migrated and control) were fixed in the fixative solution (2% formaldehyde/methanol, 0.1% glutaraldehyde in PBS) for 2 mins at room temperature. After 3 washes with PBS, the staining solution (1 mg/ml of x-gal, 5 mM of potassium ferricyanide, 5 mM of potassium ferrocyanide, 2 mM of magnesium chloride) was added to each well. The plate was incubated over night at 37°C. Images were taken under the light microscope. Three individual fields were taken for each well. This experiment was performed three times.
In situ hybridization
E15.5 embryos were fixed in 4% paraformaldehyde (PFA) overnight at 4°C then processed for OCT embedding and cryo-sectioning. Probes for PDGFc were synthesized from cDNA, followed by the cloning using pGEM-T Easy Vector System (Promega). The templates were linearized by restriction enzymes NcoI or PstI following the manufacture’s protocol (NEB). DIG labelled probes were created using T7/Sp6 DIG RNA Labeling Kit (Roche). The in situ hybridization was performed following the method described previously (Sohn et al., 2010).
Results
TGFβ signaling regulates cell migration
Our previous results showed that deletion of Tgfbr2 in Col2a expressing tissue in mice resulted in alterations in the formation of the vertebrae and IVD (Baffi et al., 2004). These mice demonstrated small vertebrae and the spinous process failed to form mimicking SBO in humans. To determine the timing and extent of the defect, we marked the sclerotome with LacZ using a Cre-reporter line that conditionally expresses LacZ in the presence of Cre by the excision of a PGKneo cassette in front of the lacZ gene. Thus, cells that express Cre can be stained blue with X-gal (Soriano, 1999). The dorsal most edge of sclerotome derived tissue was compared in control (Col2aCre+;ROSA26/+;Tgfbr2fl/wt) and Tgfbr2cko (Col2aCre+;ROSA26/+;Tgfbr2fl/fl) embryos starting at E12.5 days. No differences were observed at E12.5 or E15.5 days (not shown and Fig. 1A). Between E16.5 and E17.5 the dorsal gap decreased in both control and Tgfbr2cko embryos; however, the decrease in the gap size was less in Tgfbr2cko embryos (Fig. 1B–D). At E17.5 days a significant increase in the size of the dorsal gap was seen in control versus Tgfbr2cko embryos (Fig. 1C, D). Alcian blue staining of sections from the lumbar vertebrae of E17.5 embryos clearly showed the defective neural arch closure in the Tgfbr2cko mice (Fig. 1E, F). In addition, the dense mesenchyme normally adjacent to the dorsal part of the neural arch was thin or missing as early as E15.5 days in Tgfbr2cko embryos (Fig 1G, H, white arrows), even though Cre was not directly active in these cells (Fig. 1I, J)
Figure 1. Disrupted formation of the dorsal vertebrae in Tgfbr2cko embryos.

(A–C) Representative images of whole mount X-gal staining on control (Con) and Tgfbr2cko (Cko) embryos at E15.5, E16.5 and E17.5 days. Magnification: 25×. (D) Measurements of the gap between neural arches at the second lumbar region from E16.5 and E17.5 embryos. Data was analyzed by 2-way ANOVA; N=5; ***, P<0.001 [E17.5 Cko vs E17.5 Con]; #, P<0.05 [E17.5 Con vs E16.5 Con]; +, P<0.05 [E17.5 Cko vs E16.5 Cko]. (E, F) Alcian blue staining of sections from lumbar vertebrae of E17.5 control and Tgfbr2cko embryos. Dense mesenchyme dorsal to the neural arch is seen in the control (white arrow). Magnification: 10×. (G, H) H & E staining of cryosections of the lumbar region from E15.5 control and Tgfbr2cko embryos. Dense mesenchyme adjacent to the sclerotome-derived cartilage is visible surrounding the dorsal edge of the neural arch in control mice (G, white arrow). This mesenchyme is reduced or absent in mutants (H, white arrow). Magnification: 20×. (I) Cryosection of X-gal stained E15.5 Col2aCre+;Rosa26/+ embryo. Cre activity is seen as blue X-gal stain in sclerotome derived cells. Staining is not observed in the adjacent sclerotome-associated mesenchyme. (J) Phase contrast image of I to show unstained adjacent mesenchyme. Magnification: 20×. c: cartilage; nt: neural tube; m: sclerotome associated mesenchyme.
To determine if the reduction in the mesenchyme adjacent to the sclerotome-derived cartilage was due to alterations in proliferation, we stained sections from E15.5 and E17.5 day embryos with Ki67 and determined the percent of labelled nuclei in the cartilage and in the adjacent mesenchyme (Table 2). Proliferation in sclerotome-derived cartilage was not affected in Tgfbr2cko embryos compared to controls at either stage, confirming what we reported earlier for ventral vertebrae and long bone cartilage (Baffi et al., 2006; Baffi et al., 2004). In contrast, proliferation in mesenchymal cells adjacent to the neural arch was significantly down-regulated in Tgfbr2 mutant mice at E 15.5 days (Table 2). Since Cre is not active in the adjacent mesenchyme, the result suggests that a paracrine interaction may be involved.
Table 2.
Percentage of cell proliferation in E15.5 and E17.5 day dorsal vertebrae
| Stage | Cell Type | Control | Tgfbr2cko | P value |
|---|---|---|---|---|
| E15.5 | Chondrocytes | 75.0 ± 4.38 | 80.9 ± 2.58 | 0.10 |
| Adjacent mesenchymal cells | 82.8 ± 4.54 | 73.4 ± 1.86 | 0.02* | |
|
| ||||
| E17.5 | Chondrocytes | 84.8 ± 2.51 | 86.6 ± 4.08 | 0.56 |
| Adjacent mesenchymal cells | 66.7 ± 3.54 | 59.8 ± 3.93 | 0.08 | |
Data are expressed as mean ± SE, n = 5 fields in 3 separate slides each,
: P < 0.05
It was previously shown that migration of the mesenchyme adjacent to the sclerotome regulates closure of the neural arches (Pickett et al., 2008). To test the hypothesis that TGFβ regulates migration, we established an in vitro transwell chemotaxis assay using sclerotome and associated mesenchymal cells from E11.5 embryos. We showed that cells lacking Tgfbr2 via adenovirus Cre infection of cells isolated from Tgfbr2fl/fl mice had impaired basal migration compared with cells infected with adenovirus GFP as a control, suggesting that TGFβ regulates a basal amount of migration in these cells (Fig. 2A). Furthermore, wild type cells responded to exogenous TGFβ with increased migration at the dosages of 2.5, 5, and 10 ng/ml (Fig. 2B). The maximum response was observed at the concentration of 2.5 ng/ml. These data indicate that TGFβ regulates cell migration and suggest that TGFβ-mediated migration may play an important role in closure of the neural arches.
Figure 2. TGFβ regulates migration in sclerotome associated mesenchyme in culture.

(A) Sclerotome and associated mesenchyme was isolated from E11.5 Tgfbr2fl/fl embryos and infected with adenovirus Cre (Ad-Cre) to delete Tgfbr2 or with adenovirus GFP (Ad-GFP) as a control. Loss of Tgfbr2 resulted in reduced basal migration. ***, P<0.001. RT-PCR in the upper panel shows the deletion of the Tgfbr2 allele using Ad-Cre, 100 MOI. (B) The migration assay was performed on sclerotome and associated mesenchymal cells isolated from E11.5 wild type embryos and treated with or without TGFβ in the bottom well. TGFβ stimulated migration. ***, P<0.001. (C) Cells were isolated from Col2aCre+;Rosa26/+ mice and used in the migration assay with TGFβ in the bottom well. Sclerotome derived cells were stained blue with X-gal, unstained cells represent sclerotome associated cells. The total nonmigrated cell population demonstrated both stained and unstained cells. Migrated cells at the bottom of the chamber were not stained. Images were selected from several wells for both total and migrated cells. Magnification: 40×.
To determine which cells in the cultures, sclerotome-derived or sclerotome-associated, were migrating in response to TGFβ, we used cells isolated from Col2aCre+;Rosa26/+ mice in the migration assay. In this way, Cre activity could be visualized by blue X-gal staining in sclerotome derived cells whereas sclerotome associated cells in the culture would not express Cre and would not stain with X-gal. As a control for staining in the total cell population, we plated some of the isolated cells directly on the bottom wells of the migration chamber. As expected, the total population of cells contained a mixture of X-gal stained and unstained cells (15% X-gal stained); however, we could not detect any X-gal staining in the cells that had migrated in response to TGFβ (Fig. 2C). This result suggests that cells isolated from E11.5 embryos are mixed populations containing sclerotome and associated mesenchyme, and TGFβ induces migration of the sclerotome associated mesenchymal cells.
PDGF plays an essential role in TGFβ-mediated cell migration
A recent study showed that PDGFRα plays a central role in regulating closure of the neural arches by promoting migration of the mesenchymal cells adjacent to the sclerotome-derived cartilage through a PI3K dependent mechanism (Pickett et al., 2008). It was also shown that the PDGF receptor was expressed in the mesenchyme adjacent to the sclerotome while the PDGF ligands were expressed in the cartilage derived sclerotome (Pickett et al., 2008). The phenotype observed in the dorsal vertebrae in PdgfraPI3K/PI3K mice resembled that seen in the Tgfbr2cko mice including failure of the spinous process to form and reduced or thin mesenchyme adjacent to the cartilage. Since PDGF ligands are regulated by TGFβ in other systems (Haberstroh et al., 1993; Taylor and Khachigian, 2000), we looked at the expression of PDGF ligands at the mRNA level in cultures of sclerotome and associated mesenchyme treated with TGFβ. Relative levels of mRNA were compared using quantitative real-time RT-PCR. Our results showed that TGFβ up-regulated the expression of Pdgfb and Pdgfc, whereas Pdgfa did not appear to be regulated (Table 3). We next assayed the expression of Pdgfb and Pdgfc in cells that had been deleted for Tgfbr2. Sclerotome and associated cells were isolated from Tgfbr2fl/fl mice and infected with adenovirus Cre to delete Tgfbr2. In cells with disrupted TGFβ signaling, expression of Pdgfb and Pdgfc were decreased (Table 3), suggesting that PDGF ligands -B and/or -C could be downstream effectors of TGFβ regulating cell migration. It should be noted that both PDGF-B and PDGF-C can bind and activate PDGFRα (Bornfeldt et al., 1995), suggesting that they could act as a link between TGFβ signaling in the sclerotome and PDGFRα signaling in the adjacent mesenchyme.
Table 3.
Gene expression level tested by real-time PCR
| Gene Name | Treatment | Fold Change | Standard Error Range |
|---|---|---|---|
| Pdgfa | TGFβ1 2.5 ng/ml | 0.885 | 0.647–1.201 |
| 5 ng/ml | 0.812 | 0.587–1.110 | |
|
| |||
| Pdgfb | TGFβ1 2.5 ng/ml | 4.563 | 3.451–6.037 |
| 5 ng/ml | 3.515 | 2.736–4.801 | |
|
| |||
| Pdgfc | TGFβ1 2.5 ng/ml | 2.908 | 2.569–3.326 |
| 5 ng/ml | 2.417 | 2.186–2.676 | |
|
| |||
| Pdgfb | Ad-Cre 5 MOI | 0.678 | 0.484–0.884 |
| 10 MOI | 0.645 | 0.586–0.703 | |
| 20 MOI | 0.617 | 0.261–0.981 | |
|
| |||
| Pdgfc | Ad-Cre 5 MOI | 0.586 | 0.533–0.666 |
| 10 MOI | 0.433 | 0.379–0.487 | |
| 20 MOI | 0.291 | 0.147–0.445 | |
Data are normalized by the reference gene β2-microglobulin. The TGFβ1 treated groups are compared with the un-treated group for each gene, and the Ad-Cre treated groups are compared with the Ad-GFP (10 MOI) treated group for each gene.
We next determined the direct effect of PDGF ligands on cell migration using the chemotaxis assay. In our experiment, PDGF-AA and PDGF-BB both induced migration at distinct concentrations (Fig. 3A). PDGF-AA activates PDGFRα and PDGF-BB can activate PDGFRα and β. To determine whether PDGF signaling is required for sclerotome migration, we blocked PDGF signaling by administering AG1296 to the cells prior to the chemotaxis assay. AG1296 is a small molecule that selectively inhibits PDGF receptor kinase, thereby blocking the action of PDGF ligands (Reiterer et al., 2008). Treatment with the antagonist alone did not affect the basal level of migration in the cells. However, cells in which PDGF signaling was blocked did not respond to TGFβ1 as the untreated cells did (Fig. 3B), suggesting that PDGF signaling is required for TGFβ-mediated cell migration. We showed above that cells without Tgfbr2 have impaired migration (Fig. 2A). Migration was restored in Tgfbr2 depleted cells by adding exogenous PDGF-BB (Fig. 3C). Together, these data suggest that the effects of TGFβ on migration are mediated by PDGF signaling.
Figure 3. PDGF ligands plays an important role in TGFβ mediated cell migration.

(A) Cells were isolated from wild type embryos and were either treated or not treated with PDGF-AA or PDGF-BB at the bottom of the well. Both PDGF ligands stimulated cell migration. ***, P<0.001. (B) Cells from wild type embryos were pre-treated with AG1296 (25 μM) for 30 min. before being placed in the migration assay with or without TGFβ in the bottom well. DMSO is the solvent control. SFM is the serum free medium control. TGFβ-stimulated migration was blocked by the PDGFR antagonist. ***, P<0.001 [SFM +TGFβ1 vs SFM-TGFβ1]; ###, P<0.001 [AG1296+TGFβ1 vs DMSO +TGFβ1]. (C) Cells were isolated from Tgfbr2fl/fl embryos and infected with Ad-Cre to delete Tgfbr2 or with Ad-GFP as a control. The cells were placed in the migration assay with or without PDGF-BB in the bottom well. PDGF-BB stimulated migration in Tgfbr2 deleted cells. ***, P<0.001 [Ad-Cre vs Ad-GFP]; #, P<0.01[Ad-Cre+PDGF-BB 10ng/ml vs Ad-Cre]. Representative of two separate experiments is shown.
PDGF-B and PDGF-C are reduced in Tgfbr2cko cartilage
Pickett et al. showed that PDGFRα is primarily expressed in perichondrium and the dense mesenchyme adjacent to cartilage forming sclerotome. Disruption of Pdgfra in sclerotome-derived cartilage using Col2a-Cre had no effect on the closure of the vertebrae. However, embryos lacking Pdgfrα in the adjacent mesenchyme developed SBO (Pickett et al., 2008). In order to further determine the mechanism for the phenotype observed in Tgfbr2cko mice, we performed immunohistochemical staining for PDGF-B on sections of lumbar vertebrae from control and Tgfbr2cko E15.5 embryos (Fig. 4A–C). PDGF-B was expressed in sclerotome-derived cartilage (Fig. 4A). In Tgfbr2cko embryos, the expression of PDGF-B was reduced in the cartilage even though expression in the dorsal root ganglion (DRG) was comparable in control and Tgfbr2cko mice (Fig. 4A, B). PDGF-C protein was previously shown to be localized to sclerotome, myotome, and vertebral cartilage (Aase et al., 2002). To determine if there were alterations in the Pdgfb and Pdgfc gene expression, we used in situ hybridization on transverse sections of E15.5 day control and Tgfbr2cko embryos. Both Pdgfb (Fig. 4D–H) and Pdgfc mRNA (Fig. 4I–K) were limited in the Tgfbr2cko cartilage relative to the controls. Expression of Pdgfb mRNA was again comparable in control and Tgfbr2cko dorsal root ganglion (Fig. 4E, G). No signal was observed with the RNA sense strand controls (Fig. 4H, K). We therefore propose that TGFβ-mediated alterations in expression of the PDGF ligands in cartilage may affect PDGFRα signalling in the adjacent mesenchymal cells resulting in failure in the formation of the spinous process.
Figure 4. PDGF-B and PDGF-C expression in Tgfbr2cko mice.

(A–C) Immunostaining of PDGF-B protein on sections from control (A) and Tgfbr2cko (B) embryos at E15.5 days. Positive staining is seen as a brown DAB substrate. Negative control without primary antibody is shown in (C). c: cartilage, drg: dorsal root ganglion, Magnification: 40×. (D–H) In situ hybridization to Pdgfb mRNA in control (D, E) and Tgfbr2cko (F, G) cartilage (D, F) and dorsal root ganglion (E, G). (I-K) In situ hybridization to Pdgfc mRNA in control (I) and Tgfbr2cko (J) cartilage. Digoxigenin labelled RNA hybridization is seen as a purple NBT/BCIP substrate. Hybridization to a labelled sense probe is used as the negative control (H, K).
Discussion
The previous study in our laboratory showed that deletion of Tgfbr2 in Col2a expressing tissue in mice resulted in alterations in the formation of the vertebrae and IVD (Baffi et al., 2004). These mice demonstrated small vertebrae and the spinous process failed to form, mimicking SBO in humans. SB is one type of neural tube defect in which the backbone and spinal canal do not close before birth. SBO is the mildest and most common form of SB, a condition in which the spinous process and neural arch appear abnormal on a radiogram, but meninges remain in place and skin usually covers the defect. The underlying mechanism for SBO is not exactly clear, but accumulating evidence suggests that SBO is caused primarily by defects in the somatic mesoderm surrounding the neural tube rather than the neural tube itself (Furumoto et al., 1999; Payne et al., 1997; Pickett et al., 2008; Stottmann et al., 2006).
In this study, we demonstrated that mice with conditional deletion of Tgfbr2 have defects in formation of the dorsal vertebrae grossly evident around E16.5 days. In addition, the layer of mesenchyme adjacent to the cartilage was reduced. We used a transwell-type chemotaxis assay to show that TGFβ regulates migration of sclerotome associated mesenchyme in culture and we proposed that the failure of the neural arches to close in the Tgfbr2 mutant embryos could be due to defects in proliferation and migration of these cells in vivo. Mice with mutations in Pdgfra have overlapping phenotypes specifically in the spinous process similar to the Tgfbr2cko mice described here (Pickett et al., 2008). A broad deletion of Pdgfrα in condensed mesenchyme showed significantly affected vertebral development, in which the spinous process was absent in the lumbar region. Unlike mice deleted for Tgfbr2, vertebral bodies, vertebral arches, and pedicles developed normally. It was suggested that loss of PDGF signaling in the mesenchyme adjacent to the sclerotome led to failure of the neural arches to close since deletion of Pdgfrα from the sclerotome itself using Col2a-Cre mediated recombination did not affect vertebral development (Payne et al., 1997; Pickett et al., 2008). This may have been expected since PDGFRα is not expressed in the sclerotome but is expressed in the adjacent mesenchyme (Pickett et al., 2008). Likewise, we showed that PDGF-B and -C are expressed in sclerotome-derived cartilage and PDGFRβ (not shown) is predominantly expressed in the adjacent mesenchyme. Furthermore, immunohistochemical staining of PDGF-B and in situ hybridization of Pdgfb and Pdgfc in Tgfbr2cko embryos suggested restricted expression in cartilage in agreement with decreased expression seen in Tgfbr2cko cells in culture. Since PDGF-B and PDGF-C can bind PDGFRα (Bornfeldt et al., 1995), they could act as a link between TGFβ signaling in the cartilage and PDGFRα signaling in the adjacent mesenchyme to regulate formation of the spinous process.
It has been shown that TGFβ induces Pdgfb transcription at least partially through Smad family members (Taylor and Khachigian, 2000). Consistent with these results, we showed that Pdgfb and Pdgfc are downstream targets of TGFβ and PDGF-BB promotes cell migration. Furthermore, blockade of the PDGF signaling pathway by the drug AG1296 disrupted TGFβ-induced migration of cells in culture, while exogenous PDGF-BB restored the migration capability of cells lacking Tgfbr2. These results suggest that PDGF signalling is required for TGFβ-induced migration; however, loss of Pdgfb function in mice leads to a phenotype dramatically unrelated to the phenotype resulting from loss of Pdgfrα (Leveen et al., 1994). Previous characterization of mice with targeted deletion of Pdgfc demonstrated failure of the neural arches to close in the lower spine, similar to what is seen in the Pdgfra deleted mice (Ding et al., 2004). It was suggested that PDGF-C acts through PDGFRα to mediate this phenotype. Together the results suggest that failure of the neural arch closure in Tgfbr2cko mice is likely due to TGFβ mediated regulation of Pdgfc expression although we cannot rule out an effect due to the other PDGF ligands. Based on these observations, we propose a model in which TGFβ signaling in the sclerotome-derived cartilage regulates the closure of the neural arches (Fig. 5): First, TGFβ stimulates PDGF ligand expression in sclerotome derived cells. PDGF ligands then act in a paracrine manner to stimulate PDGFRα on the adjacent mesenchymal cells. Activation of PDGF signaling promotes proliferation and migration of these cells.
Figure 5. Proposed model for the role of Tgfbr2 in development of the spinous process.
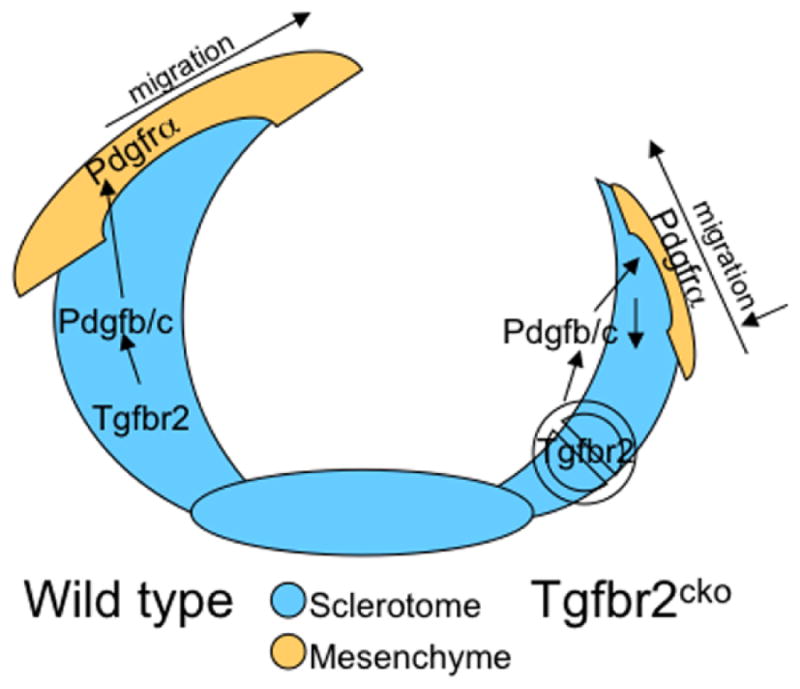
We propose the following model for TGFβ action in the development of the spinous process. In wild type embryos (left), TGFβ acts on its receptor in cartilage derived from sclerotome to stimulate PDGF ligand expression. PDGF ligands are then secreted and bind to PDGFRα on the adjacent mesenchyme. This paracrine activation of PDGF signaling promotes proliferation and migration of these cells eventually leading to the closure of the neural arches and formation of the spinous process. In Tgfbr2cko embryos (right), PDGF ligands are not made in sufficient amount to stimulate proliferation and migration of the adjacent mesenchyme. Alterations in the adjacent mesenchyme lead to failure in the formation of the spinous process.
It was previously shown that the PDGFRα responding mesenchyme secretes an unknown chondrogenic factor (Pickett et al., 2008). Expression of the chondrogenic factor was not dependent on PDGF; however, it would be predicted that the presence and proper location of the mesenchyme adjacent to the sclerotome would be necessary to direct the chondrogenic activity to the correct location at the dorsal tip of the developing neural arch and spinous process. BMPs have been found to be expressed in the dorsal part of the embryo at the time the spinous process is forming, and the effects of BMP on promoting the dorsal closure of vertebrae were demonstrated using grafts with BMP4-producing cells in chick embryos (Monsoro-Burq et al., 1996). It is possible that loss of Tgfbr2 disrupts chondrogenesis in the spinous process because of a reduction in the mesenchyme adjacent to the forming cartilage and subsequent mislocalization of BMP.
Based on our observations, we conclude that TGFβ signaling is required for closure of the neural arches and formation of the spinous process. This action is likely mediated by PDGF ligands and its paracrine role in regulating proliferation and migration of the adjacent mesenchyme. Our study is the first to analyze the mechanism of action of TGFβ signaling on development of the axial skeleton, and to show that PDGF is an important mediator in TGFβ-induced migration associated with skeletal development.
Acknowledgments
This study was supported by grants from the National Institutes of Health, R01- AR053860 to RS. YW is supported by NIH T32 Institutional Training Grant T32-AR047512 in bone biology and disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aase K, Abramsson A, Karlsson L, Betsholtz C, Eriksson U. Expression analysis of PDGF-C in adult and developing mouse tissues. Mechanisms of development. 2002;110:187–191. doi: 10.1016/s0925-4773(01)00560-3. [DOI] [PubMed] [Google Scholar]
- Baffi MO, Moran MA, Serra R. Tgfbr2 regulates the maintenance of boundaries in the axial skeleton. Dev Biol. 2006;296:363–374. doi: 10.1016/j.ydbio.2006.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffi MO, Slattery E, Sohn P, Moses HL, Chytil A, Serra R. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276:124–142. doi: 10.1016/j.ydbio.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- Bornfeldt KE, Raines EW, Graves LM, Skinner MP, Krebs EG, Ross R. Platelet-derived growth factor. Distinct signal transduction pathways associated with migration versus proliferation. Ann N Y Acad Sci. 1995;766:416–430. doi: 10.1111/j.1749-6632.1995.tb26691.x. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Christ B, Huang R, Wilting J. The development of the avian vertebral column. Anat Embryol (Berl) 2000;202:179–194. doi: 10.1007/s004290000114. [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, Hart TC, Marazita ML, Field LL, Tam PP, Nagy A. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nature genetics. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- Furumoto TA, Miura N, Akasaka T, Mizutani-Koseki Y, Sudo H, Fukuda K, Maekawa M, Yuasa S, Fu Y, Moriya H, Taniguchi M, Imai K, Dahl E, Balling R, Pavlova M, Gossler A, Koseki H. Notochord-dependent expression of MFH1 and PAX1 cooperates to maintain the proliferation of sclerotome cells during the vertebral column development. Dev Biol. 1999;210:15–29. doi: 10.1006/dbio.1999.9261. [DOI] [PubMed] [Google Scholar]
- Haberstroh U, Zahner G, Disser M, Thaiss F, Wolf G, Stahl RA. TGF-beta stimulates rat mesangial cell proliferation in culture: role of PDGF beta-receptor expression. Am J Physiol. 1993;264:F199–205. doi: 10.1152/ajprenal.1993.264.2.F199. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26:743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- Molloy T, Wang Y, Murrell G. The roles of growth factors in tendon and ligament healing. Sports Med. 2003;33:381–394. doi: 10.2165/00007256-200333050-00004. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Duprez D, Watanabe Y, Bontoux M, Vincent C, Brickell P, Le Douarin N. The role of bone morphogenetic proteins in vertebral development. Development. 1996;122:3607–3616. doi: 10.1242/dev.122.11.3607. [DOI] [PubMed] [Google Scholar]
- Paaske PB, Witten J, Schwer S, Hansen HS. Results in treatment of carcinoma of the external auditory canal and middle ear. Cancer. 1987;59:156–160. doi: 10.1002/1097-0142(19870101)59:1<156::aid-cncr2820590130>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Payne J, Shibasaki F, Mercola M. Spina bifida occulta in homozygous Patch mouse embryos. Dev Dyn. 1997;209:105–116. doi: 10.1002/(SICI)1097-0177(199705)209:1<105::AID-AJA10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Pickett EA, Olsen GS, Tallquist MD. Disruption of PDGFRalpha-initiated PI3K activation and migration of somite derivatives leads to spina bifida. Development. 2008;135:589–598. doi: 10.1242/dev.013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourquie O, Coltey M, Teillet MA, Ordahl C, Le Douarin NM. Control of dorsoventral patterning of somitic derivatives by notochord and floor plate. Proc Natl Acad Sci U S A. 1993;90:5242–5246. doi: 10.1073/pnas.90.11.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiterer G, Bunaciu RP, Smith JL, Yen A. Inhibiting the platelet derived growth factor receptor increases signs of retinoic acid syndrome in myeloid differentiated HL-60 cells. FEBS letters. 2008;582:2508–2514. doi: 10.1016/j.febslet.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MB, Chen EH, Lynch SE. A review of the effects of insulin-like growth factor and platelet derived growth factor on in vivo cartilage healing and repair. Osteoarthritis Cartilage. 2006;14:403–412. doi: 10.1016/j.joca.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Serra R, Chang C. TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res C Embryo Today. 2003;69:333–351. doi: 10.1002/bdrc.10023. [DOI] [PubMed] [Google Scholar]
- Sohn P, Cox M, Chen D, Serra R. Molecular profiling of the developing mouse axial skeleton: a role for Tgfbr2 in the development of the intervertebral disc. BMC Dev Biol. 2010;10:29. doi: 10.1186/1471-213X-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Berrong M, Matta K, Choi M, Klingensmith J. The BMP antagonist Noggin promotes cranial and spinal neurulation by distinct mechanisms. Dev Biol. 2006;295:647–663. doi: 10.1016/j.ydbio.2006.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Wang DA, Jain RK, Carie A, Paquette S, Ennis E, Blaskovich MA, Baldini L, Coppola D, Hamilton AD, Sebti SM. Inhibiting angiogenesis and tumorigenesis by a synthetic molecule that blocks binding of both VEGF and PDGF to their receptors. Oncogene. 2005;24:4701–4709. doi: 10.1038/sj.onc.1208391. [DOI] [PubMed] [Google Scholar]
- Taylor LM, Khachigian LM. Induction of platelet-derived growth factor B-chain expression by transforming growth factor-beta involves transactivation by Smads. J Biol Chem. 2000;275:16709–16716. doi: 10.1074/jbc.275.22.16709. [DOI] [PubMed] [Google Scholar]
- Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford) 2008;47(Suppl 5):v2–4. doi: 10.1093/rheumatology/ken265. [DOI] [PubMed] [Google Scholar]