

Abstract
Cardiac overexpression of the angiotensin II type 2 receptor (AT2R) attenuates left ventricular (LV) remodeling after myocardial infarction (MI) in transgenic mice. We hypothesized that a novel nonpeptide AT2R agonist, Compound 21 (C21), would attenuate post-MI LV remodeling. Fifty nine mice were studied for 28 days after 1 hour surgical occlusion-reperfusion of the left anterior descending coronary artery. Immediately thereafter, 23 mice received 0.3 mg/kg/d of C21 via Alzet osmotic mini-pump, 16 received 10 mg/kg/d of the AT1R antagonist candesartan in drinking water and 20 were untreated controls. Cardiac magnetic resonance imaging (CMR) measured ejection fraction (EF), LV end-systolic and end-diastolic volumes (ESVI, EDVI) indexed to weight serially post-MI. Infarct size was measured on day 1 by late gadolinium-enhanced CMR. At baseline, heart rate, blood pressure, EDVI, ESVI, and EF were similar between groups. Mean infarct size (42–45% of LV mass) was similar between groups. C21-treated animals demonstrated adverse LV remodeling (increased EDVI and ESVI at all post-MI time points) compared to control. Candesartan-therapy preserved LVEF at day 28 compared to the C21-treated group. Thus, direct stimulation of the AT2R by C21 at 0.3 mg/kg/d does not attenuate post-MI LV remodeling in reperfused MI in mice.
Keywords: myocardial infarction, remodeling, magnetic resonance imaging, angiotensin II, angiotensin receptors
Introduction
The deleterious role of increases in angiotensin II (Ang II) in cardiovascular disease is well established1. The role of the Ang II receptors in post-myocardial infarction (MI) left ventricular (LV) remodeling has been an object of intense study in recent years. Antagonism of type 1 receptors (AT1R) is known to be beneficial post-MI as an alternative to angiotensin converting enzyme inhibition (ACE-I)2, whereas the role of the AT2R is less clear. Cardiac magnetic resonance imaging (CMR) data from our laboratory in a transgenic mouse of cardiac overexpression of the AT2R demonstrated that baseline LV function is improved and post-MI remodeling is attenuated3. Pharmacologic antagonism of AT1Rs with losartan was not additive to AT2-R overexpression, whereas genetic knockout of AT1Rs (AT1ARs) further attenuated remodeling4. This implies that upregulation of the AT2R may be responsible for much of the benefits of AT1R antagonism. Treatment with the nitric oxide synthase inhibitor L-NAME abrogated the AT2R mediated protection against LV remodeling, suggesting that nitric oxide may underlie this benefit5. AT2R's protective effect was not directly mediated via the bradykinin subtype 2 receptor (B2R) pathway6.
Translating this potential benefit into larger animals including humans has suffered from the lack of an adequate receptor agonist. There are very few selective AT2R agonists available and those that are, such as CGP42112A, are peptides that are readily metabolized and are partial agonists/antagonists at high concentrations. Another pharmacological AT2R antagonist, PD123319, has suffered from low selectivity when applied at high doses, leading to contradictory findings7. However, a selective AT2R agonist, Compound 21 (C21) is now available that is 25,000 times more selective for the AT2R than for the AT1R, which allows investigators the opportunity to specifically stimulate the AT2R in in vitro and in vivo settings8. Previous studies in a rat model suggest potential benefit of this compound in the post-MI setting9.
We hypothesized that direct AT2R stimulation with C21 would attenuate remodeling in the post-MI mouse heart. To test this hypothesis, we used CMR to measure LV size and global function post-MI in control mice, C21-treated mice, and mice treated with the AT1R inhibitor candesartan beginning after the infarction. Infarct size was measured on day 1 to be certain these therapies were not differentially affecting infarct size. Thus, any subsequent effect would be on LV remodeling and the progression to congestive heart failure.
Methods
Mouse Model
Animal protocols were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1996) and were approved by the University of Virginia Animal Care and Use Committee. Seventy seven male wild type C57BL/6 mice, age 8 to 14 weeks, were studied before and after MI for a period of 28 days. Eighteen mice died post-operatively, leaving 59 mice in the study including 20 control, 23 C21-treated mice, and 16 candesartan-treated mice. The surgical infarction and reperfusion techniques have been previously described10. Briefly, the coronary artery was occluded for 60 minutes by tying down silk suture over a short length of PE-20 tubing with successful occlusion verified by myocardial color change and ECG changes. After 60 minutes, reperfusion was achieved by cutting the suture. In previous studies, this length of occlusion leads to an infarct that subtends approximately 65% of the risk area11. The chest wall was closed in layers and mice recovered with 100% oxygen.
Magnetic Resonance Imaging
Anesthesia was induced with 3.0% inhaled isoflurane and maintained with 1.0–1.25% isoflurane administered via nosecone during imaging at baseline before (day 0) and days 1, 7, and 28 post-MI (Figure 1). Imaging was performed on a 7-Tesla ClinScan (Bruker/Siemens, Erlangen, Germany) with an MRI-compatible physiological monitoring and gating system designed for small animals (SA Instruments, Inc., Stony Brook, NY). All mice were imaged using a 30 mm diameter cylindrical birdcage radiofrequency (RF) coil. A 2D fast, low-angle shot (FLASH) sequence was used to obtain orthogonal long-axis images. For simultaneous assessment of LV size, global and regional LV function, and infarct size, continuous true-short-axis gradient echo (blackblood) cine MRI was performed from the base to the apex with an ECG-triggered 2D cine FLASH sequence12 (Figure 1). The echo time (TE) was 1.9 ms, and the repetition time (TR) was adjusted continuously to obtain 16 equally spaced phases through each cardiac cycle. Other parameters include a 15° flip angle, 25.6 × 25.6 mm field of view (FOV), matrix of 128 × 128 pixels, 4 averages and slice thickness of 1.0 mm. This yields a spatial resolution of 200 × 200 × 1000 μm. For late gadolinium enhanced (LGE) imaging of myocardial infarct size on day 1 post-MI (Figure 2), 0.3 mmol/kg of Gd (Gadodiamide, Omniscan, GE Healthcare) was infused intraperitoneally 20 minutes prior to imaging. An inversion recovery sequence was used, with a 180° inversion pulse, TR of 3.9 ms, TE of 1.9 ms, 30° flip angle and 25.6 mm FOV.
Figure 1.

Representative end-systolic black blood cine imaging at baseline, day 0, (top row) and day 28 (bottom row) in a control mouse (left), C21-treated mouse (center), and candesartan-treated mouse (right.) Note the cavity dilatation and regional wall thinning at end-systole at day 28 most pronounced in the control and C-21 treated example.
Figure 2.
Representative day 1 late gadolinium enhancement (LGE) images of the mid-LV in a control (left), C21-treated mouse (center), and candesartan-treated mouse (right). LGE infarct areas (bright regions) are seen in the distribution of the left coronary artery in the anterolateral free wall in all mice with similar sizes. The black arrowheads point to the border between the bright infarcted tissue and the dark normal myocardium. Mean infarct sizes by LGE CMR, when summed over the entire LV, were similar between groups.
Therapy
Twenty three mice received 0.3 mg/kg/d of C21 delivered by surgically placed Alzet osmotic mini-pumps from immediately after the time of infarction to day 28 post-MI. This dose has previously been shown to attenuate post-MI remodeling in Wistar rats9. Sixteen mice received candesartan 10 mg/kg/d in drinking water, a dose validated in prior post-MI studies13, from after the time of infarction to day 28 post-MI. The remaining controls (n=20) did not have an Alzet minipump implanted nor anything added to their drinking water.
Invasive Hemodynamics
Invasive measurement of LV filling pressures was performed on all mice after the day 28 post-MI MRI. Mice were anesthetized with an intraperitoneal injection of pentobarbital sodium 50 μg/g body weight enhanced by local subcutaneous injection of 0.5% Bupivacaine 100 μl. LV pressure was obtained by direct puncture of the LV via left 5th interspace with a 27G needle connected to PE-50 tubing and pressures as well as heart rate were recorded by a MacLab recording System.
AT2R Protein Levels
Plasma membranes from whole mouse hearts were isolated using a triton solubilized 100,000 × G membrane preparation based upon the method of Nagamatsu et al with slight modifications14. Protein concentrations were quantified using a bicinchoninic acid protein (BCA) assay (Pierce) and subjected to Western blot analysis. Sodium dodecylsulfate (SDS) samples were then prepared, separated by SDS-PAGE (10% polyacrylamide gels), and transferred onto a nitrocellulose membrane by electroblotting. Membranes were incubated overnight with AT2R antibody (Chemicon 1:200) at 4°C and subsequently incubated with infrared secondary antibody (anti-rabbit IRDye 800nm; 1:15,000, Licor Biosciences). Immunoreactivity and quantitative assessment of band densities were performed using the Odyssey Infrared Imaging System (Licor Biosciences). The blots were then incubated with SimplyBlue SafeStain (Invitrogen) in order to stain total protein loaded per lane. Immunoreactivity was performed using the Odyssey Infrared Imaging System and quantitative assessment of band densities was performed using MacBiophotonics ImageJ software. Results from 14 controls, 11 candesartan-treated, and 18 C21-treated were reported as a ratio of densitometric units of AT2R to total protein loaded.
Image Analysis
Quantitative image analysis was performed using the ARGUS (Siemens Healthcare, Princeton, NJ) image analysis program to quantify end-diastolic (EDV) and end-systolic volumes (ESV) and ejection fraction (EF). EDV and ESV were indexed to body weight in grams (EDVI, ESVI). Infarct size quantification, also using the ARGUS program, was performed on the day 1 LGE images15 (Figure 2). For this calculation, LGE regions were planimetered and the total infarct area calculated as a percentage of LV mass.
Statistical Analysis
Parameters of LV size and function over time were compared between 3 groups using repeated measures analysis of variance (ANOVA) using Sigma Stat (v. 2.0 for Windows, SPSS, Chicago, IL). Infarct size, hemodynamic parameters, and AT2R protein levels were compared between groups by one way ANOVA. All values are presented as mean ± S.D; P<0.05 was considered significant.
Results
Baseline LV Size and Function
LV EDVI, ESVI, and EF were similar amongst the 3 groups at baseline as shown in Table 1, Figures 3 and 4.
Table 1.
LV end-diastolic and end-systolic volume indexed to body weight in grams and ejection fraction
| EDVI (μL/g) | ESVI (μL/g) | EF (%) | |
|---|---|---|---|
| Day 0 | |||
| Control (n=20) | 1.56±0.35 | 0.62±0.23 | 62±7 |
| C21 (n=23) | 1.84±0.45 | 0.74±0.30 | 61±8 |
| Candesartan (n=16) | 1.73±0.39 | 0.74±0.29 | 58±10 |
| Day 1 | |||
| Control | 1.98±0.50 | 1.17±0.46 | 42±10 |
| C21 | 2.39±0.44* | 1.50±0.34 | 37±7 |
| Candesartan | 2.32±0.40 | 1.47±0.44 | 37±9 |
| Day 7 | |||
| Control | 2.63±0.57 | 1.82±0.60 | 32±11 |
| C21 | 3.08±0.69* | 2.30±0.71* | 27±11 |
| Candesartan | 2.82±0.70 | 1.95±0.66 | 33±9 |
| Day 28 | |||
| Control | 2.77±0.85 | 1.96±0.78 | 29±11 |
| C21 | 3.34±0.82* | 2.43±0.74* | 27±12 |
| Candesartan | 3.03±0.78 | 1.96±0.77 | 37±10† |
All results expressed as mean±S.D.
P<0.05 vs. control,
P<0.05 vs. C21
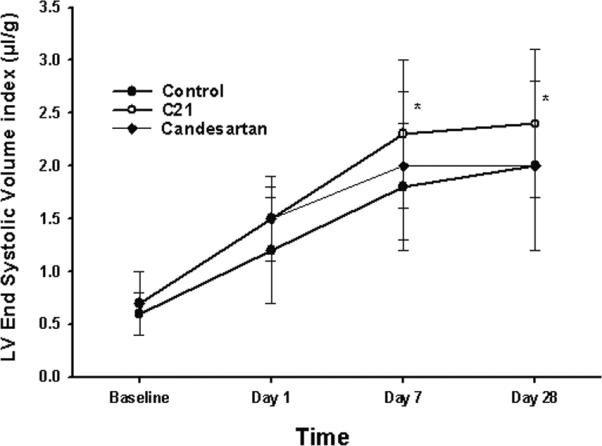
Figure 3.

LV end systolic volume index at each time point for all 3 groups. Data shown are mean±S.D.. *P<0.05 vs. control
Figure 4.

Ejection fraction at each time point for all 3 groups. Data shown are as mean±S.D.. †p<0.05 vs. C21
Infarct Size
MI size as defined by LGE CMR on day 1 post-MI was similar between control, C21-treated and candesartan-treated mice (45 ± 12%, 45 ± 11%, 42 ± 13%; respectively, p=NS) as illustrated in Figure 2.
Post-MI LV Size and Function
In all 3 groups, EDVI and ESVI increased stepwise during the course of post-MI remodeling with most of the dilatation occurring by day 7 (Table 1). Similarly, EF fell between baseline and day 1, further by day 7 and then stabilized in all groups. EDVI was higher in C21 than control at each time point after infarction. ESVI was higher in C21 than control at day 7 and day 28 post-MI. No difference in EDVI or ESVI was noted between candesartan-treated and control mice. EF was best preserved in the candesartan-treated group such that at day 28 LVEF was 37±10%, higher than C21 (27±12%) but not statistically different than control mice (29±11%).
Invasive Hemodynamics
LV end-systolic pressure was lower in the candesartan-treated group at day 28 post-MI compared to controls (Table 2) due to the vasodilatory properties of AT1R antagonism. C21 was blood pressure neutral (Table 2) as has been true in past studies8;9. Otherwise, heart rate and LV end-diastolic pressure and were similar between groups.
Table 2.
Invasive Hemodynamic Parameters on Day 28
| HR (bpm) | LVESP (mmHg) | LVEDP (mmHg) | |
|---|---|---|---|
| Control | 521 ± 38 | 101 ± 8 | 4 ± 3 |
| C21 | 552 ± 67 | 94 ± 8 | 6 ± 5 |
| Candesartan | 559 ± 25 | 80 ± 14* | 5 ± 8 |
HR, Heart Rate; LVESP, LV End-Systolic Pressure; LVEDP, LV End-Diastolic Pressure;
p<0.01 vs. control,
AT2R Protein Levels
AT2R protein levels, expressed as a ratio of densitometric units of AT2R to total protein loaded (Figure 5), were higher in C21-treated mice (1.21±0.17, mean±S.E., p<0.05) than control (0.72±0.06) suggesting AT2R upregulation by AT2R agonism in the post-MI setting. No difference was found between and the candesartan-treated group (1.11±0.16) and control.
Figure 5.

Top: Representative western blots measuring the AT2R band at 42 KD as well as total protein for each treatment group. Data represent mean ± SE. Bottom: Western blot analysis of whole cell membranes from day 28 post-MI mouse hearts (control n=14, candesartan-treated n=11, and C21 treated n=18) demonstrating higher AT2R protein expression following C-21 treatment than control. Data is presented as the ratio of AT2R protein expression over total protein loaded.
Discussion
In the present study, 59 infarcted mice were studied over 28 days post-MI, 20 of which were untreated controls, 23 treated with a novel non-peptide agonist of the AT2R (C21) and 16 treated with the AT1R inhibitor candesartan. As expected, AT1R antagonism attenuated the decline in LV ejection fraction post-MI. In contrast, AT2R agonism and receptor upregulation were not beneficial and, in fact, were associated with adverse parameters of LV remodeling compared to control as evidenced by the increase in LVEDVI and LVESVI. In this study, direct stimulation of the AT2R by C21, at the 0.03mg/kg/d dose, did not attenuate post-MI LV remodeling.
LV remodeling or the increase in LV size over time post-MI, is associated with adverse outcomes including congestive heart failure (CHF) and death. A principal goal of pharmacologic therapy post-MI is to prevent his process. ACE-I remain the standard of care in patients with post-MI LV dysfunction while AT1R antagonists are commonly used substitutes in ACE-I intolerant patients. Both therapies have been shown to be beneficial in this setting2, with the edge to ACE-I.
It is well established that Ang II in the heart causes vasoconstriction, cellular hypertrophy and interstitial fibrosis 16. However, most of the recognized effects of Ang II are mediated through the AT1R, while physiologic functions of the AT2R in myocardium remain under scrutiny. The AT2R is generally thought to inhibit ventricular growth and remodeling17 and induce vasodilatation18. A 2–3-fold increase in AT2R mRNA has been demonstrated in non-infarcted and infarcted regions of the 1-week post-MI rat heart19. Antagonism of the AT2R with selective antagonists reduces endothelial and fibroblast DNA synthesis in the heart and negatively impacts cardiac output and stroke volume in a rat model of MI 20. Our group has shown that overexpressing the AT2R in the heart leads to attenuation of post-infarct LV remodeling3 and that his process is mediated by nitric oxide5, but not by bradykinin6. Overexpression of AT2Rs was equivalent to AT1R antagonism4, suggesting that much of the benefit of AT1R antagonism could be due to AT2R upregulation. Thus, there is significant potential for a new class of agents that are direct AT2R agonists that could be additive to therapy in post-MI LV dysfunction.
Studying the effects of the AT2R remains challenging, primarily due to the lack of reliable tools to directly activate the AT2R. Previously, the only available AT2R agonists were metabolically unstable peptides (CGP42112A and p-amino-Phe6)21–23 while the only available AT2R antagonist, PD123319, is nonselective when used at higher doses7, The emergence of the first non-peptide AT2R agonist, C21, has allowed investigators to selectively stimulate this receptor in both in vitro and in vivo settings8.
Recently, it was reported that the use of C21 in post-MI Wistar rats may attenuate post-myocardial remodeling, leading to reduced scar volumes as assessed by MRI, improved hemodynamic parameters as assessed by Millar catheter, and improved LV structural changes as assessed by transthoracic echocardiography9. In fact, remodeling attenuation was reported at both the 0.03 and 0.3 mg/kg/d doses. While data from this study are encouraging, limitations in study design create potential problems with interpretation of the data. For example, induced MI's led to small overall infarct scar sizes on day 7, ranging from 14.6 ± 1.4% in the C21-treated group to 20.5 ± 1.5% in controls9. Measuring infarct size on day 7 rather than on day 1 is potentially problematic because the therapy itself may have altered infarct size and remodeling of noninfarcted myocardium, leading to incorrect sizing of the infarcts by that time point. It would have been important to ensure that infarct sizes were similar on day 1 post-MI. In addition, infarct size drives subsequent LV remodeling, so if infarct size is different, it follows that LV remodeling will be attenuated in the group with the smaller infarct. Thus, the main conclusion that can be drawn from the study of Kaschina et al is that C21 attenuates infarct size. Its effect on LV remodeling cannot be separated from the latter effect.
Benefits of AT2R agonism on infarct size have been shown in the cerebrovascular bed. When administered centrally, the AT2R agonist CGP42112A reduced infarct volume in the setting of stroke in a conscious rat model and minimized the loss of AT2R expression in the infarcted region24. The authors suggested that AT2Rs reduce oxidative stress during ischemia based on an inverse relationship between superoxide production and AT2R expression. In this study there was no tonic benefit of AT2R expression as the AT2R inhibitor PD123319 had no effect on outcomes. A major difference between this study and the present study was that the stimulation of the AT2R occurred prior to the initial injury whereas in the present experiments, therapy was not begun until after the reperfused infarction had occurred.
There are several potential reasons for the lack of benefit and tendency towards adverse effects of C21. Firstly, the 0.3mg/kg/d dose may not be the ideal dose in mice despite its proven benefits in the rat model9. Secondly, mean infarct sizes in the current study were quite large (ranging from 42–45%). The larger infarct sizes could have overridden any potential benefit of C21 at the dose studied. Future studies might include not only a range of infarct sizes, but also a range of C21 doses. It may be that the amount of increased activity with AT2R overexpression in mice that we have shown to be beneficial in this setting which is likely to be an increase of more than 50-fold3 is greater than can be achieved with direct agonism. In addition, it may be that the prime effect of AT2R agonism in this setting is reduction in infarct size as it was in prior studies in MI9 and stroke24 models.
In conclusion, AT2R activation with Compound 21 at the dose studied is ineffective in attenuation of post-infarct LV remodeling in the mouse model, especially when compared to the AT1R inhibitor candesartan. Although previous studies suggest that C21 is associated with reduction in infarct size, the present study does not support the concept that C21 reduces LV remodeling, raising doubt regarding its potential efficacy in the heart failure setting.
Acknowledgements
none
Grant Funding - Supported by AHA Mid-Atlantic Affiliate Grant-in-Aid 0855409E (CMK) and T32 EB003841 (ABJ).
Footnotes
Disclosures - none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Li H, Gao Y, Grobe JL, Raizada MK, Katovich MJ, Sumners C. Potentiation of the antihypertensive action of losartan by peripheral overexpression of the ANG II type 2 receptor. Am J Physiol Heart Circ Physiol. 2007;292:H727–H735. doi: 10.1152/ajpheart.00938.2006. [DOI] [PubMed] [Google Scholar]
- 2.Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau J, Kober L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM, The Valsartan in Acute Myocardial Infarction Trial Investigators Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med. 2003;349:1893–1906. doi: 10.1056/NEJMoa032292. [DOI] [PubMed] [Google Scholar]
- 3.Yang Z, Bove CM, French BA, Epstein FH, Berr SS, Dimaria JM, Gibson J, Carey RM, Kramer CM. Angiotensin II type 2 receptor overexpression preserves left ventricular function after myocardial infarction. Circulation. 2002;106:106–111. doi: 10.1161/01.cir.0000020014.14176.6d. [DOI] [PubMed] [Google Scholar]
- 4.Voros S, Yang Z, Bove CM, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP, Conaway MR, Matsubara H, Carey RM, Kramer CM. The interaction between AT1 and AT2 receptors during postinfarction left ventricular remodeling. Am J Physiol Heart Circ Physiol. 2006;290:H1004–H1010. doi: 10.1152/ajpheart.00886.2005. [DOI] [PubMed] [Google Scholar]
- 5.Bove CM, Yang Z, Gilson WD, Epstein FH, French B, Berr SS, Bishop SP, Matsubara H, Carey RM, Kramer CM. Nitric oxide mediates benefits of angiotensin II type 2 receptor in post-infarct remodeling. Hypertension. 2004;43:680–685. doi: 10.1161/01.HYP.0000115924.94236.91. [DOI] [PubMed] [Google Scholar]
- 6.Isbell DC, Voros S, Yang Z, DiMaria JM, Berr SS, French BA, Epstein FH, Bishop SP, Wang H, Roy RJ, Kemp BA, Matsubara H, Carey RM, Kramer CM. Interaction between bradykinin subtype 2 and angiotensin II type 2 receptors during post-MI left ventricular remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H3372–H3378. doi: 10.1152/ajpheart.00997.2007. [DOI] [PubMed] [Google Scholar]
- 7.Macari D, Bottari S, Whitebread S, de Gasparo M, Levens N. Renal actions of the selective angiotensin AT2 receptor ligands CGP 42112B and PD 123319 in the sodium-depleted rat. Eur J Pharmacol. 1993;249:85–93. doi: 10.1016/0014-2999(93)90665-5. [DOI] [PubMed] [Google Scholar]
- 8.Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, Johansson B, Holm M, Botoros M, Karlen A, Pettersson A, Nyberg F, Fandriks L, Gallo-Payet N, Hallberg A, Alterman M. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47:5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 9.Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato C, Namsolleck P, Tschope C, Hallberg A, Alterman M, Hucko T, Paetsch I, Dietrich T, Schnackenburg B, Graf K, Dahlof B, Kintscher U, Unger T, Steckelings UM. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation. 2008;118:2523–2532. doi: 10.1161/CIRCULATIONAHA.108.784868. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z, Zingarelli B, Szabo C. The crucial role of endogenous interleukin-10 production in myocardial ischemia-reperfusion injury. Circulation. 2001;101:1019–1026. doi: 10.1161/01.cir.101.9.1019. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Huo Y, Toufektsian MC, Ramos SI, Ma Y, Tejani AD, French BA, Yang Z. Activated platelets contribute importantly to myocardial reperfusion injury. American Journal of Physiology - Heart and Circulatory Physiology. 2006;290:H692–H699. doi: 10.1152/ajpheart.00634.2005. [DOI] [PubMed] [Google Scholar]
- 12.Berr SS, Roy RJ, French BA, Yang Z, Gilson WD, Kramer CM, Epstein FH. Black blood gradient echo cine magnetic resonance imaging of the mouse heart. Magn Reson Med. 2005;53:1074–1079. doi: 10.1002/mrm.20487. [DOI] [PubMed] [Google Scholar]
- 13.Lange SA, Wolf B, Schober K, Wunderlich C, Marquetant R, Weinbrenner C, Strasser RH. Chronic Angiotensin II Receptor Blockade Induces Cardioprotection During Ischemia by Increased PKC-[varepsilon] Expression in the Mouse Heart. J Cardovasc Pharmacol. 2007;49 doi: 10.1097/FJC.0b013e31802c2f77. [DOI] [PubMed] [Google Scholar]
- 14.Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE, Bell GI. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem. 1992;267:467–472. [PubMed] [Google Scholar]
- 15.Yang Z, Berr SS, Gilson WD, Toufektsian MC, French BA. Simultaneous evaluation of infarct size and cardiac function in intact mice by contrast-enhanced cardiac magnetic resonance imaging reveals contractile dysfunction in non-infarcted regions early after myocardial infarction. Circulation. 2004;109:1161–1167. doi: 10.1161/01.CIR.0000118495.88442.32. [DOI] [PubMed] [Google Scholar]
- 16.Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 17.Stoll M, Steckelings UM, Paul M, Bottari SP, Metzger R, Unger T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J Clin Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res. 1998;83:1182–1191. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- 19.Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest. 1995;95:46–54. doi: 10.1172/JCI117675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuizinga MC, Smits JFM, Arends JW, Daemen MJ. AT2 receptor blockade reduces cardiac interstitial cell DNA synthesis and cardiac function after rat myocardial infarction. J Mol Cell Cardiol. 1998;30:425–434. doi: 10.1006/jmcc.1997.0607. [DOI] [PubMed] [Google Scholar]
- 21.Chiu AT, Herblin WF, McCall DE, Ardecky RJ, Carini DJ, Duncia JV, Pease LJ, Wong PC, Wexler RR, Johnson AL. Identification of angiotensin II receptor subtypes. Biochem Biophys Res Commun. 1989;165:196–203. doi: 10.1016/0006-291x(89)91054-1. [DOI] [PubMed] [Google Scholar]
- 22.Speth RC, Kim KH. Discrimination of two angiotensin II receptor subtypes with a selective agonist analogue of angiotensin II, p-aminophenylalanine6 angiotensin II. Biochem Biophys Res Commun. 1990;169:997–1006. doi: 10.1016/0006-291x(90)91993-3. [DOI] [PubMed] [Google Scholar]
- 23.Whitebread S, Mele M, Kamber B, de Gasparo M. Preliminary biochemical characterization of two angiotensin II receptor subtypes. Biochem Biophys Res Commun. 1989;163:284–291. doi: 10.1016/0006-291x(89)92133-5. [DOI] [PubMed] [Google Scholar]
- 24.McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 Receptor Stimulation Causes Neuroprotection in a Conscious Rat Model of Stroke. Stroke. 2009;40:1482–1489. doi: 10.1161/STROKEAHA.108.531509. [DOI] [PubMed] [Google Scholar]