

Abstract
The M2 protein from influenza A is a proton channel as a tetramer, with a single transmembrane helix from each monomer lining the pore. Val27 and Trp41 form gates at either end of the pore and His37 mediates the shuttling of protons across a central barrier between the N- and C-terminal aqueous pore regions. Numerous structures of this transmembrane domain and of a longer construct that includes an amphipathic helix are now in the Protein Data Bank. Many structural differences are apparent from samples obtained in a variety of membrane mimetic environments. High-resolution structural results in lipid bilayers have provided novel insights into the functional mechanism of the unique HxxxW cluster in the M2 proton channel.
Introduction
The M2 protein from Influenza A has a long history as a drug target, indeed before it was known to be a proton channel.[1–3] However, the drug resistant M2 S31N mutation has become dominant in the recent seasonal flu seasons and recent swine flu pandemic. Today, there is no effective drug that targets the M2 protein and a major effort has been underway to characterize the detailed structure and conductance mechanism of this proton channel for lead development efforts.[4] Multiple structures of M2 constructs are now in the Protein Data Bank (PDB). A consensus is emerging with regard to the backbone structure of this channel in the closed state. However, a consensus has not been achieved for the sidechains of the unique HxxxW sequence associated with proton conductance, nor for the structure of the conducting state, although models exist.
High resolution structural biology techniques do not permit the characterization of membrane protein structures in their native membrane environment. Since structure is the result of the total set of molecular interactions experienced by the protein, its environment is important.[5] The environment of membrane proteins is very complex and heterogeneous. Small proteins experience a greater fraction of their interactions with the environment, conferring the latter with a particularly significant potential for modulating protein structures.[6] Typically, solution NMR and x-ray crystallography are dependent on the use of detergents that have limitations as a membrane mimetic for membrane protein sample preparation. Furthermore, it is not always obvious what the native lipid environment is for a protein. Hong and coworkers have pioneered the use of a model membrane environment that mimics the bulk membrane environment of the influenza viral coat.[7] However, it has recently been shown that M2 is not evenly distributed over the virion, but is localized to the neck of the budding virus where the raft-like environment meets the liquid crystalline bilayer environment.[8] Apparently, the antiviral drugs that bind M2 do not bind M2 in a raft-like lipid environment.[9] Consequently, membrane protein PDB submissions, such as those for the M2 protein may or may not reflect native functional states.[6]
Solid state NMR (ssNMR) is a technique that permits spectroscopy of proteins that do not undergo rapid isotropic motions, such as the limited motions that occur when proteins are solubilized in lipid bilayers. To date, this new approach for structural characterization has resulted in a dozen small membrane protein structures deposited in the PDB and a total of 52 ss NMR PDB submissions.[10–14] However, unlike solution NMR, it has the option to look at large structures and to characterize the structures in a variety of lipid environments.[15] This technique was used recently to characterize the M2 protein structure in synthetic bilayers of dioleoylphosphatidyl choline (DOPC) and ethanolamine (DOPE). using liposome preparations and uniformly aligned samples with approximately 50% by weight water.[12]
The M2 protein is small, having 97 amino acid residues that form an N-terminal segment of 25 residues in the viral exterior, a single transmembrane (TM) helix of 21 residues, and a C-terminal segment in the viral interior. M2 carries out at least three functions as a homo-tetramer. The TM helix and the immediately following amphipathic helix (residues 47–62), which binds to the lipid interface form the proton conductance domain. The amphipathic helix is also associated with viral budding, as influenza lacks the ESCRT proteins to facilitate budding.[8] Finally the C-terminal segment is involved in M1 binding. Early structures determined by ssNMR with or without amantadine were of the TM domain (residues 22–46), which functions as a proton selective channel (PDB IDs: 1NYJ and 2H95).[16,17] The initial crystal structure was also determined for this construct (PDB ID: 3BKD), and a refinement of the original ssNMR structure in the presence of amantadine has been accomplished (PDB ID: 2KQT).[18,19] The first solution NMR structure was of the longer conductance domain (PDB ID: 2RLF).[20] Recently, an ssNMR structure (PDB ID: 2L0J) and a solution NMR structure (PDB ID: 2KWX) of the conductance domain as well as a crystal structure of the TM domain (PDB ID: 3LBW) have been obtained.[12,21,22] Here, the focus will be on these three most recent structures for an understanding of proton conductance.
Overview of recent structural characterizations
The early ssNMR structures for the TM domain showed that the four-helix bundle was left-handed, with a helical tilt to the bilayer normal of more than 30°.[16,17] Earlier, crosslinking and modeling studies showed that the channel was formed by a tetramer and that the hydrophilic residues, Ser31, His37, and Trp41, of the TM helix all faced the aqueous pore of the tetrameric bundle.[23–25] The 2010 crystal structure of the G34A mutant (PDB ID: 3LBW; Fig. 1 magenta carbons)[22] lacks the distortions present in the earlier crystal structure[26] and has helix tilt angles similar to the ssNMR structure (PDB ID 2L0J: Fig. 1 green carbons).[12] Both solution NMR structures for the conductance domain[20,21] show significantly smaller TM helix tilt angles, and the first solution NMR structure (PDB ID: 2RLF) showed a water soluble tetrameric bundle for the amphipathic helix.[20] The ssNMR structure of the conductance domain (PDB ID: 2L0J)[12] shows a 32° helical tilt and an amphipathic helix tilt of 105°, with this later helix buried in the lipid interface. The second solution NMR structure of a V27A mutant (PDB ID: 2KWX; Fig. 1 yellow carbons)[21] has the amphipathic helices rotated by 90° so that the hydrophobic surface now interacts with the bilayer interface. The M2 backbone structures of the most recent characterizations are compared in Fig.1A, showing great similarity in the TM helix except for a somewhat smaller tilt angle for the solution NMR TM helix. A somewhat different location is also evident for the amphipathic helix of the ssNMR vs. solution NMR structure.
Fig. 1.
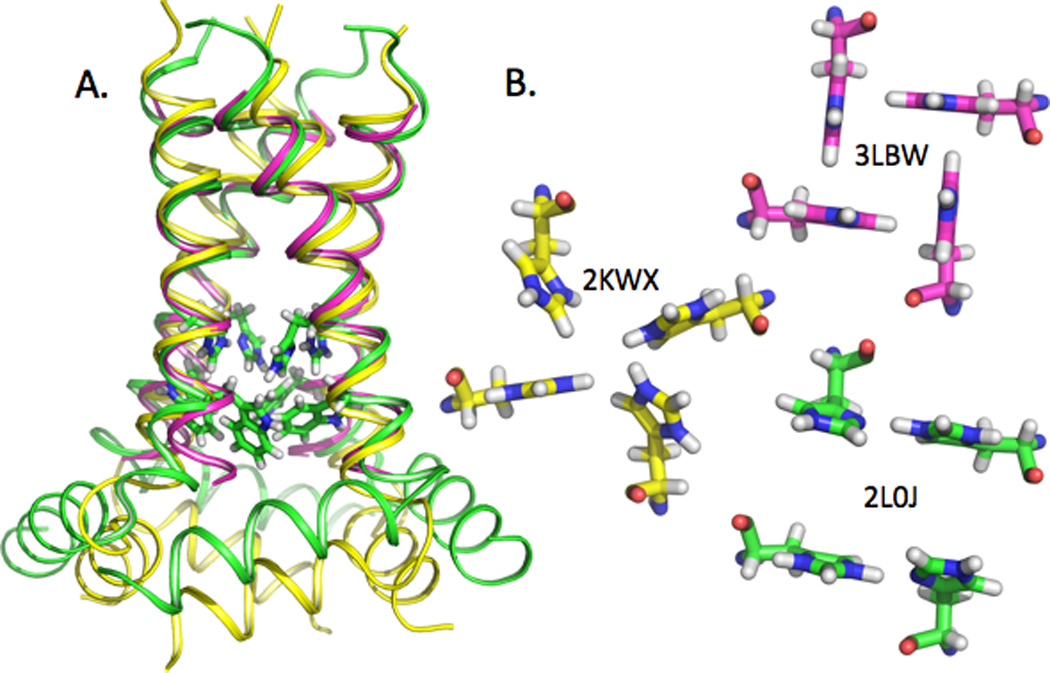
Comparison of three recent structures of M2, shown with the backbone as a helical ribbon and the His37 and Trp41 sidechains as sticks. A) 2L0J was obtained by solid state NMR spectroscopy on the M2 conductance domain (residues 22–62) in liquid crystalline lipid bilayers at pH 7.5 (green). 2KWX was obtained by solution NMR spectroscopy on the conductance domain (residues 18–60) in detergent micelles at pH 7.5 (yellow). 3LBW was obtained by x-ray crystallography on the TM domain (residues 22–46) in an octylglucoside environment at pH 6.5 (magneta). The ssNMR and x-ray structures of the TM helices superimpose very well, with a slight deviation at the C-terminus. The tilt of the solution NMR TM helix is significantly less than in the other two structures. Relative to the ssNMR structure, the amphipathic helices in the solution NMR structure have a different location with regard to both depth in the ‘membrane’ environment and lateral position. The His37 and Trp41 sidechains are shown for 2L0J. B) Comparison of the histidine sidechains in the three M2 structures displayed in A) as viewed from the viral interior (amphipathic helix side). The His37 sidechain torsion angles for 2KWX are similar to those for 2L0J, but the imidazole-imidzolium hydrogen bonds are not formed. As such the His37 sidechain conformations in 2KWX appear to be unstable due to charge repulsion. The His37 sidechain conformations in 3LBW provide an alternative, but more limited mechanism through cation-π interactions for charge dispersion and for structural stabilization.[22]
Amantadine and rimantadine were two effective pharmaceuticals against the seasonal flu prior to the widespread distribution of the S31N M2 mutation. In recent years there has been considerable debate as to the binding location for these drugs that result in their inhibition of proton conductance. Pore binding in the vicinity of Gly34 and Ser31 had been supported by initial structural efforts on the TM domain (PDB ID: 2H95 & 3BKD),[17,18] but an external binding site was elucidated with the first solution NMR structure (PDB ID: 2RLF).[20] This latter binding site is adjacent to the native position for these drugs in the lipid bilayer, where they are preferentially soluble.[27,28] However, in performing an MD refinement on the 2H95 structure, it was shown that amantadine bound in the pore below the secondary Val27 gate.[29] Furthermore, in refining the 2H95 structure by measuring distances from the protein to the drug, a similar strong binding site in the pore and a weak binding site adjacent to the lipid environment were identified.[19] Moreover, the stoichiometry was determined to be one drug per tetramer.[30] Finally, in solving the structure of the conductance domain in liquid crystalline lipid bilayers, it was found that two hydrophobic amino acid sidechains from the amphipathic helix filled the binding pocket adjacent to the lipid environment, thereby eliminating the binding site adjacent to the lipid environment in this preparation.[12] It should be noted that, in the solution NMR structure 2RLF,[20] the pore itself was more restricted due to the smaller tilt angle of the TM helices (Fig. 1A). Therefore, it now seems clear that the effective binding site of amantadine and rimantadine is in the pore.
History of mechanistic studies
Functionally, measurements of reversal potentials have demonstrated that voltage-clamped M2-transfected cells transport protons in preference to sodium, potassium, and chloride by a factor of ~10, in spite of ~five orders of magnitude concentration handicap.[31] The proton transport rate is ~200 s−1 in transfected oocytes[32,33] and proteoliposomes.[33] Although the direction of channel current is reversible with changes in membrane potential,[2,31] as is common in (but not exclusive to) electrodiffusion through an aqueous pore, the low transport rate and high selectivity against alkali metal cations strongly suggest a dehydrated selectivity step rather than an open aqueous pore. Replacement of His37 with other residues enhances conductance[34] and reduces selectivity,[31] suggesting that the His37 tetrad is the location for this selectivity step. M2 channels display evidence of ligand-gating. Namely, unless the exterior bulk solution is acidic, proton transport in either direction is inhibited.[31] Effects of site mutations on proton efflux and the accessibility of interior Cu+2 to coordinate and block the H37 tetrad indicate that the W41 sidechains form the primary gate.[35] V27 appears to form a secondary gate at the N-terminal entrance to the pore. [12,22,29]
In 2006 the individual pKas of the His37 tetrad were determined from liposome preparations of the TM domain.[36] The protonation of two of the four histidine residues occurs at a proton concentration (pH 8.2) nearly two orders of magnitude lower than that necessary for protonating histidines exposed to a bulk aqueous environment. These two pKas suggest cooperative binding of protons, generating two charges in close proximity in a low dielectric environment, against potentially destabilizing electrostatic repulsion. The solution to this conundrum was the observation of strong hydrogen bonds between pairs of histidine residues distributing the charges and hence greatly reducing the electrostatic repulsion suggested by the two high pKas. The channel activation pH is approximately 6,[31] coincident with the approximate value of the third pKa, and so it appears that the third protonation activates the channel.
Quantum mechanics/molecular mechanics (QM/MM) calculations of the +2 charged HxxxW quartet restrained by the backbone geometry of 2L0J led to a dimer-of-dimers structure for the His37 residues (Fig. 1B green).[12] The stabilization of charge is achieved not only through the sharing of a proton between rings (resulting in a strong hydrogen bond), but one of the residues in each histidine pair hydrogen bonds back to the backbone carbonyl and the other forms a cation-π interaction with the indole of the tryptophan residues. Furthermore, it has been suggested that hydrogen bond exchange could occur among the four His37 residues, resulting in overall stability enhancement of the tetramer.[12] The dispersion of charge and formation of inter and intra-helix hydrogen bonds fully explains how the full length M2 protein can have a tetramer stability (as measured by Kapp) that is three orders of magnitude higher at pH 6 than at pH 9.[37] When a third proton is added to the histidine tetrad, an activated state results by forming an additional cation-π interaction with Trp41 while one of the imidazole-imidazolium hydrogen bonds is broken along with the unique charge stabilization mechanism, and hence the activated state is thought to be an unstable configuration. As long as the indoles form cation-π interactions with the imidazoles there is no transport of the protons through the pore. Motions of the helices (such as kinking) could result in the breaking of these interactions and hence the opening of the tryptophan gate.[22,38,39]
Recently, clear evidence for the dimer-of-dimers structure extending to the polypeptide backbone has been obtained by MAS ssNMR data from multiple backbone sites,[40] supporting the above conductance mechanism. It is anticipated that the structural differences in the backbone between the monomers of a dimer is small and that the monomers comprising the two dimers interconvert on a millisecond timescale. The dimer-of-dimers conformation and the histidine pKas are crucial elements for understanding the proton conductance mechanism. While a mechanism has been outlined here, this is still a contentious field based on the diversity of the His tetrad conformations (Fig. 1B).[7,22,41,42]
Channel conductance measurements[12,37,43,44] provide the ultimate, quantitative test of any conductance mechanism. For the M2 proton channel, that test has been made possible by a mathematical model for predicting the rate of proton transport (Fig. 2A).[45] The model envisions that a permeant proton entering the channel pore from the exterior bulk solution would obligatorily bind to the +2 charged His37 tetrad. When the Trp41 gate is open, the permeant proton would then be released to the interior bulk solution. The rate of proton transport is determined by several factors: (i) the rate constant for the proton, starting from the external side, passing through the V27 gate, and binding to the His37 tetrad; (ii) the probability that the Trp41 gate is open; and (iii) the rate constant for the proton to be released to the internal side when the gate is open. These quantities can be estimated, based on the structure of the channel and its dynamic fluctuations observed experimentally or through molecular dynamics simulations. The mathematical model has been able to quantitatively reproduce measured current-voltage relations and dependence of proton transport rate on pH (Fig. 2B).[45] Moreover, it has now[46] explained the H2O/D2O isotope effect of the proton transport rate observed by Mould et al.[47] When the solvent is changed from H2O to D2O, two parameters of the mathematical model are changed: the diffusion constant of the permeant ion (and hence the rate constants for binding to the His37 tetrad and for being released to the intracellular side) is decreased by ~40%; and the pKa for the permeant ion to dissociate from the His37 tetrad is upshifted by ~0.4 pH units (corresponding to a further 2.5-fold decrease in the rate constant for being released to the intracellular side). The net effect of these two changes quantitatively reproduce the observed isotope effect.
Fig. 2.
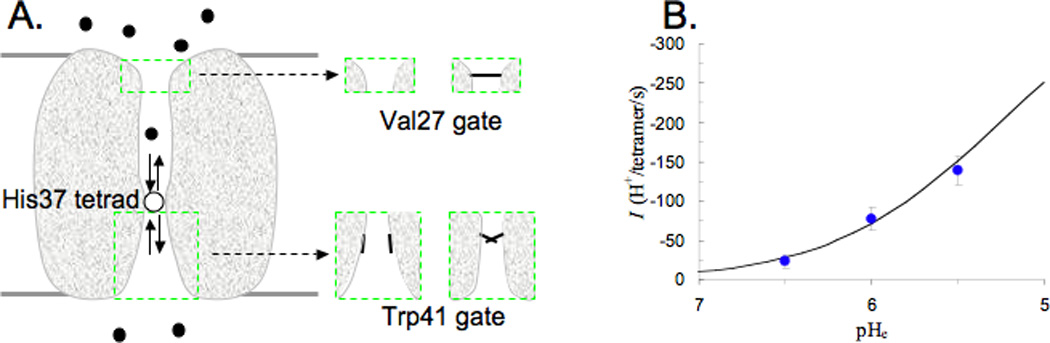
Mathematical model for calculating the rate of proton transport. A) Illustration of the model. A permeant proton, starting from the exterior bulk solution, diffuses toward the N-terminal pore, passes through the Val27 gate, and binds to the +2 charged His37 tetrad. A proton then dissociates from the His37 tetrad, either back to the N-terminal pore, or into the C-terminal pore, from which it passes by the Trp41 gate and diffuses into the interior bulk solution. The insets illustrate the fluctuating Val27 and Trp41 gates. B) Comparison of calculated (curve) and measured (symbols) rates of proton transport as functions of the exterior pH, pHe. The experimental data are from Ref. 13 (the membrane potential was -114 mV and the interior pH was 8), In the calculations, the rate constant for binding to the His37 tetrad for a proton from the interior side is 1.2 × 108 M−1s−1, the rate constant for releasing the proton from the His37 tetrad to the interior side is 3.6 × 103s−1, and the probability for the Trp41 gate to be open is ~5%.
Conclusions
Important structural information has been achieved for M2 from many experimental methods. Molecular dynamics simulations and QM/MM calculations are rapidly improving and have provided unique insights where experimental data have been difficult or cannot be obtained. Through all of these efforts a great deal has been learned not only about the M2 channel, but also about the biophysics of membrane proteins and the need to model the membrane environment well in order to obtain functionally relevant data. It is reassuring that a near consensus has been achieved for the backbone of the M2 conductance domain in the closed state, but even so we do not have a high-resolution backbone structure of the dimer of dimers, nor detailed information on the structure as a function of pH, nor a detailed characterization of the dynamics associated with the dimer-of-dimers interconversion, helix kinking, or the His37 and Trp41 sidechain motions. In addition there are many fascinating conductance properties associated with mutations that have yet to be explained. So while excellent progress has been made on characterizing the M2 structure and functional mechanism there remains much more to be fathomed on this small membrane protein that displays considerable sensitivity to the membrane mimetic environment.
Highlights.
Characterization of M2 in lipid bilayers has revealed a native-like structure
The tetrameric M2 structure is a dimer of dimers conformation in the +2 charged state
The functionally significant amantadine and rimantadine binding site is in the pore
The M2 structure from influenza A is sensitive to its membrane mimetic environment
A Mathematical model for M2 H+ transport is consistent with much conductance data
Acknowledgment
This research has been supported in part by the National Institutes of Health through AI-023007 and the National Science Foundation through Cooperative Agreement 0654118 between the Division of Materials Research and the State of Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Hao Dong, Email: donghaonj@gmail.com.
Mukesh Sharma, Email: sharmas@magnet.fsu.edu.
David D. Busath, Email: david_busath@byu.edu.
Huan-Xiang Zhou, Email: hzhou4@fsu.edu.
References
- 1.Pinto LH, Lamb RA. Influenza virus proton channels. Photochem Photobiol Sci. 2006;5:629–632. doi: 10.1039/b517734k. [DOI] [PubMed] [Google Scholar]
- 2.Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Qiu JX, Soto C, DeGrado WF. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr Opin Struct Biol. 2011;21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, Degrado WF, Pinto LH. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- **6. Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. X-ray, solution and ssNMR structures of M2 are compared, clearly illustrating the influence of different membrane mimetic environments on the structures of this protein.
- *7. Hu F, Luo W, Hong M. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science. 2010;330:505–508. doi: 10.1126/science.1191714. Novel ssNMR spectroscopy of the TM domain in a cholesterol rich mimetic environment of the viral envelope.
- **8. Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. For the first time it is shown that the M2 protein has a specific role in the viral budding process and that this is associated with the amphipathic helix. Furthermore, it is shown that M2 is not dispersed throughout the viral coat.
- 9.Cady S, Wang T, Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J Am Chem Soc. 2011;133:11572–11579. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ketchem RR, Hu W, Cross TA. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 11.De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **12. Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. Structure of the M2 conductance domain in a lipid bilayer. The oriented sample data was collected and the structure refined in a liquid crystalline lipid bilayer, resulting in a novel functional mechanism and an explanation for the protein's unique structural stability.
- 13.Marassi FM, Das BB, Lu GJ, Nothnagel HJ, Park SH, Son WS, Tian Y, Opella SJ. Structure determination of membrane proteins in five easy pieces. Methods. 2011 doi: 10.1016/j.ymeth.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci U S A. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park SH, Casagrande F, Chu M, Maier K, Kiefer H, Opella SJ. Optimization of Purification and Refolding of the Human Chemokine Receptor CXCR1 Improves the Stability of Proteoliposomes for Structure Determination. Biochim Biophys Acta. 2012;1818:584–591. doi: 10.1016/j.bbamem.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura K, Kim S, Zhang L, TA Cross. The Closed State of a H+ Channel Helical Bundle: Combining Precise orientational and Distance Restraints from Solid State NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 17.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *19. Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. ssNMR distance restraints identified two amantadine binding sites: a high affinity site in the pore and a low lipid-exposed affinity site observable only at high drug concentrations.
- 20.Schnell JR, Chou JJ. Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *21. Pielak RM, Chou JJ. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem Biophys Res Commun. 2010;401:58–63. doi: 10.1016/j.bbrc.2010.09.008. Solution NMR structure of the M2 conductance domain showing the amphipathic helix interacting with the lipid bilayer.
- *22. Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci U S A. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. A crystal structure of M2 TM domain that is free of the distorting influences of crystal contacts and monomeric detergents found in an earlier structure.
- 23.Bauer CM, Pinto LH, Cross TA, Lamb RA. The influenza virus M2 ion channel protein: probing the structure of the transmembrane domain in intact cells by using engineered disulfide cross-linking. Virology. 1999;254:196–209. doi: 10.1006/viro.1998.9552. [DOI] [PubMed] [Google Scholar]
- 24.Sugrue RJaH AJ. Structural Characteristics of the M2 Protein of Influenza A Viruses: Evidence that it forms a Tetrameric Channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. A Functionally Defined Model for the M2 Proton Channel of Influenza A Virus Suggests a Mechanism for its Ion Selectivity. Proc. Natl.Acad. Sci. U.S.A. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stouffer AL, Ma C, Cristian L, Ohigashi Y, Lamb RA, Lear JD, Pinto LH, DeGrado WF. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Schnell JR, Chou JJ. Amantadine partition and localization in phospholipid membrane: a solution NMR study. Biochem Biophys Res Commun. 2004;324:212–217. doi: 10.1016/j.bbrc.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 28.Li C, Yi M, Hu J, Zhou HX, Cross TA. Solid-state NMR and MD simulations of the antiviral drug amantadine solubilized in DMPC bilayers. Biophys J. 2008;94:1295–1302. doi: 10.1529/biophysj.107.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi M, Cross TA, Zhou HX. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma M, Li C, Busath DD, Zhou HX, Cross TA. Drug sensitivity, drug-resistant mutations, and structures of three conductance domains of viral porins. Biochim Biophys Acta. 2011;1808:538–546. doi: 10.1016/j.bbamem.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol. 1996;494:329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH. Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:31038–31050. doi: 10.1074/jbc.M003663200. [DOI] [PubMed] [Google Scholar]
- 33.Lin T, Schroeder C. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J. Virol. 2001;75:3647–3656. doi: 10.1128/JVI.75.8.3647-3656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Lamb RA, Pinto LH. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 36.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci U S A. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma C, Polishchuk AL, Ohigashi Y, Stouffer AL, Schon A, Magavern E, Jing X, Lear JD, Freire E, Lamb RA, et al. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2009;106:12283–12288. doi: 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi M, Cross TA, Zhou HX. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc Natl Acad Sci U S A. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou HX. Diffusion-Influenced Transport of Ions across a Transmembrane Channel with an Internal Binding Site. J Phys Chem Lett. 2010;1:1973–1976. doi: 10.1021/jz100683t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *40. Andreas LB, Eddy MT, Pielak RM, Chou J, Griffin RG. Magic angle spinning NMR investigation of influenza A M2(18–60): support for an allosteric mechanism of inhibition. J Am Chem Soc. 2010;132:10958–10960. doi: 10.1021/ja101537p. High Resolution ssNMR data of M2 conductance domain in liposomes showing that the dimer-of-dimers conformation extends over much of the TM domain.
- 41.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado WF, Klein ML. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phongphanphanee S, Rungrotmongkol T, Yoshida N, Hannongbua S, Hirata F. Proton transport through the influenza A M2 channel: three-dimensional reference interaction site model study. J Am Chem Soc. 2010;132:9782–9788. doi: 10.1021/ja1027293. [DOI] [PubMed] [Google Scholar]
- 43.Pielak RM, Chou JJ. Kinetic analysis of the M2 proton conduction of the influenza virus. J Am Chem Soc. 2010;132:17695–17697. doi: 10.1021/ja108458u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leiding T, Wang J, Martinsson J, DeGrado WF, Arskold SP. Proton and cation transport activity of the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2010;107:15409–15414. doi: 10.1073/pnas.1009997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *45. Zhou HX. A theory for the proton transport of the influenza virus M2 protein: extensive test against conductance data. Biophys J. 2011;100:912–921. doi: 10.1016/j.bpj.2011.01.002. A mathematical model for calculating the proton flux through the M2 channel is tested against an extensive set of conductance data.
- 46.Zhou HX. Mechanistic insight into the h(2)o/d (2)o isotope effect in the proton transport of the influenza virus m2 protein. J Membr Biol. 2011;244:93–96. doi: 10.1007/s00232-011-9402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mould JA, Li H-C, Dudlak CS, Lear JD, Pekosz A, Lamb RA, Pinto LH. Mechanism for proton conduction of the M2 Ion Channel of Influenza A Virus. J. Biol. Chem. 2000;275:8592–8599. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]