

Abstract
This work describes the proteomic characterization of a novel in vitro prostate cancer model system, the clonal Prostatic Human Epithelial Cancer (PHEC) cell lines. The model is composed of three cell lines representing the three progressive cancer states found in vivo: non-tumorigenic, tumorigenic, and metastatic. The cell lines were evaluated for differential protein expression between states using two dimensional liquid:liquid chromatographic separation followed by mass spectral identification. The proteins from cellular extracts were first separated using liquid:liquid primary separation based on their isoelectric points and molecular masses. The resulting peptide fractions were applied to liquid chromatography mass spectrometry (LCMS) separation for mass determination and protein identification based on Mascot database inquiry. Over 200 proteins that change expression over the course of progression of this in vitro prostate cancer model were discovered during the comparative analysis of the three cell lines. The importance of these proteins on prostate cancer progression remains to be elucidated with further characterizations. The combination of the two dimensional liquid:liquid separation and mass spectral identifications was used to successfully analyze differential protein expression between multiple cell lines.
Keywords: PF2D, Prostate Cancer Progression
1. Introduction
Prostate cancer (PRCA) is the most common non-skin cancer in men, affecting 1 in 6 men. According to the American Cancer Society, there were more than 217,730 new cases diagnosed, and approximately 32,050 deaths, in 2010 alone1. Mortality rates have decreased due to earlier detection through the prevalence of screening with the prostate specific antigen (PSA) test. The protein PSA, initially characterized in 19712, is a serine protease (EC 3.4.21.77) that is neither specific nor solely prostatic in origin. PSA is also found in breast3, lung4, uterine5 and renal6 cancer. Though not specific as a predictor of PRCA, PSA in patients diagnosed with PRCA is an excellent marker for the disease attaining a metastatic state independent of treatment. To facilitate the understanding of PRCA etiology and provide an earlier and more specific diagnostic tool for prostate cancer detection, improved model systems for PRCA biomarker research are required.
The development of cancer model systems is valuable to understand the progression of the disease. However, few systems distinctly present all states of the disease progression within one model. Most model systems are developed using a single state of cancer or disease, such as the LNCaP prostate cancer metastasis model. The few models that do represent cancer progression are limited by difficulties in tracking all of the perturbations caused by disease progression. These systems are excellent at modeling the effects of a single perturbation on a “closed” system. However, being able to follow the progression of a disease through one consistent model allows a systems approach, providing a closer emulation of true disease progression. Close proximity of any model system to true disease physiology is essential for the evaluation of therapeutic targets. Here, we report the proteomic characterization of one uniform model system of prostate cancer progression, the Prostatic Human Epithelial Cancer (PHEC) cell lines, to develop a refined system for studying human PRCA.
1.1. Human Prostate Cancer Model
The PHEC cell lines, first developed by Cunha7, represent a unique cell line for the evaluation of PRCA progression. Various model systems have been developed to characterize PRCA. However, only a minority of these demonstrate cancer progression to metastasis, including TRAMP, Pten-null, Noble rat, and the Lobund-Wistar rat. Examples of those that do not include metastasis are Myc-null, LPG-TAg or “LADY”, Nkx3.1 null, TbR-II-knockout (KO), Prb-AR-transgenic, IGF transgenics, (urogenital sinus mesenchyme) UGM+mPrERb−/−, and carcinoma associated fibroblasts+hPrE TRs. The drawbacks of these models are that the carcinomas are usually of non-human (rodent) origin, and they lack an analogous phenotype, the development of prostatic intraepithelial neoplasia (PIN)8.
Our PHEC model is derived from the common ancestor line of other human models (BPH-1)9. Unlike these other models, however, the PHEC model progresses through all three cancer states seen in vivo, as well as developing the pre-cancerous PIN. While the etiology of PRCA remains unknown, the steroid hormones testosterone and estradiol-17β are known to be involved in carcinogenesis of the prostate, likely through reaction with the androgen receptor10. These cell lines stimulate progression through these constitutive physiological human hormones, and it is the only model system that undergoes human epithelial malignant transformation and metastasis, and thus represents all stages of human carcinogenesis.
These cell lines originate from tissue recombination of the non-tumorigenic human prostate epithelial cell line (BPH-1) with embryonic stroma-urogenital mesenchyme (UGM) isolated from rat seminal vesicles. Tissue recombinants grown in control animals developed into orderly and benign prostate-like tissue (PHECNT), whereas grafts grown in hormone-treated hosts developed into cancer at 2 months (PHECT). At 4 months, growth progressed to malignant cancer with metastases (PHECM) to lymph nodes, lung, and liver. These cells now have the ability to be grown in vitro, with three distinct cell lines created at sequential time points to represent the three states of cancer progression, and have been shown to be human in origin11. They model many of the same cell types, progression markers, and epigenetic behavior as human cancer progression. Therefore, it serves as an exceptional model system for the analysis of the physiological changes of PRCA progression at the cellular level.
1.3. Liquid:Liquid Separation System
We have applied the use of a PF2D protein fractionation system (Beckman Coulter) to our analysis of the PHEC cell line. This system has an advantage over conventional two-dimensional electrophoresis (2DE) as it allows identification of a new fraction of the proteome, which overlaps with the 2DE proteome by approximately 20%12. In this system, samples are fractionated based on their pI and hydrophobicity, in contrast to the 2DE separations based on pI and molecular weight. Separation by PF2D employs a liquid:liquid system, based on liquid chromatography. The PF2D involves alternate physical separation properties and does not use a gel, so the resulting subset of proteins can be isolated to identify many proteins not seen with other methods. The PF2D system also preserves post-translational modifications routinely stripped during other separation techniques. Overall, the PF2D combines the benefits of liquid-based and gel-based separations to generate an alternate sample for further characterizations.
2. Materials and Methods
2.1.Culture and Lysate Collection of PHEC lines
Lysates analyzed in these studies were generated from the PHEC in vitro cell lines as described previously13. Briefly, PHECNT, PHECT, and PHECM cells were grown in RPMI 1640 medium containing 25 mM HEPES and L-glutamine (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 1X primocin (Invivogen). Cells were grown at 37°C and 5% CO2. When cells reached approximately 75% confluency in a T-150 flask, they were harvested using 0.25% trypsin containing EDTA (Invitrogen). Trypsin was inactivated by adding 10% FBS, and the cells were pelleted via centrifugation at 1500×g for 5 minutes at 4°C. The pellet was rinsed with HBSS (Invitrogen) and stored at −80°C until analysis. Frozen cell pellets, prepared from approximately 1×108 cells, were lysed in a buffer of 6 M urea (Sigma), 2 M thiourea (Sigma), 10% glycerol (EM Science), 50 mM Tris-HCl pH 7.8–8.2 (JT Baker), 2% n-octylglucoside (Calbiochem), and 1 mM protease inhibitor (Sigma). The resulting samples were then vortex agitated for 30–60 seconds, incubated for 30 minutes at room temperature, and centrifuged at 20,000×g for 1 hour at 4°C to pellet the particulate matter. The cell lysate supernatant was exchanged into 3.5 mL Start Buffer (Beckman Coulter) using a PD10 buffer exchange column (GE Healthcare).
2.2. PF2D Liquid/Liquid Proteome Fractionation
Total protein concentrations of the lysates were measured at 280 nm using a Nanodrop spectrophotometer (Nanodrop, Wilmington, DE, USA), and 5 mg of total protein solubilized in PF2D loading buffer was injected into the PF2D system. The first dimension separated by chromatofocusing (CF) using a proprietary eluent buffer (Beckman Coulter) flowing isocratically at 0.2 mL/min on a Beckman Coulter CF column. Fraction collection occurred in intervals of 0.3 pH units within a range of pH 8.5–4.0, and then every 8.5 min outside this pH range, with fractions collected in a 96-well deep-well plate in a chilled fraction collector. These samples were then re-injected automatically for the second dimension analysis, taking 250 µL per well. The separation of the second dimension incorporated standard reversed-phase chromatography, performed on a non-porous C18 reverse phase column (Beckman Coulter) with gradient flow of 0.75 mL/min from 0%B to 100%B over 30 minutes using 0.1% TFA in water for mobile phase A and 0.08% TFA in acetonitrile for mobile phase B. Second dimension fractions were collected into 96-well plates continuously from 6–24 minutes run time in 30 second intervals, and fractions were stored in the plates at −80°C until subsequent analysis by mass spectrometry. The second dimension elution parameters represent the standard in the field, and are routinely used to perform post-translational modification analysis for phosphorylations, methylations, dimethylations, and acetylations.
Results were visualized in Mapping Tools software (Beckman Coulter), where differential analysis was performed in the “DeltaVue” mode. Mapping Tools is limited in its ability to overlay samples, only allowing two samples to be overlaid at a time, creating a need for a new technique to view the three states of our model system simultaneously. To visualize plots of the three PRCA states of our model, the data files were read into Excel. We generated a spreadsheet to transfer the values from the Mapping Tools files, converting raw data values into absorbance and time values, which were then plotted and labeled by fraction number and pI.
2.3. Bottom-up Proteomics
Samples were selected for subsequent analysis based on the criteria of a two-fold change in intensity or a substantial change in peak shape. Although peaks were assumed to contain multiple species, a 2X increase in UV signal would reflect an increase in one or more of the peak components. This criteria was qualitative, serving only as a selection parameter for further analysis. Addresses for samples were determined using a spreadsheet generated internally based on timing and patterning of fraction collection in the second dimension. For each well selected for analysis, the corresponding wells in the other two cell lines were also analyzed to provide a true comparison of the full proteomic progression model. However, if a selected well address in one of the other cell lines did not contain a UV signal exceeding 0.01 AU in the primary chromatogram, the analysis of this sample was not performed. Repeated analysis determined individual peaks with signal intensities < 0.01 AU to be below the lower limit of detection for reliable mass spectrometric identification.
For samples selected, the wells were rinsed with acetonitrile, and the samples evaporated with a speedvac until 20 µL sample remained. Samples evaporated below 20 µL were adjusted to 20 µL with 100 mM ammonium bicarbonate solution. Samples were reduced with 3 µL of 10 mM DTT (Calbiochem) and alkylated with 2 µL of 55 mM IAA (Sigma) before digestion with 375 ng trypsin (Promega). Digestion was stopped by the addition of 3 µL of 1% TFA to a final concentration of 0.1% TFA, and digested samples were evaporated to dryness before reconstitution in 10 µL of 5% acetonitrile with 0.1% formic acid in water. Samples were loaded into autosampler vials and placed in a cooled autosampler for LC-MS/MS analysis. LC separation was performed on an Ultimate 3000 (Dionex) plumbed for nanoflow parameters. Separations were achieved on a 75 µm × 15 cm C18 PepMap column (3 µm, 100 Å pore size) from LC Packings. Peptides were eluted with a gradient of 5–50% B over 35 minutes followed by 50–90% B for 10 minutes at a flow of 400 nL/min. MS/MS analysis was performed in-line with the LC on a Micro-TOF QII (Bruker Daltonics).
For MS/MS analysis, a survey scan was performed, followed by fragmentation (MS/MS) of the top three most abundant peaks in the survey spectrum. A spectral rate of 1.3 Hz was used for MS spectra and 0.7 Hz for MS/MS spectra. Argon was used as a collision gas, and the RF of the collision cell was used in sweep mode to ensure detection of low m/z ions. Peptides were identified via the MASCOT database (Matrix Science) by searching individual peptide mass data against the NCBInr database for human proteins with carbamidomethyl cysteine as a fixed modification and oxidized methionine as a variable modification14. MuDPit scoring was used, so peptides with scores ≥ 38 were considered to have high homology, or a positive identification (p<0.05). Peptide identifications were accepted if the false discovery rate was less than 5% and a minimum of one peptide met the identity threshold.
2.4. Western Blot Analysis
Cells were grown and harvested as described above, and pelleted cells were lysed in 1X RIPA buffer. Protein concentration was determined using Amersham’s 2D quant kit, and 50 µg protein for each sample was used for Western Blot analysis. Here, proteins were separated on a 1D SDS-PAGE gel (4–20% TRIS-Glycine from BioRad) then transferred to PVDF membrane using an iBlot System (Invitrogen). Membranes were blocked with 10% milk in TBST. Primary antibodies were either α-tubulin for loading control or maspin (Cell Signaling). Detection occurred through HRP-coupled secondary antibodies.
3. Results and Discussion
We undertook the study of the PHEC cell lines using the PF2D system to define the differences between the three states of our cancer progression model. Our investigations have accomplished the bottom-up proteomic analysis of the three PHEC states and have identified several species of interest corresponding to specific cancer states. In doing so, these studies also allowed an evaluation of the PF2D liquid separation format.
3.1. PF2D-Differential Liquid:Liquid Separation of PHEC Cell Line
Output from PF2D separations occurs initially as a series of single chromatograms of the different separations from the two-dimensional HPLC system, as shown in Figure 1. We extracted the 214 nm UV data signal for each chromatogram and compared each as in Figure 1C. These chromatograms are then converted to pixelated graphs, visualized using the software “Mapping Tools” (Beckman Coulter) to produce an image similar to that generated when analyzing two-dimensional gels. This “pseudo” 2D gel image was then compared between multiple proteomes for differences, with color intensity in the image corresponding to concentration of protein eluted from the columns. For each proteome analyzed, 5 mg protein was injected, allowing for direct comparison between proteomes based on changes in protein expression in the 2D images. Figure 2 illustrates the digital UV output from the PF2D system of the proteomes for PHECNT, PHECT, and PHECM cell lysates. Between each pair of individual state images is a comparison spectrum. Circles have been added to highlight the differences between the proteomes of the two cell lines being compared. These comparison spectra are referred to as the “difference spectrum” between the proteomes of PHECNT vs. PHECT, and PHECT vs. PHECM. Though UV spectral comparisons are done to select fractions for MS/MS identification, it is understood that each UV peak likely contains multiple proteins. Mass spectral analysis is used for all comparative conclusions regarding single proteins and characterizations of expression throughout the cancer progression model.
Figure 1.

Chromatographic separation of the three cell lines of the PHEC model. Equivalent amounts of protein from the lysis of the three individual cell lines were separated by pI in the first dimension and hydrophobicity (reversed-phase) in the second dimension. Fractions were collected in the first dimension in the range of pI 4.0–8.3 and in the second dimension for retention times 6–24 minutes, with UV detection at 214 nm for both dimensions. A) A UV chromatogram is acquired for each fraction within a given sample. B) Mapping Tools is used to yield a composite image showing a “pseudo” 2D gel for each sample. Each lane represents one fraction. Fractions can then be compared between samples (boxed section). C) Relative quantitation can be determined by overlaying the fraction chromatograms from the three samples.
Figure 2.

Proteomic difference maps for the comparison of the PHECNT, PHECT and PHECM cell lines show the PF2D profiles for the respective cell lines from the pI range of 4.0 to 8.3. Between each single cell line profile is the composite difference map comparing two cell lines: red and black for PHECNT vs. PHECT, and black and blue for PHECT vs. PHECM. In the profiles, the intensity of the band color gives a relative intensity for that sample, with a darker band correlating to a higher protein concentration as compared to another spot of lower color intensity. The difference maps also show relative intensity, with the color indicating the sample of origin. Circles indicate samples that were recovered for mass spectrometry analysis and identification, with numbers corresponding to information in Table 1.
3.2. Identification of Altered Protein Production Between Progression States
Based on comparative analysis of the proteomes of the three PHEC cell lines, the liquid fractions were identified for further characterization using for bottom-up proteomics. Retention time was used to determine the well address for each fraction of interest, and comparisons were made only between the exact two-dimensional positions between proteomes. The selected samples underwent tryptic digestion and mass spectrometry for protein identification. In Figure 2, the circled differences were identified in this manner, and a condensed summary of proteins identified is given as Table 1. Full quantitative assessment of total expression levels of proteins was not performed. The identification and expression of proteins discussed refers to relative qualitative expression between cancer progression states.
Table 1.
A summary of proteins identified by mass spectrometry after separation by 2D HPLC. Information pertaining to location in first and second dimension plates of the PF2D run, MASCOT score, accession number and number of identified peptides is also given for each protein. Mascot scores >38 indicate homology or identity in the database searching lead to an identification of a protein of interest. Spot number refers to the location number circled in Figure 2.
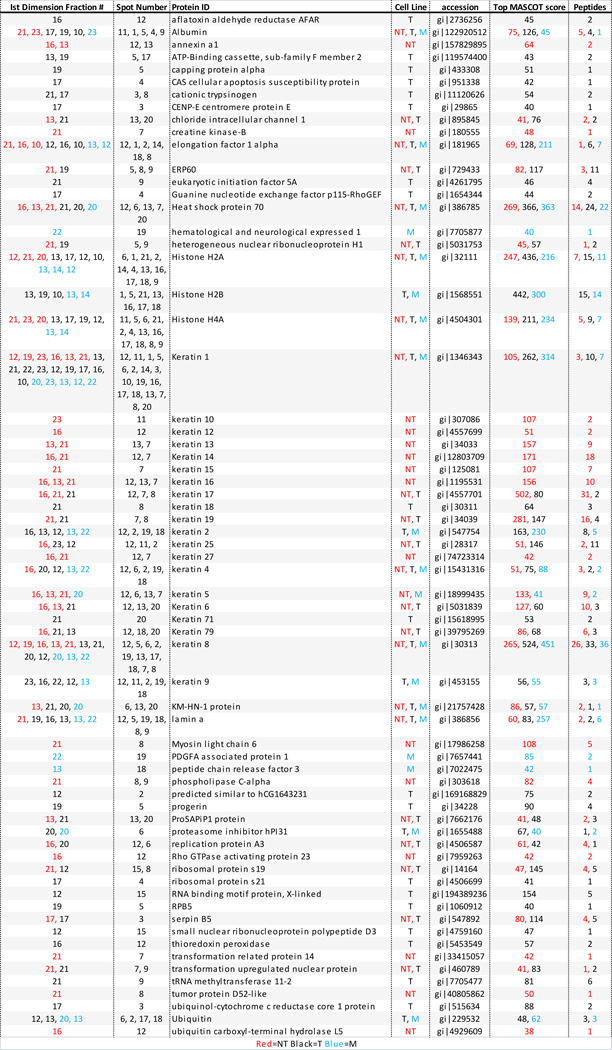 |
An overview of Table 1 reveals that the some proteins have been identified in more than one sample, or at multiple elution points during the 2D chromatography separation process. This can be understood by classifying the identifications and localizations into three general groups: 1) proteins that are localized to the same first dimension fraction but have different retention times in the second dimension, categorized as hydrophobic effects, 2) proteins in adjacent first dimension fractions having similar retention times in the second dimension, categorized as focusing effects, and 3) proteins with the same identification that eluted in different fractions in both first and second dimensions, categorized as modification effects. As these separations use liquid chromatography to resolve complex protein mixtures, the resulting protein fractions have a range of elution points, similar to a normal distribution curve. The fractionation ranges possible due to hydrophobic and focusing effects are compounded by the two separate dimensions of the PF2D separation, creating mixtures of proteins with multiple final endpoints.
Proteins eluted from the PF2D were fractionated by defined intervals, using pH units for the first dimension and time for the second dimension. If a protein began to elute at a pH or time near the end of one fraction qualifier, it could finish in a second fraction qualifier, thereby splitting the protein into multiple fractions. This can be confounded further if the protein elutes into two fractions in the first dimension and two in the second dimension, essentially allowing it to be identified in four separate fractions. Depending on the biochemical and conformational properties of the protein, elution can occur across a very narrow fraction range or a wider one. In this study, one example of impact of these effects was the protein eukaryotic translation elongation factor (eEF1A1). EEF1A1 was found in fractions 13, 17 and 18 (Figure 3), indicating elution over a 1.5 minute period, from retention times 16.5–18 minutes. Since the second dimension collects a fraction every 30 seconds, eEF1A1 was identified in 3 separate fractions. This is also an example of a protein with focusing effects, as it is found in first dimension fractions 12 and 13 (pI 7.4–7.7 and 7.1–7.4, respectively).
Figure 3.

Sites of PF2D where eukaryotic translation elongation factor (eEF1A1) Proteomic difference maps for the comparison of the PHECNT, PHECT and PHECM cell lines show the PF2D profiles for the respective cell lines from the pI range of 4.0 to 8.3. Between each single cell line profile is the composite difference map comparing two cell lines: red and black for PHECNT vs. PHECT, and black and blue for PHECT vs. PHECM. In the profiles, the intensity of the band color gives a relative intensity for that sample, so a darker band correlates to a higher protein concentration in that spot as compared to another spot of lower color intensity. The difference maps also show relative intensity, with higher color intensity from one sample versus the other indicating a higher in abundance in this sample than the one to which it is compared. Circles indicate samples that EEF1A1 was recovered.
Proteins with multiple elution points can also originate from modification effects, the third classification. This general term refers to proteins with structural or conformational changes that impact the chemistry of separation. Included in this category are proteins with various post-translational modifications (PTMs) different isoforms of one protein, analogous proteins, and proteins with point mutations. PTMs and point mutations can lead to subtle or drastic changes in pI, depending on the amount and nature of the alteration15. Proteins that have non-covalently bound species, such as histones, experience an overall change in pI due to molecular complexation. Protein degradation products are also commonly identified in proteomic analyses. While samples were lysed with buffer containing protease inhibitors to prevent proteolysis during sample preparation, protein degradation occurs regularly within cells, so degradation products were expected. Degradation can lead to changes in tertiary or quaternary structures, so a single degraded protein could have several conformational structures present within a sample. Each conformation interacts with the chromatographic column matrix and mobile phases with slight variations. Depending on the level of degradation present, a single protein could be identified, but with multiple elution points due to changes in chemical interactions with the columns used in the PF2D separation. Similarly, each fraction contains multiple proteins in the same eluent, so all comparative analyses are based upon MS/MS identification of single proteins within complex mixtures.
Further analysis of the results in Table 1 indicates that, in most cases, more than one protein was identified in each fraction. This can be attributed to the biochemical properties of the protein separation and fraction collection methods. Because the separation involves two-dimensional chromatography, as opposed to a traditional single dimensional separation, the elution parameters cannot be optimized simultaneously for both separation chemistries. However, as the chromatography was designed to separate proteins for the subsequent identification by mass spectroscopy, the separation parameters do not need to resolve individual species per peak or fraction collected. Multiplicity of proteins within peaks is balanced by the power of the detection system, so the separation method serves only to enhance the resolution and sensitivity of the subsequent identification.
There are multiple advantages to this technique over more routine methods, such as single dimension chromatography or gel electrophoresis. The basis of the separation is liquid chromatography, so samples are injected in solution and collected as fractions of eluent. This allows the proteins to remain in solution throughout the full separation process, eliminating dry-down and reconstitution steps that can lead to sample loss. The liquid-based separation procedure allows for preservation of PTMs for subsequent identification. Collecting fractions in solution leads to characterization of modifications directly by mass spectrometry, providing a more accurate picture of the nature of the proteins within the cells of origin. This creates a more reliable picture of the proteins within the cells than can be generate from gel-based separation techniques routinely used in proteomic evaluations.
In addition to the advantages for characterizing whole proteins, the use of two-dimensional chromatography for proteomic separations introduces a different set of proteins into the proteomes being studied. While gel-based separations exclude high mass, aggregated, and hydrophobic proteins, such as those found in cell membranes and within subcellular architecture, solution-based separations allow the analysis of all cell lysate proteins injected into the chromatograph. As such, a different subset of the proteome becomes available for identification and characterization.
The comparative analysis of the three proteomes of the PHEC cell lines showed identifications of proteins from a variety of cellular locations. As shown in Figure 4, proteins typically associated with the nucleus increased as the cells progressed toward metastasis. The overall complexity of distribution of protein localizations within the cell changed dramatically as cancer progressed. Cells in the non-tumorigenic state localized to 9 sites, while tumorigenic cellular proteins occurred in 12 locations. Interestingly, the metastatic cellular proteins identified were localized to only 5 cellular places. The combination of the inclusion of higher mass and hydrophobic proteins with the ability to evaluate PTMs and binding partners introduces a powerful tool to the understanding of biomolecular interactions within the cell membrane and within the architecture of the cell. This information would not be available with gel-based methodologies.
Figure 4.

Subcellular localization breakdown of identified proteins. Proteins in categories with one or more (+) in them are uniquely counted in that category and not the individual categories comprise up that category. A) Subcellular localization of total proteins identified from all three states combined. B) Localization of proteins identified from PHECNT lysates. C) Localization of proteins identified from PHECT lysates. D) Localization of proteins identified from PHECM lysates.
With these techniques, we generated a proteomic comparison of the three cancer progression states of the PHEC model. The highest frequencies of proteins identified in the fractions of interest between the three cell lines were members of the cytokeratin family (KRT), filamentous protein polymers responsible for the structural integrity of epithelial cells. Others have shown evidence for the correlation between changes in cytokeratins and cancer progresses to a malignant state. In PRCA, cytokeratins have shown differential expression from human prostate tissue depending on the progression state. A tumor-associated species, specifically present in tumors and absent from normal and most benign prostatic hyperplasia samples, was identified to be a proteolytic fragment of KRT8 that showed a mass reduction and a change in pI value16. Extensive fragmentation of KRT molecules by these proteases has been suggested to support the participation of cytokeratins in malignancy17.
In the PHEC model, cancer progression across the three states showed decreased total expression of keratins, with 42 identifications in 21 fractions of the non-tumorigenic cells, 41 identifications in the same fractions of tumorigenic cells, and 17 identifications in those of metastatic cells. Of the identifications made in the chosen fractions, the non-tumorigenic cells displayed 30 different types of keratins, while only 15 and 9 types of keratins were displayed in the tumorigenic and metastatic cells, respectively, as shown in Figure 5. Their identities are given in Table 1. This level of change in keratin production and variety as cells morph from a healthy state into metastatic cancer cells poses an interesting point of speculation regarding the role of keratins in cancer progression. Keratins perform a predominantly structural role, aligning into large aggregate assemblies within cells. Our data showed the progression to malignancy in the PHEC cell lines leads to altered and inconsistent morphology within tumorigenic and metastatic cell cultures. The observed morphological changes could be the result of a limited expression of keratins, or to the introduction of keratin expression types not typically found in the normally replicating cells. These findings support extensive reports in the literature of the phenotypic changes cells undergo as they transform into fully malignant cancer forms.
Figure 5.
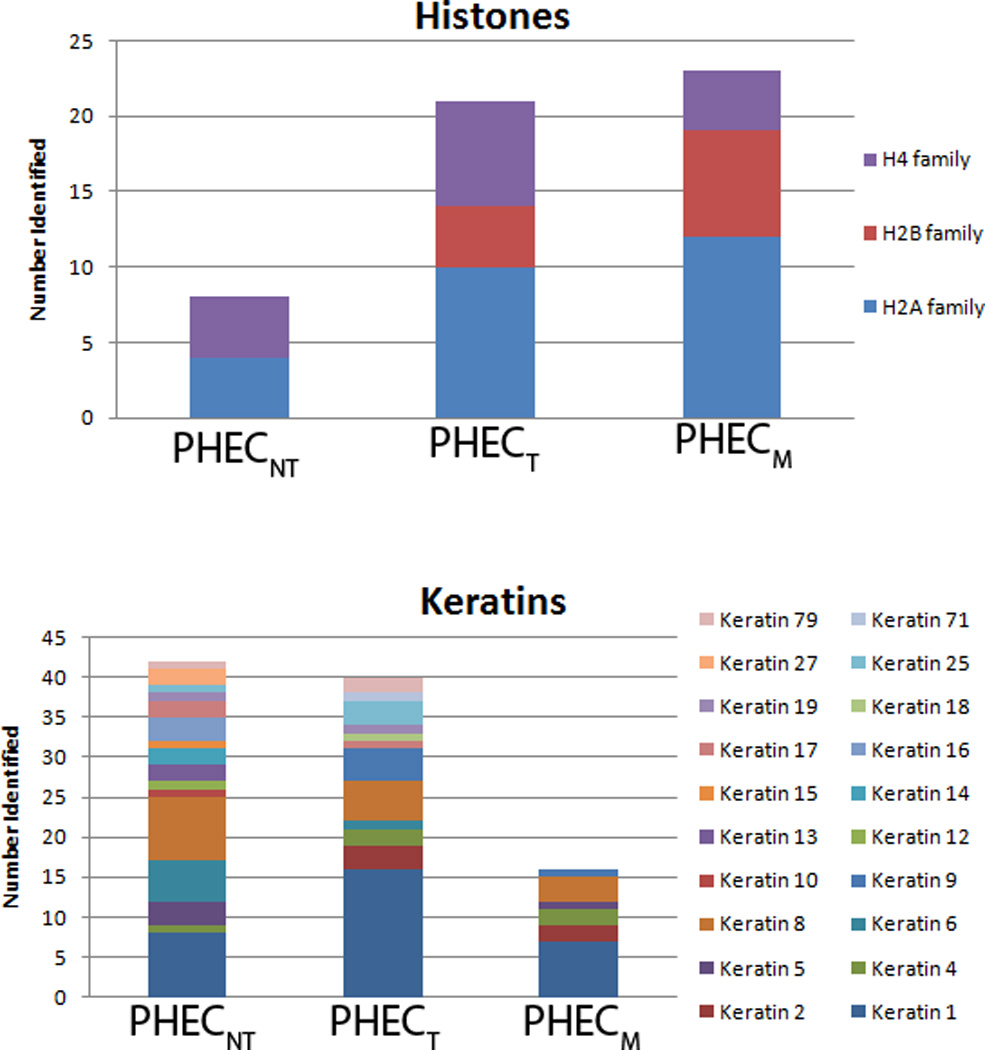
A comparative analysis of the distribution of histones and keratins within the three PHEC model cell lines. The colors within each bar represent the number of identifications made in the fractions characterized for each cell line. The size of each colored segment represents the number of identifications of a species made within each of the separate cell lines.
The second most prevalent protein family found in the comparison of the three progression cell lines was the histone family. These proteins showed a trend opposite to that of the keratin family, with histone expression and variety increasing across the progression states toward metastasis, illustrated in Figure 5. Seven individual histones representing 6 histone types were identified in the non-tumorigenic cells, as opposed to 20 identifications representing 9 types in the tumorigenic state and 23 identifications representing 10 types in the metastatic state. Histones are predominantly responsible for packaging chromosomes. Most importantly, histones play a crucial role in epigenetic variations, and they are drastically altered in cancer formation. One of the key mechanisms of tumor formation is the silencing of genes associated with cancer by epigenetic regulators18, and epigenetic variations have been suggested to be indicative of disease prognosis, including prostate cancer19. Histones are able to alter the degradation processes and signaling mechanisms of the healthy cell through their own modifications, fundamental cellular changes that could lead to cancer progression.
Of those proteins identified from the comparison of the three cancer states, individual proteins of interest have shown to be differentially expressed between the progression states and have previous associations with cancer in the literature. Found in fraction 3, maspin (SERPINB5) is a member of the serpin super family of proteins, known for their antiprotease activity. This protein was discovered through its tumor suppressive properties. Maspin is a non-inhibitory serpin showing importance in preventing metastasis in breast20 and prostate21 cancers. In the PHEC system, the maspin protein elutes at only one pI (6.1–6.4), indicating only one form present. Maspin shows a clear reduction in concentration as the cells develop into cancer and metastasize (Figure 6). The initial reduction in UV peak area seen in the PF2D was confirmed using immunoblot analysis, as shown in Figure 6. This behavior reflects that observed in the literature and substantiates the potential of maspin as an indicator of the transformation from tumorigenic to metastatic cancer.
Figure 6.
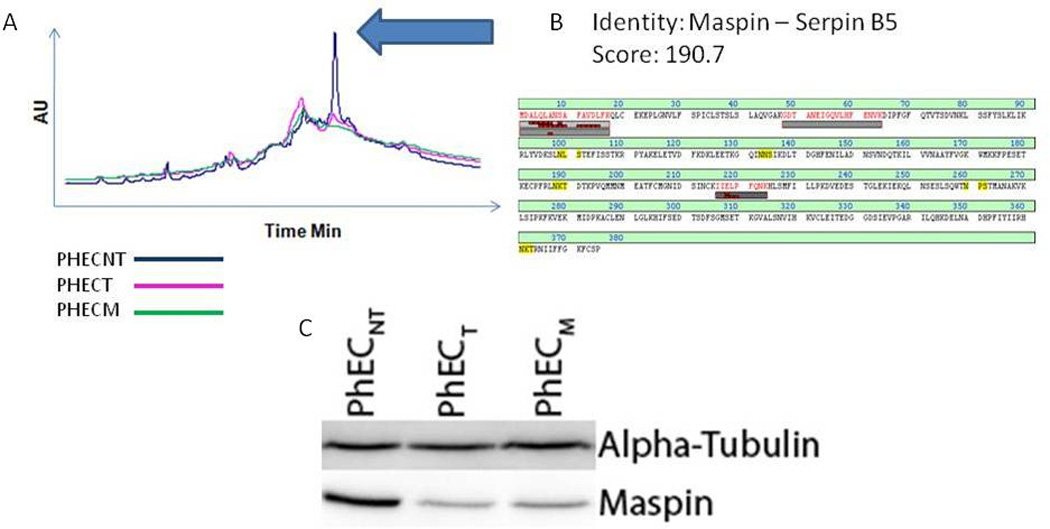
Identification and verification of a difference in abundance of the serine protease inhibitor MASPIN. A) Overlaid chromatograms of the fraction containing MASPIN in the three samples. The arrow indicates the peak associated with the identity. B) Sequence coverage for the LC-MS/MS identification of MASPIN with the corresponding MASCOT score. C) Western blot analysis to verify the difference in abundance observed by LC-MS/MS. Alpha-tubulin was used as a loading control.
Another protein differentially expressed between the three cancer states is HSP70. The HSP70 proteins are a family of proteins, including the two major cytoplasmic isoforms heat shock protein 70 (HSP70) and heat shock 70 kDa protein 8 (HSC70), that assist in a broad set of functions as part of the cellular machinery for protein folding and protection of cells from stress. As one of the chaperone proteins, HSP70 may have a role in cancer because it has been found in increased abundance in cancer cells, though not as a reliable marker to predict the reoccurrence of cancer22. The poor diagnostic ability of HSP70 in cancer evaluations may be caused by the failure to monitor the precise species. In the PHEC system, both HSP70 and HSC70 were identified. In the PHECNT cells, HSP70 and HSC70 were identified separately in multiple fractions. In the PHECT cells, HSP70 and HSC70 were identified together in the same two fractions. However, in the PHECM cells, only one fraction contained both HSP70 and HSC70. The presence of HSC70 in the metastatic cells corroborates previous findings that HSC70 is over-expressed in the metastatic state23. Overall, the presence of multiple fractions indicates differing forms of the HSP proteins, suggesting there might be post-translational modifications on these proteins that could change their cellular functions throughout the progression of prostate cancer.
Elongation factor 1 alpha (eEF1A) is also found in several places in the comparative proteomes of the three PHEC cell lines. Physiologically, eEF1A has shown expression in human prostate tumors, while absent in normal human prostate tissue24, and recently has been shown to interact with phospho-Akt to regulate proliferation, survival and motility of breast cancer cells25. In addition, overexpression of eEF1A has been demonstrated in cell lines modeling pancreatic cancer, leukemia and osteosarcoma26. In our PHEC model of prostate cancer progression, eEF1A occurs in all three progression states at multiple pI and hydrophobicity levels, as shown in Figure 3. Many PTMs have been shown on eEF1A27, which could explain the various forms eluted in the separations.
The identification of these selected proteins of interest from the comparative analysis of the cancer progression showed one striking feature among all them: the presence of multiple elution points from the two-dimensional chromatography separations employed. As previously described, this could indicate the presence of post-translational modifications, leading to considerations of the purpose of these PTMs. Proteins generally undergo PTMs for regulation of cellular processes. The PHEC model undergoes the full progression of PRCA from non-tumorigenic cells to metastatic cells, so changes in the modifications of proteins across this progression could be suggestive of the regulatory role of the proteins evaluated in the development of cellular metastasis.
The key to understanding the mechanism of cancer metastasis lies in the ability to capture a snapshot of the cell at rapid time intervals as the cells actively convert to the metastatic state, introducing a possible kinetic model of cancer metastasis not previously reported. This allows the disassembly and evaluation of individual aspects of the cell at each time point to consider individually the changes occurring simultaneously physiologically, rather than following only a single molecule or group of species of interest. Gathering such a “cellular snapshot” provides a more complete analysis, allowing a characterization of the molecules and organelles performing together to lead the cell to a metastatic endpoint. While this scenario sounds ideal, it is not trivial experimentally. Such an undertaking requires the ability to capture cells directly at successive time points, from which the lysates undergo total cellular analysis for protein changes. In addition, the cells studied must be capable of transforming fully from healthy prostate cells to metastatic cancer cells.
The purpose of this work is to introduce the model and methodologies by which a “cellular snapshot” study of human prostate cancer metastasis will begin. The PHEC model provides the necessary cellular component – humanized in vitro cell lines with identical genetic backgrounds that provide the full cancer progression to metastasis. The origin of all three PHEC cell lines from the same genetic source provides uniformity to the analysis, eliminating the variable background proteins unavoidable when multiple cell lines are used to cover the full cancer progression. This allows much more sensitive detection of subtle changes in proteins, such as PTMs and isoforms, that could be key facets to understanding metastatic mechanisms.
The combination of the PHEC human prostate cancer model system with an alternate proteomic analysis using two-dimensional HPLC/MS/MS introduces a new aspect to cancer research. This concept could provide a fundamentally unique perspective on the manner in which cellular mechanics are studied. More importantly, these tools together provide a model for human prostate cancer research that has not been studied previously, visualizing differences in individual proteins from the milieu of cellular components changing as cells metastasize.
Highlights.
-
➢
Proteomic characterization of a novel in vitro prostate cancer model system
-
➢
Progressive cancer states in vivo: non-tumorigenic, tumorigenic, and metastatic
-
➢
differential protein expression using PF2D followed by mass spectral identification
Supplementary Material
Acknowledgement
We thank Michelle Friedman for insightful discussion and editing and the University of Rochester Proteomics Center for instrument time. PF2D instrumentation was funded by FA9550-04-1-0430 (DOD) AEF derived funding from (NIH), 1 UL1 RR024160-1, National Institute of Environmental Health Sciences Training Grant ES07026, and Center Grant ES01247 and U of R start-up funds (AEF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Hara M, Koyanagi Y, Inoue T, Fukuyama T. Some physico-chemical characteristics of " - seminoprotein", an antigenic component specific for human seminal plasma. Forensic immunological study of body fluids and secretion. VII. Nihon Hoigaku Zasshi. 1971;25(4):322–324. [PubMed] [Google Scholar]
- 3.Poh BH, Jayaram G, Sthaneshwar P, Yip CH. Prostate-specific antigen in breast disease. Malays J Pathol. 2008;30(1):43–51. [PubMed] [Google Scholar]
- 4.Zarghami N, D'Costa M, Tsuyuki D, Asa SL, Diamandis EP. Expression of the prostate specific antigen gene by lung tissue. Clin Cancer Res. 1997;3(7):1201–1206. [PubMed] [Google Scholar]
- 5.Clements J, Mukhtar A, Ehrlich A, Yap B. Glandular kallikrein gene expression in the human uterus. Braz J Med Biol Res. 1994;27(8):1855–1863. [PubMed] [Google Scholar]
- 6.Rae F, Bulmer B, Nicol D, Clements J. The human tissue kallikreins (KLKs 1–3) and a novel KLK1 mRNA transcript are expressed in a renal cell carcinoma cDNA library. Immunopharmacology. 1999;45(1–3):83–88. doi: 10.1016/s0162-3109(99)00059-4. [DOI] [PubMed] [Google Scholar]
- 7.Ricke WA, Ishii K, Ricke EA, Simko J, Wang Y, Hayward SW, Cunha GR. Steroid hormones stimulate human prostate cancer progression and metastasis. Int J Cancer. 2006;118(9):2123–2131. doi: 10.1002/ijc.21614. [DOI] [PubMed] [Google Scholar]
- 8.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107(1):1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 10.Cunha GR, Lung B. The importance of stroma in morphogenesis and functional activity of urogenital epithelium. In Vitro. 1979;15(1):50–71. doi: 10.1007/BF02627079. [DOI] [PubMed] [Google Scholar]
- 11.(a) Greenberg NM, DeMayo FJ, Sheppard PC, Barrios R, Lebovitz R, Finegold M, Angelopoulou R, Dodd JG, Duckworth ML, Rosen JM, et al. The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice. Mol Endocrinol. 1994;8(2):230–239. doi: 10.1210/mend.8.2.8170479. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Colby JK, Yang P, Fischer SM, Newman RA, Klein RD. The resistance to the tumor suppressive effects of COX inhibitors and COX-2 gene disruption in TRAMP mice is associated with the loss of COX expression in prostate tissue. Carcinogenesis. 2008;29(1):120–128. doi: 10.1093/carcin/bgm226. [DOI] [PubMed] [Google Scholar]; (c) Voelkel-Johnson C, Voeks DJ, Greenberg NM, Barrios R, Maggouta F, Kurtz DT, Schwartz DA, Keller GM, Papenbrock T, Clawson GA, Norris JS. Genomic instability-based transgenic models of prostate cancer. Carcinogenesis. 2000;21(8):1623–1627. [PubMed] [Google Scholar]
- 12.Billecke C, Malik I, Movsisyan A, Sulghani S, Sharif A, Mikkelsen T, Farrell NP, Bogler O. Analysis of glioma cell platinum response by metacomparison of two-dimensional chromatographic proteome profiles. Mol Cell Proteomics. 2006;5(1):35–42. doi: 10.1074/mcp.M500124-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Cunha GR, Ricke W, Thomson A, Marker PC, Risbridger G, Hayward SW, Wang YZ, Donjacour AA, Kurita T. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J Steroid Biochem Mol Biol. 2004;92(4):221–236. doi: 10.1016/j.jsbmb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 15.(a) D'Antonio M, Borrelli F, Datola A, Bucci R, Mascia M, Polletta P, Piscitelli D, Papoian R. Biological characterization of recombinant human follicle stimulating hormone isoforms. Hum Reprod. 1999;14(5):1160–1167. doi: 10.1093/humrep/14.5.1160. [DOI] [PubMed] [Google Scholar]; (b) Smith-Beckerman DM, Fung KW, Williams KE, Auersperg N, Godwin AK, Burlingame AL. Proteome changes in ovarian epithelial cells derived from women with BRCA1 mutations and family histories of cancer. Mol Cell Proteomics. 2005;4(2):156–168. doi: 10.1074/mcp.M400157-MCP200. [DOI] [PubMed] [Google Scholar]; (c) Saldanha RG, Xu N, Molloy MP, Veal DA, Baker MS. Differential proteome expression associated with urokinase plasminogen activator receptor (uPAR) suppression in malignant epithelial cancer. J Proteome Res. 2008;7(11):4792–4806. doi: 10.1021/pr800357h. [DOI] [PubMed] [Google Scholar]; (d) Davis AJ, Carr JM, Bagley CJ, Powell J, Warrilow D, Harrich D, Burrell CJ, Li P. Human immunodeficiency virus type-1 reverse transcriptase exists as post-translationally modified forms in virions and cells. Retrovirology. 2008;5:115. doi: 10.1186/1742-4690-5-115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Itkonen O, Helin J, Saarinen J, Kalkkinen N, Ivanov KI, Stenman UH, Valmu L. Mass spectrometric detection of tyrosine sulfation in human pancreatic trypsinogens, but not in tumor-associated trypsinogen. FEBS J. 2008;275(2):289–301. doi: 10.1111/j.1742-4658.2007.06200.x. [DOI] [PubMed] [Google Scholar]
- 16.Alberti I, Barboro P, Barbesino M, Sanna P, Pisciotta L, Parodi S, Nicolo G, Boccardo F, Galli S, Patrone E, Balbi C. Changes in the expression of cytokeratins and nuclear matrix proteins are correlated with the level of differentiation in human prostate cancer. J Cell Biochem. 2000;79(3):471–485. [PubMed] [Google Scholar]
- 17.(a) Buccheri G, Ferrigno D. Lung tumor markers of cytokeratin origin: an overview. Lung Cancer. 2001;34 Suppl 2:S65–S69. doi: 10.1016/s0169-5002(01)00347-6. [DOI] [PubMed] [Google Scholar]; (b) Linder S. Cytokeratin markers come of age. Tumour Biol. 2007;28(4):189–195. doi: 10.1159/000107582. [DOI] [PubMed] [Google Scholar]
- 18.Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med J. 2009;50(4):455–463. doi: 10.3349/ymj.2009.50.4.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper CS, Foster CS. Concepts of epigenetics in prostate cancer development. Br J Cancer. 2009;100(2):240–245. doi: 10.1038/sj.bjc.6604771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Alvarez M, Slamon DJ, Koeffler P, Vadgama JV. Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer. 10(1):32. doi: 10.1186/1471-2407-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall DC, Johnson-Pais TL, Grubbs B, Bernal R, Leach RJ, Padalecki SS. Maspin reduces prostate cancer metastasis to bone. Urol Oncol. 2008;26(6):652–658. doi: 10.1016/j.urolonc.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 22.(a) Glaessgen A, Jonmarker S, Lindberg A, Nilsson B, Lewensohn R, Ekman P, Valdman A, Egevad L. Heat shock proteins 27, 60 and 70 as prognostic markers of prostate cancer. APMIS. 2008;116(10):888–895. doi: 10.1111/j.1600-0463.2008.01051.x. [DOI] [PubMed] [Google Scholar]; (b) Maehara Y, Oki E, Abe T, Tokunaga E, Shibahara K, Kakeji Y, Sugimachi K. Overexpression of the heat shock protein HSP70 family and p53 protein and prognosis for patients with gastric cancer. Oncology. 2000;58(2):144–151. doi: 10.1159/000012091. [DOI] [PubMed] [Google Scholar]
- 23.Deichmann M, Polychronidis M, Benner A, Kleist C, Thome M, Kahle B, Helmke BM. Expression of the heat shock cognate protein HSP73 correlates with tumour thickness of primary melanomas and is enhanced in melanoma metastases. Int J Oncol. 2004;25(2):259–268. [PubMed] [Google Scholar]
- 24.Shen R, Su ZZ, Olsson CA, Fisher PB. Identification of the human prostatic carcinoma oncogene PTI-1 by rapid expression cloning and differential RNA display. Proc Natl Acad Sci U S A. 1995;92(15):6778–6782. doi: 10.1073/pnas.92.15.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pecorari L, Marin O, Silvestri C, Candini O, Rossi E, Guerzoni C, Cattelani S, Mariani SA, Corradini F, Ferrari-Amorotti G, Cortesi L, Bussolari R, Raschella G, Federico MR, Calabretta B. Elongation Factor 1 alpha interacts with phospho-Akt in breast cancer cells and regulates their proliferation, survival and motility. Molecular cancer. 2009;8:58. doi: 10.1186/1476-4598-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selga E, Oleaga C, Ramirez S, de Almagro MC, Noe V, Ciudad CJ. Networking of differentially expressed genes in human cancer cells resistant to methotrexate. Genome medicine. 2009;1(9):83. doi: 10.1186/gm83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Zobel-Thropp P, Yang MC, Machado L, Clarke S. A novel post-translational modification of yeast elongation factor 1A. Methylesterification at the C terminus. J Biol Chem. 2000;275(47):37150–37158. doi: 10.1074/jbc.M001005200. [DOI] [PubMed] [Google Scholar]; (b) Rosenberry TL, Krall JA, Dever TE, Haas R, Louvard D, Merrick WC. Biosynthetic incorporation of [3H]ethanolamine into protein synthesis elongation factor 1 alpha reveals a new post-translational protein modification. J Biol Chem. 1989;264(13):7096–7099. [PubMed] [Google Scholar]; (c) Lamberti A, Longo O, Marra M, Tagliaferri P, Bismuto E, Fiengo A, Viscomi C, Budillon A, Rapp UR, Wang E, Venuta S, Abbruzzese A, Arcari P, Caraglia M. C-Raf antagonizes apoptosis induced by IFN-alpha in human lung cancer cells by phosphorylation and increase of the intracellular content of elongation factor 1A. Cell Death Differ. 2007;14(5):952–962. doi: 10.1038/sj.cdd.4402102. [DOI] [PubMed] [Google Scholar]; (d) Dapas B, Tell G, Scaloni A, Pines A, Ferrara L, Quadrifoglio F, Scaggiante B. Identification of different isoforms of eEF1A in the nuclear fraction of human T-lymphoblastic cancer cell line specifically binding to aptameric cytotoxic GT oligomers. Eur J Biochem. 2003;270(15):3251–3262. doi: 10.1046/j.1432-1033.2003.03713.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.