

Abstract
A genome-wide association study in Japan identified the C-C chemokine receptor type 6 gene (CCR6) as associated with rheumatoid arthritis (RA). This finding has not been validated in other Asian populations. A case–control study involving 996 subjects, comprising 440 controls and 556 RA patients, was done to determine their anticyclic citrullinated peptide (anti-CCP) antibody status and CCR6 polymorphism (rs3093024) genotype. Three hundred eighty-seven patients were anti-CCP positive and 153 anti-CCP negative. Logistic regression showed that allele A was likely to increase the risk of developing RA among females via a recessive model (odds ratio [OR]=1.55, 95% confidence interval [CI]=1.01, 2.39), whereas the risk effect appeared to be reduced among males via an additive model (OR=0.60, 95% CI=0.42, 0.85). Considering only subjects who are anti-CCP positive, allele A increased RA risk among females via a recessive model (OR=1.68, 95% CI=1.07, 2.64) but decreased the risk among males via an additive model (OR=0.59, 95% CI=0.39, 0.89). We showed that CCR6 polymorphism was a risk factor among females but a protective factor among males. Functional studies are warranted to unravel the pathophysiological relevance of the gene variant and other linked variants with RA.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that targets mainly the synovial joints. The prevalence of RA is 0.5%–1.0% in many European and North-American populations. However, in Southeast Asia, including Japan and China, the frequency is slightly lower (0.2%–0.3%) (Silman and Pearson, 2002). RA has a higher prevalence and impact on women than men, starting usually around the age of 40–50 for women and later for men (Alamanos et al., 2006).
The pathogenesis of RA has not been fully elucidated. A combination of multiple genetic and environmental factors may contribute to its development. An increasing number of genetic risk factors related to RA have been identified (Stahl et al., 2010). A recent genome-wide association study (GWAS) from Japan has reported the association of the C-C chemokine receptor type 6 gene (CCR6) with RA. This gene is preferentially expressed by immature dendritic cells and memory T cells and is involved in maturation of B cells and in antigen-driven B-cell differentiation. In addition, it may regulate the migration and recruitment of dendritic and T cells during immunological responses (Nanki et al., 2009). The discovery of a triallelic dinucleotide polymorphism of CCR6 (CCR6DNP), demonstrating its significance in the transcription process and its role as a surface marker for Th17 cells, has critically implicated CCR6 in IL-17 human autoimmunity disorders (Kochi et al., 2010). Mutations in its highly conserved cysteine residues have resulted in an influx of intracellular CCR6, leading to the reduced activity and signaling of the receptor, thereby affecting the receptor–ligand interaction (Ai and Liao, 2002). In studies from Kochi et al. (2010) and Stahl et al. (2010), the allele A of CCR6 gene variant was shown to increase the risk of RA. As independent replication is the litmus test for the validity of genetic association studies, and as there have been no previous Asian studies outside Japan, we conducted a case–control study to determine the association of CCR6 gene with RA in our Asian cohort.
Materials and Methods
A total of 996 subjects, comprised of 440 controls and 556 RA cases, were included in the study. RA patients attending the outpatient clinic of the Department of Rheumatology, Allergy, and Immunology at Tan Tock Seng Hospital (TTSH) were recruited. All patients were at least 18 years of age at study entry and fulfilled the 1987 American College of Rheumatology revised criteria for RA (Arnett et al., 1988). Informed consent was obtained from patients or their legal guardians according to the Helsinki Declaration. The work was approved by the Institutional Review Ethics Board. The following data were collected: sociodemographic profile, clinical data including date of onset of symptoms, comorbidities, and extra-articular manifestations. The anticyclic citrullinated peptide (anti-CCP) antibody level was measured by ELISA using the Euroimmune kit. The positive anti-CCP level was defined as >5 IU/mL. Healthy controls without evidence of RA were recruited from the community.
Genotyping analysis
Genomic DNA was extracted from both control and RA patient blood samples according to standard QIAGEN DNA extraction protocol and quantified using a NanoDrop spectrophotometer. Genotyping of CCR6 single-nucleotide polymorphism (SNP) (rs3093024) was performed using TaqMan® SNP Genotyping Assay and TaqMan Universal PCR Master Mix according to the manufacturer's standard protocol on a 96-well plate. Fluorescent signals of VIC® dye-labeled and FAM™ dye-labeled probes were analyzed at end-point; allele call and genotype were generated automatically on Applied Biosystems 7500 Real-Time PCR System.
Statistical analysis
The minor allele frequency (MAF) and 95% confidence interval (CI) were reported separately for RA cases and controls. Fisher's exact test was performed to compare the allele frequency between these two groups. It was also used to verify the Hardy–Weinberg equilibrium (HWE) of CCR6 gene polymorphism (rs3093024) among controls.
Odds ratio (OR) and 95% CI were estimated to investigate the role of this SNP in RA by means of logistic regression. The role of this SNP was appropriately assessed based on the relationship of OR1 (AA vs. GG) and OR2 (AG vs. GG) to reflect a biological model of gene effect (Bagos and Nikolopoulos, 2007).
(1) Recessive model:
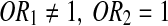 ;
;(2) Dominant model:
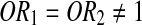 ;
;(3) Additive model:
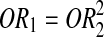 .
.
Once the best genetic model was identified, the three genotypes were collapsed into two groups for dominant (AG+AA vs. GG) and recessive (AA vs. AG+GG) models, or estimation of per-allele risk was performed if additive model is more suitable. ORs were also adjusted by age and gender to minimize their effects. All analyses were done using R version 2.11.1 (www.R-project.org).
Taking RA prevalence of 0.2% and risk allele frequency of 0.45, to test the recessive effect with OR of 1.5, 530 case/control female pairs will be required to have 80% power at a significance level of 0.05, whereas 270 case/control male pairs are needed to test the per-allelic risk of 1.5 with a protective allele frequency of 0.45. As ours is a replication study, statistical significance was defined at p<0.05.
Results
A total of 996 subjects comprised of 440 controls and 556 RA cases (387 anti-CCP positive, 153 anti-CCP negative, and 16 anti-CCP status unknown) were included in the analysis. CCR6 gene polymorphism (rs3093024) was in HWE among controls (all controls: p=0.3836; female controls: p=0.4711; male controls: p=0.6036).
The summary profile of age and gender of controls and RA cases is listed in Table 1. Fisher's exact test showed that MAF was significantly lower in male RA cases than in their counterpart controls (p=0.0049). Similarly, MAF was significantly lower in male anti-CCP-positive cases than male controls (p=0.0128). However, MAF was higher in female anti-CCP-positive cases than female controls (p=0.0295; Table 2).
Table 1.
Demographics (Age and Gender) of Study Subjects
| Control | Case | |
|---|---|---|
| Age | 57 (40, 96) | 44 (20, 81) |
| Gender | ||
| Female | 198 (45.0%) | 456 (82.0%) |
| Male | 242 (55.0%) | 100 (18.0%) |
Table 2.
Demographics (Minor Allele Frequency) of Study Subjects
| |
MAF (95% CI) |
|
|
|---|---|---|---|
| Control | Cases | p-Value | |
| All | 44.0 (40.7, 47.3) | 44.9 (42.0, 47.8) | 0.7165 |
| Female | 42.4 (37.7, 47.3) | 47.4 (44.1, 50.6) | 0.1032 |
| Male | 45.2 (40.9, 49.7) | 33.5 (27.3, 40.3) | 0.0049 |
| Anti-CCP positive | |||
| All | 44.0 (40.7, 47.3) | 46.8 (43.3, 50.3) | 0.2555 |
| Female | 42.4 (37.7, 47.3) | 49.5 (45.7, 53.4) | 0.0295 |
| Male | 45.2 (40.9, 49.7) | 33.1 (25.6, 41.5) | 0.0128 |
| Anti-CCP negative | |||
| All | 44.0 (40.7, 47.3) | 42.8 (37.4, 48.4) | 0.7384 |
| Female | 42.4 (37.7, 47.3) | 44.6 (38.5, 50.9) | 0.6214 |
| Male | 45.2 (40.9, 49.7) | 35.9 (25.3, 48.2) | 0.1811 |
MAF, minor allele frequency; CI, confidence interval; CCP, cyclic citrullinated peptide.
Logistic regression showed that allele A was likely to increase the risk of developing RA among females via a recessive model (OR=1.55, 95% CI=1.01, 2.39), whereas the risk effect appeared to be reduced among males via an additive model (OR=0.60, 95% CI=0.42, 0.85) (Table 3). Considering only subjects who are anti-CCP positive, allele A increased RA risk among females via a recessive model (OR=1.68, 95% CI=1.07, 2.64) but decreased the risk among males via an additive model (OR=0.59, 95% CI=0.39, 0.89) (Table 4). Although there was no significant effect of allele A among RA subjects without the anti-CCP antibody, this allele had shown to decrease the risk of developing RA among males via an additive model in our study (OR=0.67, 95% CI=0.38, 1.16) (Table 5).
Table 3.
Genotype Distribution in Rheumatoid Arthritis Cases and Controls
| Genotype | Controls | Cases | OR |
|---|---|---|---|
| GG | 133 | 176 | Reference |
| AG | 227 | 261 | 0.87 (0.65, 1.16) |
| AA | 80 | 119 | 1.12 (0.78, 1.61) |
| Female only | |||
| GG | 63 | 132 | |
| AG | 102 | 216 | 1.01 (0.69, 1.48) |
| AA | 33 | 108 | 1.56 (0.96, 2.55) |
| Recessive | 1.55 (1.01, 2.39) | ||
| p-Value of recessive model | 0.0412 | ||
| Male only | |||
| GG | 70 | 44 | |
| AG | 125 | 45 | 0.57 (0.34, 0.95) |
| AA | 47 | 11 | 0.37 (0.17, 0.79) |
| Additive | 0.60 (0.42, 0.85) | ||
| p-Value of additive model | 0.0038 | ||
OR, odds ratio.
Table 4.
Association with Anticyclic Citrullinated Peptide-Positive Rheumatoid Arthritis
| Genotype | Controls | Cases | OR |
|---|---|---|---|
| Anti-CCP positive only | |||
| GG | 133 | 113 | Reference |
| AG | 227 | 186 | 0.96 (0.70, 1.32) |
| AA | 80 | 88 | 1.29 (0.87, 1.92) |
| Recessive | 1.32 (0.94, 1.86) | ||
| p-Value of recessive model | 0.1044 | ||
| Female only | |||
| GG | 63 | 84 | |
| AG | 102 | 157 | 1.15 (0.77, 1.74) |
| AA | 33 | 81 | 1.84 (1.09, 3.10) |
| Recessive | 1.68 (1.07, 2.64) | ||
| p-Value of recessive model | 0.0212 | ||
| Male only | |||
| GG | 70 | 29 | |
| AG | 125 | 29 | 0.56 (0.31, 1.01) |
| AA | 47 | 7 | 0.36 (0.15, 0.89) |
| Additive | 0.59 (0.39, 0.89) | ||
| p-Value of additive model | 0.0106 | ||
Table 5.
Association with Anticyclic Citrullinated Peptide-Negative Rheumatoid Arthritis
| Genotype | Controls | Cases | OR |
|---|---|---|---|
| GG | 133 | 53 | Reference |
| AG | 227 | 69 | 0.76 (0.50, 1.16) |
| AA | 80 | 31 | 0.97 (0.58, 1.64) |
| Female only | |||
| GG | 63 | 40 | |
| AG | 102 | 54 | 0.83 (0.50, 1.40) |
| AA | 33 | 27 | 1.29 (0.68, 2.46) |
| Male only | |||
| GG | 70 | 13 | |
| AG | 125 | 15 | 0.65 (0.29, 1.44) |
| AA | 47 | 4 | 0.46 (0.14, 1.49) |
| Additive | 0.67 (0.38, 1.16) | ||
| p-Value of additive model | 0.1476 | ||
p-Values are provided only for those groups that fit the genetic models.
We compared the distribution of CCP status among different genotypes using Fisher's exact test, but there was no significant difference found (all: p=0.472; females only: p=0.343; males only: p=0.910).
Discussion
Twin studies suggest 50% relative genetic contribution or heritability of RA (MacGregor et al., 2000); the remaining cases could be due to environment and chance. With the advancement of technology, large-scale GWAS has become one of the popular means of detecting genetic risk factors. Identification of multiple genetic risk alleles in the major histocompatibility complex (MHC) region and non-MHC loci accounted for about 23% of the genetic burden of RA, implying that more risk alleles remain to be discovered (Stahl et al., 2010). Despite this, current genetic discoveries only explain approximately 16% of disease variance (Stahl et al., 2010). Data from various associated genes studies of RA (Raychaudhuri et al., 2008; Klareskog et al., 2009; Kochi et al., 2010; Stahl et al., 2010) provided only a glimpse of the whole story. The modest OR for most of the individual risk factors may not be sufficient to predict true disease risk. It is therefore more useful to identify molecular pathways in which these genes work together in a coordinated manner during development of RA (Klareskog et al., 2009).
In this study, we showed that allele A of CCR6 increased the risk of RA particularly in females, whereas lower risk was observed in males. This may either correspond to true gender-specific effects or be due to other unidentified causes (such as environmental or lifestyle factors) (Joanna and Andrew, 2009). Sex-specific genetic differences have been shown to associate with many human common diseases and complex traits (detailed review in Ober et al., 2008). We also showed that allele A increased the risk of RA in anti-CCP-positive RA cases (overall OR: 1.32; female OR: 1.68) (Table 4) when compared with no association observed in anti-CCP-negative RA cases (Table 5). The actual causative variant may lie in linkage disequilibrium with the reported CCR6DNP. The difference observed between subjects with anti-CCP-positive and anti-CCP-negative autoantibodies RA cases probably indicate actual etiological difference or other unknown contributory factors. The association of the CCR6 variant with anti-CCP-positive subtypes may also suggest that the association may be related to distinct subtypes of RA.
The CCR6 gene is associated with other immune diseases, for instance, Graves' and Crohn's disease (Stahl et al., 2010). Stahl et al. (2010) reported that a CCR6 polymorphism was in LD with an SNP implicated in Crohn's disease, although it was found to be weakly associated with RA. This might suggest a shared signaling pathway or gene–gene interactions of these diseases via the CCR6 gene. A study by Raychaudhuri et al. (2008) also provided strong evidence for the involvement of the CD40 signaling pathway through NF-κB activation in RA cases. However, the mechanism behind CCR6 gene interactions with other diseases has to be yet elucidated.
It is possible that environmental factors such as diet, nutrition, and even the occupation of an individual can exert an effect on the development of RA. However, it is important to note that the genetic contribution to RA may differ across disease subgroups. Although the disease is more prevalent in females, its incidence and severity may be declining over time (Silman and Pearson, 2002). A comprehensive knowledge of the differences in genetic contribution between genders and those with different disease types should therefore be considered to target subgroups with increased genetic risk for linkage and association studies (Klareskog et al., 2009).
Our study has some limitations. First, our study findings were based on one cohort. Second, our replication study used a significance level of 0.05, which might be liberal. Hence, replication of sex-specific effect in another independent cohort will be needed to provide more convincing evidence that such an effect exists.
Conclusions
In conclusion, this is the first replication Asian study of the GWAS-linked CCR6 variant outside Japan. We showed that CCR6 polymorphism was a risk factor among females but a protective factor among males. A multicenter cohort study in Asia to address the biological impact of CCR6 and gender is necesary. Functional studies may be warranted to unravel the pathophysiological relevance of the gene variant and others with RA.
Acknowledgments
This study was supported by the National Healthcare Group Small Innovative Grant II/08004. The authors thank the National Medical Research Council, Singapore General Hospital, and Tan Tock Seng Hospital for their support. The authors also thank Ms. Ng Shing Jia, Michelle Teng, and the TTSH RA Study Group for their assistance. The TTSH RA Study Group members include T.Y. Lian, H.S. Howe, K.O. Kong, T.C. Lau, W.G. Law, H.H. Chng, J. Tan, G. Chan, and F. Chia.
Disclosure Statement
No competing financial interests exist.
References
- Ai L.S. Liao F. Mutating the four extracellular cysteines in the chemokine receptor CCR6 reveals their differing roles in receptor trafficking, ligand binding, and signaling. Biochemistry. 2002;41:8332–8341. doi: 10.1021/bi025855y. [DOI] [PubMed] [Google Scholar]
- Alamanos Y. Voulgari P.V. Drosos A.A. Incidence and prevalence of rheumatoid arthritis based on the 1987 American College of Rheumatology criteria: a systematic review. Semin Arthritis Rheum. 2006;36:182–188. doi: 10.1016/j.semarthrit.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Arnett F.C. Edworthy S.M. Bloch D.A., et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Bagos P.G. Nikolopoulos G.K. A method for meta-analysis of case-control genetic association studies using logistic regression. Stat Appl Genet Mol Biol. 2007;6 doi: 10.2202/1544-6115.1281. Article17. [DOI] [PubMed] [Google Scholar]
- Joanna J.Z. Andrew P.M. Assessment of sex-specific effects in a genome-wide association study of rheumatoid arthritis. BMC Proc. 2009;3(Suppl 7):S90. doi: 10.1186/1753-6561-3-s7-s90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L. Catrina A.I. Paget S. Rheumatoid arthritis. Lancet. 2009;373:659–672. doi: 10.1016/S0140-6736(09)60008-8. [DOI] [PubMed] [Google Scholar]
- Kochi Y. Okada Y. Suzuki A., et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet. 2010;42(6):515–519. doi: 10.1038/ng.583. [DOI] [PubMed] [Google Scholar]
- MacGregor A.J. Snieder H. Rigby A.S., et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Nanki T. Takada K. Komano Y., et al. Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2009;11:R149. doi: 10.1186/ar2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C. Loisel D.A. Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9:911–922. doi: 10.1038/nrg2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S. Remmers E.F. Lee A.T., et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet. 2008;40:1216–1223. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silman A.J. Pearson J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–S227. doi: 10.1186/ar578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl E.A. Raychaudhuri S. Remmers E.F., et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]