

Abstract
Telomere attrition unleashes genomic instability, promoting cancer development. Once established, however, the malignant clone often re-establishes genomic stability through overexpression of telomerase. In two papers, one in this issue of Cell and one in the subsequent issue, DePinho and colleagues explore the consequences of telomerase re-expression and its validity as a therapeutic target in mouse models of cancer.
Telomere attrition induces a particularly invidious form of genomic instability. Once these protective caps have eroded, the naked ends of the chromosomes appear as double-stranded DNA (dsDNA) breaks to cell, yet there is no correct DNA repair solution other than restitution of an intact telomere. In the absence of p53, the cell attempts to repair this phantom “break,” or naked end of the chromosome. If this repair is performed in G2 phase of the cell cycle, after DNA replication, a common solution is to join the original and the replicated chromosomes together. This generates an end-to-end chromosome fusion, the cytogenetic hallmark of telomere attrition (Gisselsson et al., 2001; Maser et al., 2007; O’Hagan et al., 2002). During mitosis, the two centromeres are pulled to opposite daughter cells, with the intervening chromosomal material ultimately broken during cytokinesis. This leads to two daughter cells with genomic rearrangements and a whole slew of authentic dsDNA breaks. In the short term, this genomic evolution can repeat in both daughter cells with every cell cycle, driving rapid genome evolution in exponentially increasing numbers of competing subclones (Bignell et al., 2007). In the medium term, natural selection weeds out the subclones with deleterious rearrangements and fosters those with enhanced malignant potential. In the long term, however, unchecked genomic instability resulting from telomere attrition is disadvantageous: mice born with depleted telomere reserves fail to thrive, exhibit organ atrophy, and display poor proliferative response among epithelial and hematological lineages (Lee et al., 1998).
Surprisingly, though, many cancers re-express telomerase in advanced stages of malignancy (Gisselsson et al., 2001; Hashimoto et al., 2008), and this reactivation may reduce the devastation wreaked by end-to-end chromosome fusions (Campbell et al., 2010). Having painstakingly dissected the multitudinous effects of telomere erosion in mouse models of cancer and aging over the last 10–15 years, DePinho’s laboratory now presents two studies on the flip side, that of telomerase (mTert) re-expression (Ding et al., 2012; Hu et al., 2012).
In the first study, Ding et al. start with a mouse model prone to prostate cancer through targeted ablation of p53, Pten, and mTert (telomerase reverse transcriptase). They then engineer an inducible version of the mTert gene to allow reactivation of telomerase. Control mice, which are missing p53 and Pten but maintain telomere function, universally develop invasive adenocarcinoma of the prostate by 24 weeks of age. In contrast, the prostate tumors that developed in p53/Pten-deficient mice are smaller, less aggressive, and less invasive when telomerase is lost and telomere erosion is established. Thus, deletion of these two key tumor suppressor genes pushes cells a long way toward prostate cancer, and the disadvantages of ongoing genomic instability mediated by irreparably short telomeres outweigh any potential gains from further genomic evolution. When Ding et al. re-express telomerase in the prostates of mice with depleted telomeres, however, an especially virulent cancer develops, which is bulky, aggressive, invasive, and capable of metastasis to the lumbar spine. Genome profiling of these super-aggressive tumors reveals many copy number aberrations, overlapping with those seen in human prostate cancers (Beroukhim et al., 2010). Among these aberrations, loss of Smad4 is particularly prevalent, and mice with loss of p53, Pten, and Smad4 in the prostate recapitulate the more aggressive phenotype seen with telomerase re-expression, including the propensity to bone metastasis (Figure 1).
Figure 1. The Telomere Crisis Model of Cancer Evolution.
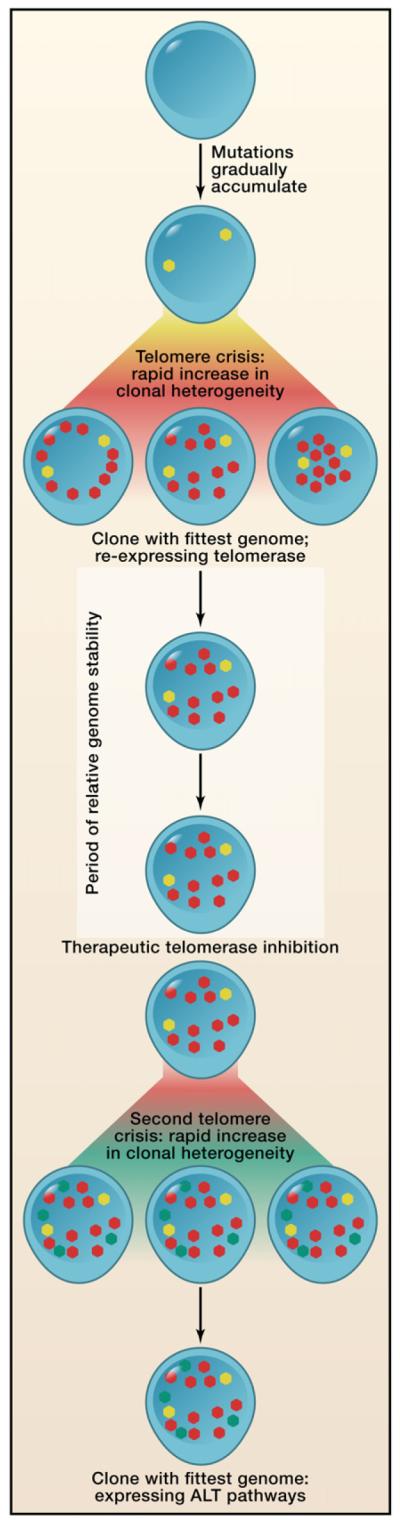
Cancers initially evolve slowly, gradually acquiring spontaneous mutations (yellow dots). With increasing numbers of cell divisions, however, telomeres erode, and this induces a rapid increase in both the number of mutations (red dots) and the subclonal heterogeneity in the organ. Out of these competing subclones, one emerges with more malignant potential. As Ding et al. (2012) show, it is to this clone’s selective advantage to re-establish genomic stability through re-expression of telomerase. A period of relative genomic stability may follow, but this equilibrium can be disrupted by inhibition of telomerase. Hu et al. (2012) find that, after initial therapeutic benefit, such inhibition induces a second telomere crisis, again with rapid acquisition of new mutations (green dots) and subclonal heterogeneity.
The model of telomere erosion that emerges from these findings is rather intriguing, not unlike trying to climb up a downward escalator. Presumably, Smad4 deletion and other advantageous lesions were indeed developing in isolated p53/Pten-deficient prostate cells with depleted telomeres, but the relentless downward effects of unfettered genome instability prevented these clones from ever reaching the summit of invasive and metastatic malignancy. When this downward pressure was switched off through re-expression of telomerase, however, the clones could escalate their malignant potential to new levels.
These findings raise the important question of whether reactivation of telomerase is a valid therapeutic target, andit is this hypothesis that is addressed in the second of the papers reported here. Hu et al. introduce a tamoxifen-inducible mTert allele into Atm-deficient mice that is susceptible to T cell lymphoma. They uncover patterns of malignancy akin to those described above for prostate cancer: lymphomas in mice with telomere erosion plus Atm deficiency are slower to evolve than tumors with only Atm deficiency. However, as Ding et al. found, re-expression of telomerase increases malignant potential, spread of the tumor, and the frequency of clonal copy number alterations.
To model what potential benefits might arise from inhibiting telomerase reactivation in human tumors, Hu et al. then serially xenograft 11 tumor lines from this mouse model into donor mice with or without concomitant tamoxifen. It took three generations of xenografts for the re-established telomeres to erode again in mice with no tamoxifen, but once they did, six lines completely fail to develop tumors in recipients, and 3 lines exhibit much slower engraftment than when tamoxifen (and therefore telomerase) is maintained throughout. Strikingly, these three lines re-attain full malignant potential upon a fourth serial transplant, even in the absence of tamoxifen. The implication is that tumors that are dependent on telomerase reactivation are indeed sensitive to loss of telomerase and telomere attrition, but this may be bypassed by other pathways. In particular, Hu and colleagues proceed to show that the lines escaping from the crisis induced by withdrawal of tamoxifen activate a pathway known as alternative lengthening of telomeres. This activation is associated with enhanced signaling through networks involving mitochondrial function and reactive oxygen species, centered on genomic amplification and overexpression of the PGC-1β gene. Inhibition of this gene by small hairpin RNA causes significant reduction of tumor potency in those lymphomas with activated pathways for alternative lengthening of telomeres but causes minimal effect on lymphomas, which had steady telomerase levels throughout xenografting.
These elegant studies illustrate the perfidy of cancer in the face of well-intentioned therapeutic strategies. Clones that re-express telomerase can overcome the genomic crisis induced by telomere erosion; inhibition of this very telomerase is then bypassed in the cancer through alternative pathways of telomere maintenance. Telomerase inhibitors have entered phase I/II clinical trials in several malignancies. The data presented here show that there may well be considerable therapeutic benefit from such agents, but they may ultimately induce the very instability that evolves the cancer genome to a drug-resistant state. Yet, as Hu et al. show, with every therapeutic bypass, the clone makes sacrifices, and these become themselves rational therapeutic targets. And so, like an ever more sophisticated arms race, the battle between cancer and clinician escalates.
REFERENCES
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Santarius T, Pole JC, Butler AP, Perry J, Pleasance E, Greenman C, Menzies A, Taylor S, Edkins S, et al. Genome Res. 2007;17:1296–1303. doi: 10.1101/gr.6522707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Wu C-J, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu G, Wang G, Lu X, Labrot E, et al. Cell. 2012;148 doi: 10.1016/j.cell.2012.01.039. Published on-line February 16, 2012. 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselsson D, Jonson T, Petersén A, Strömbeck B, Dal Cin P, Höglund M, Mitelman F, Mertens F, Mandahl N. Proc. Natl. Acad. Sci. USA. 2001;98:12683–12688. doi: 10.1073/pnas.211357798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Murakami Y, Uemura K, Hayashidani Y, Sudo T, Ohge H, Fukuda E, Shimamoto F, Sueda T, Hiyama E. J. Gastrointest. Surg. 2008;12:17–28. doi: 10.1007/s11605-007-0383-9. [DOI] [PubMed] [Google Scholar]
- Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H, et al. Cell. 2012;148:651–663. doi: 10.1016/j.cell.2011.12.028. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, II, Greider CW, DePinho RA. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, et al. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, DePinho RA. Cancer Cell. 2002;2:149–155. doi: 10.1016/s1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]