

Abstract
We discovered and reported JAZ as a unique dsRNA binding zinc finger protein that functions as a direct, positive regulator of p53 transcriptional activity to mediate G1 cell cycle arrest in a mechanism involving upregulation of the p53 target gene, p21. We now find that JAZ can also negatively regulate the cell cycle in a novel, p53-independent mechanism resulting from the direct interaction with E2F1, a key intermediate in regulating cell proliferation and tumor suppression. JAZ associates with E2F1's central DNA binding/dimerization region and its C-terminal transactivation domain. Functionally, JAZ represses E2F1 transcriptional activity in association with repression of cyclin A expression and inhibition of G1/S transition. This mechanism involves JAZ-mediated inhibition of E2F1's specific DNA binding activity. JAZ directly binds E2F1 in vitro in a dsRNA-independent manner, and JAZ's dsRNA binding ZF domains, which are necessary for localizing JAZ to the nucleus, are required for repression of transcriptional activity in vivo. Importantly for specificity, siRNA-mediated “knockdown” of endogenous JAZ increases E2F transcriptional activity and releases cells from G1 arrest, indicating a necessary role for JAZ in this transition. Although JAZ can directly inhibit E2F1 activity independently of p53, if functional p53 is expressed, JAZ may exert a more potent inhibition of cell cycle following growth factor withdrawal. Therefore, JAZ plays a dual role in cell cycle regulation by both repressing E2F1 transcriptional activity and activating p53 to facilitate efficient growth arrest in response to cellular stress, which may potentially be exploited therapeutically for tumor growth inhibition.
Key words: JAZ, p53, E2F1, G1 arrest and growth factor depletion
Introduction
p53 is a central negative regulator of cell growth that functions by integrating numerous cellular stress signals (reviewed in refs. 1–3). DNA damage, growth factor depletion, chromosomal aberrations, telomere erosion, oncogene activation and hypoxia activate p53 to induce growth arrest, differentiation or apoptosis.4 We previously identified JAZ (just another zinc finger protein) by screening a murine interleukin-3 (IL-3)-dependent NFS/N1.H7 myeloid cell cDNA library.5 JAZ is the mammalian founding member of a new class of C2H2-type ZFPs that preferentially bind to double-stranded (ds) RNA rather than DNA.5–7 JAZ is ubiquitously expressed and primarily localized in the nucleus, including the nucleolus, but can be cargoed to the cytosol by exportin-5.5,8 Forced expression of JAZ was shown to induce apoptosis in murine fibroblasts.5 This process requires intact ZF domains of JAZ not only for dsRNA binding, but also for its nuclear localization and ability to induce apoptosis.5 Furthermore, interleukin-3 (IL-3) is a multipotential hematopoietic growth factor that induces cell signaling responsible for growth and survival of hematopoietic cells.9–13 Recently, we discovered that interleukin-3 withdrawal upregulates the expression of JAZ in association with p53 activation in factor-dependent hematopoietic cells, and that JAZ functions as a direct, positive regulator of p53 transcriptional activity to mediate G1 cell cycle arrest and apoptosis.14 The mechanism involves the direct binding of JAZ to p53's C-terminal (negative) regulatory domain to activate “latent” p53 in response to non-genotoxic stress signals (such as IL-3 growth factor withdrawal).14
Regulation of the G1 phase of the cell cycle is necessary for normal growth, and dysregulation of this process contributes to neoplastic transformation.15 Members of the Rb and E2F families regulate G1/S transition by trans-activating or repressing expression of genes necessary for S-phase entry (reviewed in refs. 16–18). E2F1 is the founding member of the E2F family and functions as a primary transcription activator to promote G1/S progression and proliferation, and Rb has been well established as the principle negative regulator of E2F1 in a mechanism involving its phosphorylation.19–23 E2F1 can act as an oncogene by promoting proliferation and transformation and overexpression of E2F1, and its target genes, such as cyclin A, have been reported to occur in various human cancers.16,24–31 However, E2F1 has also been reported to behave as a tumor suppressor, because overexpression can mediate apoptosis in either a p53-dependent or -independent and a cell type-specific manner.16–18,32,33 While p53 and E2F1 may act independently, evidence demonstrates “crosstalk” between p53 and E2F1 to affect cell fate.32,33 Furthermore, a number of cellular factors may be involved in the p53-E2F1 crosstalk, including MDM2, ARF and SirT1.34–40 However, while such “crosstalk” may play a vital role in cell growth regulation and tumor suppression, the molecular mechanism(s) by which p53 and E2F1 are interconnected have not been fully elucidated.32,33 Here we report that JAZ, a recently discovered, direct regulator of p53,14 can also directly interact with E2F1 to inhibit its transcriptional activity. Thus, JAZ may represent a novel cellular component that functionally interconnects p53 and E2F1 pathways to negatively regulate G1/S transition in response to cellular stress, including growth factor deprivation.
Results
JAZ-GFP represses E2F1-mediated expression of cyclin A and G1/S transition in a p53-independent manner.
We previously demonstrated that expression of JAZ-GFP can stimulate p53 transcriptional activity and mediate G1 cell cycle arrest in wild-type p53 expressing NIH3T3 murine fibroblast cells in association with upregulation of p21 expression, dephosphorylation of Rb and repression of cyclin A expression.14 We reported that JAZ-GFP expression also does not affect expression of other G1 regulatory proteins tested, including p27, cyclins D and E.14 Thus, we proposed that JAZ may mediate p53-dependent G1 cell cycle arrest by activating p53 to induce expression of its target gene, p21, a cyclin-depependent kinase inhibitor that can induce dephosphorylation of Rb. The hypophosphorylated Rb then can bind and inhibit E2F transcriptional activity with repression of cyclin A, an E2F target gene that functions in G1/S cell cycle progression.16–18,41–43 Since p21 is a key player in p53-mediated G1 arrest,1,44 we recently directly tested a role for p21 in JAZ-mediated repression of cyclin A. For this purpose we used a p21-specific siRNA “knockdown” strategy to block any upregulation of p21 expression that occurs following JAZ-GFP transfection (Fig. 1A, lanes 1–4). Results reveal that “knockdown” of p21 upregulation increases the expression of cyclin A under these conditions (lanes 3 and 4), indicating a role for p21 upregulation in JAZ-GFP-mediated repression of cyclin A. However, while cyclin A expression is increased when p21 upregulation is blocked, its expression level is still lower than that shown in the GFP-only control cells (Fig. 1A, lanes 1 and 4). This indicates that “knockdown” of p21 upregulation can only partially but not completely inhibit JAZ-GFP-mediated repression of cyclin A, suggesting that JAZ may also function to mediate repression of cyclin A independently of any p21 upregulation that can be induced by p53.
Figure 1.

JAZ expression negatively regulates E2F1-mediated expression of cyclin A and G1/S transition. (A) JAZ-GFP or GFP alone was transfected into NIH3T3 cells with or without co-transfection with the p21 or control siRNA. After 48 h, JAZ-GFP or GFP-positive cells were FACS (fluorescence activated cell sorting)-sorted, followed by protein gel analysis using antibodies against GFP, cyclins E and A, p21 and Tubulin (as a loading control). G-GFP; J-G-JAZ-GFP. (B) JAZ-GFP or GFP alone was transfected into p53−/− MEFs with or without HA-E2F1/pcDNA3 for 48 h followed by FACS-sorting and protein gel analysis using antibodies against HA, cyclin A, p21 and Tubulin (top part). The transfected cells were also harvested for flow cytometry analysis to determine cell cycle distribution (bottom part). *p = 0.003; **p = 0.001 vs. the GFP-only control.
Since cyclin A is known to be transcriptionally targeted by E2F1,45 HA-tagged E2F1 was either transfected alone or cotransfected with JAZ-GFP into p53−/− mouse embryonic fibroblasts (MEFs) to directly test a p53-independent role for JAZ in repressing cyclin A expression. Results of Protein gel blot analysis reveal that, while, as expected, HA-E2F1 alone increases the expression of cyclin A (Fig. 1B, top part, lanes 1 and 3), co-expression of JAZ-GFP blocks HA-E2F1-mediated cyclin A upregulation (lanes 3 and 4). It should be noted that expression of HA-E2F1 and/or JAZ-GFP did not alter p21 expression (lanes 1–4). Furthermore, flow cytometry analysis reveals that while HA-E2F1 alone promotes G1/S transition in p53−/− MEFs (as indicated by a 26% decrease in the G1/G0 population when HA-E2F1 is present in the GFP control, Fig. 1B, bottom part), JAZ-GFP inhibits the stimulatory effect of HA-E2F1 (by a 22% increase in G1/G0). These data indicate that JAZ can negatively regulate E2F1's function in the cell cycle in a p53-independent mechanism.
JAZ represses E2F1 transcriptional activity in a mechanism that does not require p53.
Since E2F1's transcriptional function is essential for its role in promoting G1/S transition,16 we tested whether JAZ may modulate E2F transcriptional activity. NIH3T3 cells were co-transfected with FLAG-JAZ and a pE2F-TA luciferase reporter vector for 48 h. Results show that expression of FLAG-JAZ can repress E2F transcriptional activity in NIH 3T3 cells by 61% when compared with the empty vector control (Fig. 2A). Also, as a negative control, FLAG-JAZ has minimal if any effect on the activity of pTA control reporter vector (Fig. 2A), indicating specificity. Next, we tested whether p53 is required/involved in any JAZ-mediated repression of E2F transcriptional activity, because JAZ can bind and activate p53 as reported previously in reference 14. p53+/+ and p53−/− isogenic MEFs were used to tease out this possibility. Results reveal that while FLAG-JAZ represses E2F transcriptional activity in p53+/+ MEFs by 62%, FLAG-JAZ can also decrease E2F activity by 33% in p53−/− MEFs (Fig. 2B). These data indicate that, while p53 may play an enhancing role in JAZ-mediated repression of E2F activity, p53 is not required for the inhibitory function of JAZ.
Figure 2.

JAZ expression represses E2F1 transcriptional activity. (A) 1.0 µg of FLAG-JAZ/pcDNA3 was co-transfected with 0.1 µg of pE2F luciferase reporter vector (pE2F-TA-luc) or a pTA control reporter vector into NIH3T3 cells. After 48 h, cells were lysed and assayed for luciferase activity. V-pcDNA3; F-J-FLAG-JAZ/pcDNA3. *p = 0.003 vs. the vector control. (B) FLAG-JAZ was co-transfected with pE2F-TA-luc into p53+/+ and p53−/− MEFs. 0.1 µg of pAc-β-GAL internal control vector was also included to normalize transfection efficiencies. *p = 0.002; **p = 0.004. (C) 0–1.0 µg of FLAG-JAZ were co-transfected with 0.1 µg of HA-E2F1/pcDNA3 plus 0.1 µg of pE2F-TA-luc and pAc-β-GAL (the amount of DNA was kept constant using pcDNA3) into p53−/− MEFs followed by luciferase assay and normalization. *p < 0.01. Protein gel analysis was performed to verify protein expression.
We next directly determined the effect of JAZ on E2F1-mediated transcriptional activation by co-transfecting HA-E2F1 and FLAG-JAZ along with the E2F reporter vector into p53−/− MEFs. Results show that while, as expected, HA-E2F1 alone increases E2F transcriptional activity by greater than 20-fold in p53−/− cells, FLAG-JAZ can inhibit HA-E2F1-mediated reporter activity in a dose-dependent manner (by up to ∼70%, Fig. 2C). These results clearly indicate that FLAG-JAZ can repress E2F1-dependent transcriptional activation independently of p53.
JAZ associates with E2F1 in mammalian cells.
To investigate whether JAZ may repress E2F1 transcriptional activity through a direct mechanism, we tested whether JAZ may interact with E2F1. Co-imunoprecipitation (IP) studies were performed in several mammalian cell lines. Results reveal that while exogenously expressed FLAG-JAZ and HA-E2F1 can be co-IPed in p53−/− MEFs (Fig. 3A), FLAG-JAZ can also associate with endogenous E2F1 in NIH3T3 cells (Fig. 3B). Moreover, endogenous JAZ and E2F1 associate in both IL-3-dependent NFS/N1.H7 myeloid and p53-deficient M1 myeloid leukemic cells (Fig. 3C). The specificity is evidenced by the observation that the co-IP can be achieved only with the JAZ-specific antibody (JAZ111) but not with the control antibody for JAZ111 that was produced by antigen competition.14 It should also be noted that only a small percentage of endogenous (or exogenously expressed) JAZ is co-IPed with E2F1. While the nature of the association and its “stability” following detergent lysis could account for this, a small fraction of E2F1 association has been observed for other E2F1 regulators depending on the cell context.35,40,46 Furthermore, results from subcellular fractionation and co-IP studies performed using p53+/+ MEFs indicate that both endogenous JAZ and E2F1, as expected, associate in the nucleus (Fig. 3D), where these proteins are known to be functionally expressed.5,18 However, JAZ does not co-IP with PCNA (a nuclear protein as a control, Fig. 3D). These results indicate that nuclear JAZ-E2F1 association is specific and strongly support a potentially direct interaction between JAZ and E2F1 in vivo.
Figure 3.

JAZ associates with E2F1 in mammalian cells. (A) FLAG-JAZ/pcDNA3 (pcDNA3 as a control) was transfected alone or co-transfected with HA-E2F1/pcDNA3 into p53−/− MEFs. After 24 h, cells were lysed, and co-immunoprecipitation (IP) followed by protein gel analysis was performed using antibodies against FLAG and HA. V-pcDNA3; F-J-FLAG-JAZ/pcDNA3; 20% input 50 µg of the cell lysate. (B) FLAG-JAZ/pcDNA3 (pcDNA3 as a control) was transfected into NIH3T3 cells. After 24 h, cells were lysed and IPed using a FLAG antibody, followed by protein gel analysis using antibodies against FLAG and E2F1. The IgG control antibody did not co-IP FLAG-JAZ or E2F1 (data not shown). (C) The JAZ antibody (JAZ111) was used to IP endogenous JAZ from 1 mg of whole cell lysate from IL-dependent NFS/N1.H7 murine myeloid cells or p53-deficient M1 murine leukemic cells. JAZ-associated endogenous E2F1 was assessed by protein gel analysis using an antibody against E2F1. To facilitate detection of endogenous JAZ protein, H7 or M1 cells were deprived of IL-3/serum for 2 h to increase JAZ expression.14 5% input 50 µg of the cell lysate; “+”, JAZ111; “−”, the control Ab for JAZ111, as described in Materials and Methods. (D) Subcellular fractionation of p53+/+ MEF cells was performed. JAZ 111 was used to IP endogenous JAZ from 1 mg of the lysate of the nuclear or cytoplasmic fraction, followed by protein gel analysis using antibodies against E2F1 and PCNA (a nuclear protein as a control). Nuc., nuclear fraction; Cyto., cytoplasmic fraction; 5% input 50 µg of the nuclear or cytoplasmic lysate.
JAZ directly binds E2F1 in vitro, and the interaction involves E2F1's central DNA binding and dimerization region and C-terminal transactivation domain.
We tested whether JAZ can directly interact with E2F1 in vitro using a GST “pull-down” assay. Results show that purified GST-E2F1 not the GST control can “pull down” purified His-tagged JAZ, indicating a potentially direct binding (Fig. 4A, lane 3). Since JAZ is a dsRNA binding ZFP,5 we then tested whether JAZ-E2F1 binding may be dependent on dsRNA. RNase V1 was used, which can specifically cleave dsRNAs associated with recombinant JAZ in vitro.8,14 Purified His-JAZ and GST-E2F1 were first incubated with RNase V1, whose activity was verified as described in reference 14. Results reveal that nuclease treatment fails to influence JAZ-E2F1 binding (Fig. 4A, lanes 3 and 4), indicating a dsRNA-independent manner.
Figure 4.

JAZ directly binds E2F1 in vitro. (A) GST or GST-E2F1 glutathione-Sepharose beads were incubated with 100 ng of purified, recombinant His-tagged JAZ in the presence or absence of RNase V1 that specifically cleaves dsRNA. Protein gel analysis was performed using an anti-6xhis antibody. (B) The GST-E2F1 fragments (containing E2F1's central DNA binding/dimerization region or C-terminal transaction domain) can bind FLAG-JAZ. GST or GST-E2F1 [full-length or fragments (113–284) and (388–437)] beads were incubated with the same amount (250 µg) of COS-7 cell lysate containing FLAG-JAZ in the “pull-down” assay. COS-7 cell lysate containing HA-Rb was also used a positive control for GST-E2F1 binding. Protein gel analysis was performed using a FLAG or HA antibody. The bottom part represents Fast-green staining of GST-E2F1 proteins (arrows).
Since E2F1's central DNA binding and dimerization region and C-terminal transactivation region are essential for its transcription factor function,16–18 we tested whether JAZ can interact with these two regions (113–284 and 388–437) in vitro. Results reveal that the GST-E2F1 mutants containing either (113–284) or (388–437) of E2F1 can “pull down” FLAG-JAZ (Fig. 4B, lanes 4 and 5). However, no binding occurs for the GST protein control (lane 2). Also as a positive control for the functionality of purified GST-E2F1 proteins, HA-Rb is also shown to be “pulled down” by wild-type GST-E2F1 and the GST-E2F1 mutant (388–437) that contains the C-terminal transactivation domain, which is known to be essential for Rb binding and function (Fig. 4B, lanes 3 and 5).19–23 Of note, binding between FLAG-JAZ and the GST-E2F1 mutant (388–437) appears to be stronger than that seen with wt GST-E2F1, since more of FLAG-JAZ is “pulled down” (lanes 3 and 5). Interestingly, such an apparent avid binding was also observed between HA-Rb and the GST-E2F1 (388–437) mutant (lanes 3 and 5). Any functional significance of such an avid binding in vitro remains to be verified.
JAZ inhibits E2F1's specific DNA binding in vitro.
Since JAZ can bind to E2F1's central DNA binding and dimerization region, we tested whether JAZ may affect E2F1's specific DNA binding using an electrophoretic mobility shift assay (EMSA). The NIH3T3 nuclear extracts (NE), which were prepared from the cells transfected with an empty vector or HA-E2F1 or HA-E2F1 plus FLAG-JAZ, were mixed with a 32P-labeled DNA probe containing the wt E2F binding site. As shown in Figure 5A, the nuclear extract (the empty vector as a control) can induce moderate bandshifts (lane 2), probably because NIH cells express endogenous E2F1 and/or other E2F family members that may form the complexes with the E2F probe. However, while, as expected, the NIH3T3 NE containing HA-E2F1 alone significantly enhances the bandshifts (lane 3), the nuclear extract with co-expressed HA-E2F1 and FLAG-JAZ completely blocks such bandshift enhancement (lane 4). These data support the notion that JAZ can inhibit E2F1-mediated DNA binding.
Figure 5.

JAZ inhibits E2F1's specific DNA binding in vitro. (A) HA-E2F1/pcDNA3 (pcDNA3 as a control) was transfected alone or co-transfected with FLAG-JAZ/pcDNA3 into NIH3T3 cells (the amount of DNA was kept constant using pcDNA3). After 24 h, cells were harvested, and nuclear extracts were prepared and subjected to EMSA analysis with a 32P-labeled DNA probe that contained a wild-type E2F binding site. (B) An EMSA analysis was also performed using Raji nuclear extract containing endogenous E2F1 with the 32P-labeled E2F probe in the presence or absence of an E2F1 antibody [for induction of supershift(s)] as described in Materials and Methods. Purified GST-JAZ (GST as a control) or His-JAZ recombinant protein was added in the EMSA assay to assess the effect of JAZ on E2F1's DNA binding. Excessive amounts of unlabeled wild-type or mutant E2F oligonucleotides were also added in the controls to verify the binding specificity.
To verify the specificity of JAZ's inhibitory effect on the DNA binding of E2F1, we employed a commercial EMSA kit that includes the Raji nuclear extract containing endogenous E2F1. Results reveal that mixing the Raji NE with the 32P-labeled probe induces bandshifts that can be blocked by adding “cold” WT but not mutant E2F oligonucleotide (Fig. 5B, lanes 4–6), indicating specific E2F DNA binding. Furthermore, addition of the E2F1 antibody induces a supershift that can be blocked by added GST-JAZ but not GST (lanes 7–9), indicating that JAZ inhibits E2F1-mediated specific DNA binding in vitro. This inhibitory effect of JAZ is also confirmed by addition of purified His-JAZ (lane 10). Of note, in the absence of the Raji NE, GST-JAZ does not induce any bandshift (Fig. 5B, lanes 2 and 3), as the GST control does, indicating that GST-JAZ does not bind to the E2F DNA binding site. This is consistent with the previous finding that JAZ preferentially binds dsRNA rather than DNA in vitro.5 Therefore, these data indicate that JAZ represses E2F1 transcriptional activity in a mechanism involving inhibition of E2F1's DNA binding activity.
The dsRNA binding ZF domains of JAZ are not necessary for E2F1 binding in vitro but are required for JAZ-mediated repression of E2F1 in vivo by dictating JAZ's nuclear localization.
Since we previously discovered that JAZ's ZF domains are not only required for dsRNA binding but also for nuclear localization,5 we tested whether the ZF domains may play a role in JAZ interaction with E2F1. Results indicate that mutation of any individual ZF domain (H91A, H152A, H203A or H257A) has no or minimal effect on the interaction of GST-JAZ with HA-E2F1 in in vitro “pull-down” studies (Fig. 6A, lanes 3–7). Furthermore, a GST-JAZ NF mutant in which all four ZF domains are mutated, rendering JAZ unable to bind dsRNA,5 can also “pull down” HA-E2F1 (lane 8). Of note, the GST-JAZ H91A and NF mutants appear to “pull down” less FLAG-JAZ than the wild-type protein (lanes 3, 4 and 8), probably due to smaller amounts of these two proteins present, as shown by Fast-green staining of GST-proteins (Fig. 6A). Therefore, JAZ can directly interact with E2F1 in a mechanism that is independent of JAZ's dsRNA binding in vitro.
Figure 6.

The ZF domains of JAZ are not necessary for Rb and E2F1 binding in vitro but are required for JAZ-repression of E2F1 in vivo. (A) The GST-JAZ beads containing the individual ZF mutation (H91A, H152A, H203A or H257A) or the NF mutant in which all the four zinc fingers are point mutated were used to “pull down” HA-E2F1 from the NIH3T3 cell lysate followed by protein gel analysis using an anti-HA antibody. The bottom part represents Fast-green staining of GST-JAZ proteins. (B) Wt JAZ-GFP and its ZF mutants14 were co-transfected with HA-E2F1/pcDNA3 and pE2F-TA-luc vectors into p53−/− MEFs. After 48 h, cells were FACS (fluorescence activated cell sorting)-sorted and assayed for luciferase activity. *p < 0.01.
Next, we tested a role of the ZF domains in JAZ-repression of E2F1 transcription activity in vivo. Results again reveal that, while mutation of any single JAZ ZF domain fails to affect JAZ-GFP-mediated repression of HA-E2F1 activity, mutation of more than two such loci, which will lead to loss of nuclear localization,5 profoundly reduces/abolishes JAZ-GFP's repression effect (Fig. 6B). In addition, JAZ-GFP deletion mutants J(1–171) and J(167–294), which lose the specific nuclear localization (data not shown), also cannot repress E2F1 (Fig. 6B). These findings support the conclusion that nuclear JAZ is required to interact with and repress E2F1 activity.
siRNA “knockdown” of JAZ increases E2F transcriptional activity, attenuates growth factor deprivation-mediated G1 arrest and accelerates G1/S transition upon serum stimulation.
Endogenous JAZ and E2F1 associate in the nucleus (Fig. 3D). We therefore tested whether endogenous JAZ is required to regulate E2F transcriptional activity. For this, the expression of JAZ was “knocked down” using siRNA (Fig. 7A, top part).14 Results of the luciferase reporter assay reveal that “knockdown” JAZ increases E2F transcriptional activity in either the p53+/+ or p53−/− cells by 66% and 41%, respectively (Fig. 7A, middle part). Furthermore, when serum is removed as an added stress, a more significant increase in E2F activity is seen in p53+/+ and p53−/− cells by 355% and 192%, respectively. In addition, flow cytometry analysis shows that following serum withdrawal, “knockdown” of JAZ decreases the G1/G0 population in either the p53+/+ or p53−/− cells by 20% and 16%, respectively (Fig. 7A, bottom part). These data support a necessary role for JAZ in repressing E2F1 activity and in inducing a G1 arrest in a mechanism that does not require (but may be enhanced by) p53. This notion is supported by results from hematopoietic cells demonstrating that “knockdown” of JAZ expression attenuates interleukin-3 (IL-3) or serum deprivation-induced G1 cell cycle arrest in both p53-expressing, factor-dependent H7 or p53-deficient M1 cells, respectively (Sup. Fig. 1).
Figure 7.

(A) “Knockdown” of JAZ by siRNA increases E2F transcriptional activity and attenuates serum withdrawal-induced G1 arrest in both p53+/+ and p53−/− MEFs. The JAZ-siRNA was transfected into p53+/+ or p53−/− MEFs. After 48 h, cells were lysed for protein gel analysis using JAZ111 and a Tubulin antibody (top part). Endogenous JAZ expression was “knocked down” by ∼70%, as determined by densitometry.14 The JAZ-siRNA was also co-transfected into the MEFs with pE2F-TA-luciferase vector and a carrier vector pEGFPN1 (GFP). After 48 h, cells were grown with or without serum for 24 h and then assayed for luciferase activity (middle part). *p < 0.01; **p < 0.001 vs. the control siRNA. In addition, the JAZ-siRNA had no effect on the activity of the pTA-luciferase control vector (data not shown). Furthermore, after siRNA transfection for 48 h followed by serum deprivation for 24 h, flow cytometry analysis was performed (bottom part). *p < 0.001; **p = 0.001 vs. the control siRNA. (B) “Knockdown” of JAZ expression accelerates serum-stimulated G1/S progression in M1 cells. p53-deficient M1 leukemic cells in which JAZ was stably “knocked down” (Sup. Fig. 1sB) were synchronized in the G1 phase by incubation with 1.5 mM hydroxyurea for 18 h. Cells were then washed and repleted with fresh growth media containing 10% FBS. At 0, 3, 6, 9 and 12 h following serum stimulation, cells were harvested for flow cytometry analysis (top part). The changes in G1/G0 (decreased percentage) at 3 h and 6 h after serum stimulation are shown (bottom part). *p = 0.008; **p = 0.003 vs. the control siRNA.
We next tested whether JAZ may play a role in slowing or modulating serum stimulation-induced entry into S phase. For this, M1 cells in which endogenous JAZ expression is stably “knocked down” (Sup. Fig. 1B) were synchronized in the G1 phase with hydroxyurea and then stimulated to enter the cell cycle by adding back fresh growth medium-containing serum.47 Flow cytometry analysis reveals that “knockdown” of JAZ decreases the G1/G0 population more rapidly compared with the control siRNA and thereby accelerates the G1/S transition following serum stimulation (Fig. 7B). These findings strongly support the notion that JAZ is necessary for negative regulation of G1/S transition in the leukemic cells in a mechanism independent of p53.
Discussion
In addition to a direct stimulatory role of p53 involving JAZ's direct interaction with and activation of p53,14 we now find that JAZ, in a p53-independent mechanism, can directly repress E2F1-dependent transcription and lead to cell cycle arrest (Figs. 1–3). This dual effect of JAZ potentially represents a more efficient mechanism for mediating G1 cell cycle arrest in response to the stress of growth factor withdrawal, which may induce upregulation and/or activation of JAZ (as summarized in Fig. 8).14 It is also known that other cellular proteins are involved in regulating both p53 and E2F1 pathways to affect the cell cylce.32,33 For example, the ARF tumor suppressor is a key p53 regulator that mediates G1 cell cycle arrest in response to oncogenic stress by inhibiting MDM2, which stabilizes and activates p53.48,49 ARF has also been reported to regulate the E2F pathway in a p53-independent manner and directly bind and inhibit E2F1 transcriptional activity.36–39 Interestingly, as observed for JAZ,14 ARF can also selectively repress expression of cyclin A but not cyclin E, but the mechanism is not clear yet.50 Now, results here indicate that JAZ can directly bind and repress E2F1 activity in a mechanism independent of ARF, since JAZ represses E2F1 activity in NIH3T3 cells that lack the INK4a/ARF locus (Figs. 1A and 2A).51 Thus, we propose that JAZ-mediates repression of E2F1 in a novel mechanism that efficiently negatively regulates cell cycle progression.
Figure 8.
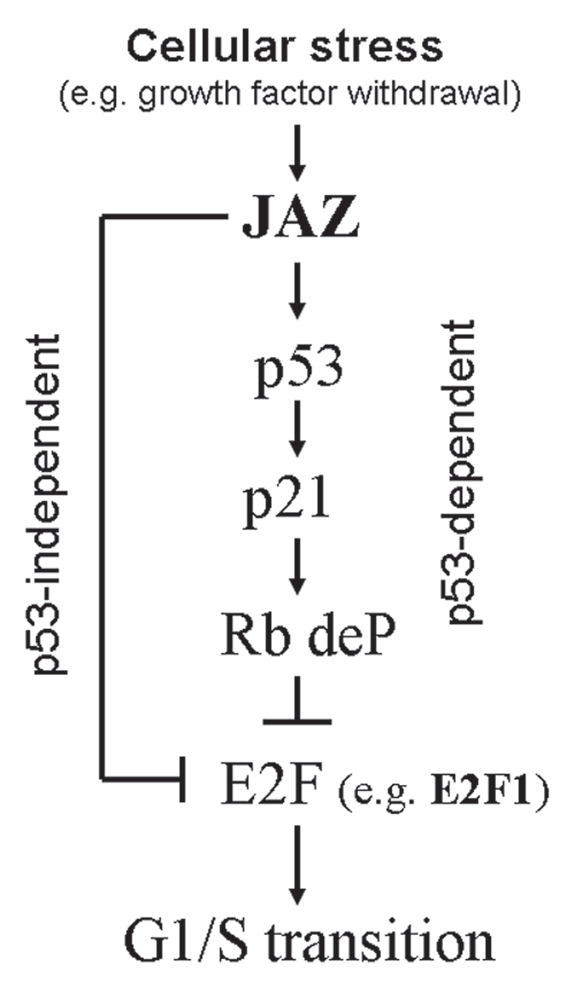
A model of how JAZ negatively regulates E2F activity to inhibit G1/S transition. Cellular stress (such as growth factor withdrawal) upregulates/activates JAZ that interacts with and activates p53. Transcriptionally activated p53 induces upregulation of p21 expression, which leads to dephosphorylation of Rb, and thus inhibition of E2F activity, results in G1 cell cycle arrest. JAZ can also directly interact with E2F1 to inhibit G1/S transition in a p53-indpendent manner.
Interaction with DNA is essential for E2F1's transcription factor function.16 A role for an E2F1 complex to bend DNA structure was reported in mediating E2F1's transcription activation.52 We find that JAZ can directly bind E2F1's central DNA binding and dimerization region and specifically inhibit E2F1's DNA in vitro (Figs. 4 and 5). This supports the notion that JAZ represses E2F1 transcriptional activity in a mechanism that involves JAZ's direct binding and inhibition of E2F1's DNA binding activity. However, since JAZ can also bind to E2F1's C-terminal transactivation domain, to which Rb and other cellular proteins can also bind (Fig. 4B),16 JAZ may repress E2F1 transcriptional activity by additional possible mechanism(s) as well. For example, binding of JAZ to E2F1's transactivation domain may potentially disrupt its functional interaction with other activation and/or co-activation proteins, including p300/CBP.53 Whether JAZ may be involved in any Rb-mediated repression of E2F1 in a p53-independent manner is not known yet. In addition, other E2F family members, especially E2F2, E2F3a and E2F3b, also contain the conserved domain structure similar to E2F1.16–18 Whether JAZ may potentially interact with these E2F protein(s) to play a role in regulating G1/S transition remains to be investigated.
Since JAZ is a dsRNA binding ZFP,5 we considered the possibility that JAZ-E2F1 interaction may involve JAZ's binding to dsRNA. However, results reveal that JAZ and E2F1 can directly interact in a mechanism independent of dsRNA binding, at least in vitro (Figs. 4A). This result is similar to the previous finding that JAZ and p53 can directly interact independently of JAZ's dsRNA binding.14 Furthermore, others reported that JAZ can bind a dsRNA binding protein ILF3 independently of dsRNA, even though its interaction with and export from the nucleus by exportin-5 does depend on both dsRNA and RanGTP.8 Collectively, these results indicate potentially different roles for dsRNA in JAZ's interaction with cellular proteins. We previously showed that JAZ's ZF domains are not only required for its dsRNA binding properties, but are also essential for nuclear localization.5 Results here indicate that JAZ's dsRNA binding ZF domains are also apparently not required for its binding to E2F1 in vitro (Fig. 6A). Because the ZF domains are necessary for JAZ's nuclear localization and thus repression of E2F1 transcriptional activity (Fig. 6B), we conclude that nuclear JAZ functionally interacts with and regulates E2F1's activity.
In summary, findings here demonstrate that in addition to acting as a direct regulator of p53, JAZ can also directly bind and repress E2F1 transcriptional activity in a p53-independent manner to mediate inhibition of G1/S cell cycle progression. Results support a necessary role for JAZ in negative growth signaling that results in G1 cell cycle arrest following growth factor removal from growing cells. Thus, JAZ can inhibit E2F1 activity and, if functional p53 is also expressed, thus may be a more potent negative regulator of the cell cycle following physiologic stress(es), including growth factor withdrawal. A model proposes that JAZ's dual role may represent a novel interconnection between the p53 and E2F1 pathways and, as such, may be a new molecular target that can be exploited in cancer therapy (Fig. 8).
Materials and Methods
Cell culture and transfection.
p53+/+ and p53−/− isogenic mouse embryonic fibroblasts (MEFs), NIH3T3 murine fibroblasts and COS-7 monkey kidney cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and cultured in a humidified incubator at 37°C and 5% CO2. M1 murine myeloid leukemic cells were grown as described in reference 14. Transfection was performed using LipofectAMINE or LipofectAMINE2000 (Invitrogen) according to the manufacturer's instruction.
Immunoprecipitation, protein gel blot analysis and subcellular fractionation.
All antibodies used were obtained from Santa Cruz Biotechnology, except p21 (Ab-1, Oncogene Research Products), FLAG (M2, Sigma-Aldrich), HA (12CA5, Roche) and JAZ111. JAZ111 is an affinity-purified rabbit polyclonal antibody against JAZ, and the control Ab for JAZ111 was produced by antigen (peptide) competition.14
Co-immunoprecipitation (IP) and protein gel blot analysis were performed similarly as described in reference 14. For subcellular fractionation, the nuclei of p53+/+ MEFs were isolated, and both nuclear and cytoplasmic lysates were prepared.14 For JAZ-GFP or GFP transfected cells, fluorescence-activated cell sorting (FACS) was used to sort out GFP positive cells for protein gel analysis.14
In vitro GST “pull-down” assay.
The E2F1 and Rb expression constructs used were kindly provided by W. Douglas Cress (Moffitt Cancer Center).54
GST-JAZ and GST-E2F1 glutathione-Sepharose beads were prepared and used as described in reference 14 and 54. The GST protein beads were incubated at 4°C for 1 h with cell lysates containing ectopically expressed HA-tagged E2F1 or FLAG-tagged JAZ (in the pcDNA3 vector). Following GST “pull down,” protein gel analysis was performed using an antibody against HA or FLAG. His-tagged, recombinant JAZ protein was also prepared and purified.5 GST-E2F1 beads were incubated with His-JAZ in the presence or absence of RNase V1 similarly as described in reference 8 and 14. GST-JAZ ZF mutants were also prepared and used to determine the effect of the ZF motifs on the binding of JAZ to HA-E2F1.
GST-E2F1 mutant (113–284) and (388–437) beads were prepared and incubated at 4°C for 2 h with 250 µg of the lysate of COS-7 monkey kidney cells (that were transfected by HA-Rb or FLAG-JAZ/pcDNA3), followed by protein gel analysis using an HA or FLAG antibody.14,54
Luciferase assay.
pE2F-(containing the E2F response elements)-TA and pTA-control luciferase reporter vectors were obtained from BD CLONTECH.55 To determine the effect of JAZ on E2F transcriptional activity, 1.0 µg of FLAG-JAZ/pCDNA3 was co-transfected with 0.1 µg of pE2F-TA-luc vector into NIH3T3 cells, p53+/+ or p53−/− MEFs that were grown in 6-well culture plates. 0–1.0 µg of FLAG-JAZ/pcDNA3 and 0.1 µg of HA-E2F1/pcDNA3 were co-transfected with 0.1 µg of pE2F-TA-luc vector into p53−/− MEFs. After 48 h, cells were harvested for luciferase assays using the Luciferase Assay System (Promega). 0.1 µg of pAc-β-GAL vector containing a β-galactosidase gene under control of a β-Actin promoter was also included as an internal control as described in references 14 and 56.
EMSA analysis.
The electrophoretic mobility shift assay (EMSA) was performed using Nushift™ Cell Cycle Regulators kit (Active Motif) according to the manufacturer's protocol. The radioactive E2F oligo WT probe (containing the conserved E2F DNA binding sequence) was prepared using [γ-32P]ATP and T4 polynucleotide kinase (Invitrogen) and purified. According to the manufacturer's recommendation, 10 µg of Raji nuclear extract (containing endogenous E2F1) was added in the reaction mixtures (for induction of bandshifts, formation of oligonuccleotide/protein complexes). The specific E2F DNA binding was verified by bandshift competition in the presence of unlabeled WT or mutant E2F oligonucleotides. To induce supershift(s), 4 µl of the E2F1 antibody was added. 100 ng of purified GST-JAZ (GST as a control) or His-JAZ was also included in the mixtures to determine the effect of JAZ on E2F1's specific DNA binding. The samples were then loaded onto 5% native polyacrylamide gel and run for 3 h at 150 V and 4°C in buffer containing 22.5 mM Tris borate, pH 8.0 and 0.5 mM EDTA, followed by autoradiography.
Alternatively, 10 µg of NIH3T3 nuclear extract that contains exogenously expressed HA-E2F1 alone or co-expressed HA-E2F1 and FLAG-JAZ was used in the EMSA analysis. To prepare the NIH3T3 nuclear extract, 8 µg of pcDNA3 or FLAG-JAZ/pcDNA3 was co-transfected with 2 µg of HA-E2F1/pcDNA3 (pcDNA3 as a control) into NIH3T3 cells that were grown in 10-cm-diameter culture plates. After 24 h, cells were harvested, and the nuclei were isolated and extracted as described previously in reference 5.
siRNA interference and flow cytometry analysis.
JAZ-specific and control siRNAs were used as described previously in reference 14. To assess the effect of the JAZ-siRNA on E2F transcriptional activity, 20 pmol of the JAZ-siRNA was co-transfected with 0.1 µg of pE2F-TA-luc (plus 0.3 µg of pEGFPN1 (GFP) as a carrier vector) into p53+/+ and p53−/− MEFs using LipofectAMINE. After 48 h, cells were grown with or without serum for another 24 h and harvested for luciferase assay and flow cytometry analysis (the GFP-positive cells were gated and then analyzed for cell cycle distribution).57
M1 murine myeloid leukemic cells that were stably transfected with a JAZ siRNA or control siRNA expressing plasmid were incubated with 1.5 mM hydroxyurea (in DMEM supplemented with 10% FBS) for 18 h and then washed and repleted with fresh medium containing 10% FBS.14,47 At 0 to 12 h post-serum stimulation, cells were harvested for flow cytometry analysis.
Student's t-test (p-value) was performed in analyzing data obtained from at least three independent experiments done for luciferase activity and flow cytometry measurements.
Supplementary Material
Acknowledgments
This work was supported by a NIH grant CA44649 (to W.S.M.) and grants from the American Cancer Society (Florida Division, Inc.) and Stop! Children's Cancer, Inc. (to M.Y.). We would like to thank Dr. Douglas Cress (Moffitt Cancer Center, Tampa, Florida) for providing the E2F1 and Rb constructs.
Abbreviations
- ds
double-stranded
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HA
hemagglutinin
- IL-3
interleukin-3
- IP
immunoprecipitation
- JAZ
just another zinc finger protein
- MEFs
mouse embryonic fibroblasts
- PAGE
polyacrylamide gel electrophoresis
- siRNA
small interference RNA
- ZF
zinc finger
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 4.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 5.Yang M, May WS, Ito T. JAZ requires the doublestranded RNA binding zinc finger motifs for nuclear localization. J Biol Chem. 1999;274:27399–27406. doi: 10.1074/jbc.274.39.27399. [DOI] [PubMed] [Google Scholar]
- 6.Finerty PJ, Bass BL. A xenopus zinc finger protein that specifically binds dsRNA and RNA-DNA hybrids. J Mol Biol. 1997;271:195–208. doi: 10.1006/jmbi.1997.1177. [DOI] [PubMed] [Google Scholar]
- 7.Méndez-Vidal C, Wilhelm MT, Hellborg F, Qian W, Wiman KG. The p53-induced mouse zinc finger protein wig-1 binds double-stranded RNA with high affinity. Nucleic Acids Res. 2002;30:1991–1996. doi: 10.1093/nar/30.9.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen T, Brownawell AM, Macara IG. Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin-5. Mol Cell Biol. 2004;24:6608–6619. doi: 10.1128/MCB.24.15.6608-19.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.May WS. Control of apoptosis by cytokines. Adv Pharmacol. 1997;41:219–246. doi: 10.1016/S10543589(08)61060-1. [DOI] [PubMed] [Google Scholar]
- 10.Johnson DE. Regulation of survival pathways by IL-3 and induction of apoptosis following IL-3 withdrawal. Front Biosci. 1998;3:313–324. doi: 10.2741/a276. [DOI] [PubMed] [Google Scholar]
- 11.Ito T, Jagus R, May WS. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci USA. 1994;91:7455–7459. doi: 10.1073/pnas.91.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb E, Oren M. p53 facilitates pRb cleavage in IL-3-deprived cells: novel pro-apoptotic activity of p53. EMBO J. 1998;17:3587–3596. doi: 10.1093/emboj/17.13.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blandino G, Scardigli R, Rizzo MG, Crescenzi M, Soddu S, Sacchi A. Wild-type p53 modulates apoptosis of normal, IL-3 deprived, hematopoietic cells. Oncogene. 1995;10:731–737. [PubMed] [Google Scholar]
- 14.Yang M, Wu S, Su X, May WS. JAZ mediates G1 cell cycle arrest and apoptosis by positively regulating p53 transcriptional activity. Blood. 2006;108:4136–4145. doi: 10.1182/blood-2006-06-029645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 16.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 17.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 18.Tsantoulis PK, Gorgoulis VG. Involvement of E2F transcription factor family in cancer. Eur J Cancer. 2005;41:2403–2414. doi: 10.1016/j.ejca.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Helin K, Lees JA, Vidal M, Dyson N, Harlow E, Fattaey A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–350. doi: 10.1016/00928674(92)90107-N. [DOI] [PubMed] [Google Scholar]
- 20.Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 21.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flemington EK, Speck SH, Kaelin WG., Jr E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci USA. 1993;90:6914–6918. doi: 10.1073/pnas.90.15.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin WG., Jr The transcription factor E2F-1 is a downstream target of RB action. Mol Cell Biol. 1995;15:742–755. doi: 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh P, Wong SH, Hong W. Overexpression of E2F-1 in rat embryo fibroblasts leads to neoplastic transformation. EMBO J. 1994;13:3329–3338. doi: 10.1002/j.1460-2075.1994.tb06635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gala S, Marreiros A, Stewart GJ, Williamson P. Overexpression of E2F-1 leads to cytokine-independent proliferation and survival in the hematopoietic cell line BaF-B03. Blood. 2001;97:227–234. doi: 10.1182/blood.V97.1.227. [DOI] [PubMed] [Google Scholar]
- 26.Reichert M, Saur D, Hamacher R, Schmid RM, Schneider G. Phosphoinositide-3-kinase signaling controls S-phase kinase-associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer Res. 2007;67:4149–4156. doi: 10.1158/00085472.CAN-06-4484. [DOI] [PubMed] [Google Scholar]
- 27.Baldini E, Camerini A, Sgambato A, Prochilo T, Capodanno A, Pasqualetti F, et al. CyclinA and E2F1 overexpression correlate with reduced disease-free survival in Node-negative breast cancer patients. Anticancer Res. 2006;26:4415–4421. [PubMed] [Google Scholar]
- 28.Lao-Sirieix P, Lovat L, Fitzgerald RC. Cyclin A immunocytology as a risk stratification tool for Barrett's esophagus surveillance. Clin Cancer Res. 2007;13:659–665. doi: 10.1158/1078-0432.CCR-06-1385. [DOI] [PubMed] [Google Scholar]
- 29.Mrena J, Wiksten JP, Kokkola A, Nordling S, Haglund C, Ristimäki A. Prognostic significance of cyclin A in gastric cancer. Int J Cancer. 2006;119:1897–1901. doi: 10.1002/ijc.21944. [DOI] [PubMed] [Google Scholar]
- 30.Holm C, Ora I, Brunhoff C, Anagnostaki L, Landberg G, Persson JL. Cyclin A1 expression and associations with disease characteristics in childhood acute lymphoblastic leukemia. Leuk Res. 2006;30:254–261. doi: 10.1016/j.leukres.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Poikonen P, Sjöström J, Amini RM, Villman K, Ahlgren J, Blomqvist C. Cyclin A as a marker for prognosis and chemotherapy response in advanced breast cancer. Br J Cancer. 2005;93:515–519. doi: 10.1038/sj.bjc.6602735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 33.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 34.Martin K, Trouche D, Hagemeier C, Sørensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z, Wang H, Li M, Rayburn ER, Agrawal S, Zhang R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene. 2005;24:7238–7247. doi: 10.1038/sj.onc.1208814. [DOI] [PubMed] [Google Scholar]
- 36.Eymin B, Karayan L, Séité P, Brambilla C, Brambilla E, Larsen CJ, et al. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene. 2001;20:1033–1041. doi: 10.1038/sj.onc.1204220. [DOI] [PubMed] [Google Scholar]
- 37.Mason SL, Loughran O, La Thangue NB. p14ARF regulates E2F activity. Oncogene. 2002;21:4220–4230. doi: 10.1038/sj.onc.1205524. [DOI] [PubMed] [Google Scholar]
- 38.Datta A, Nag A, Raychaudhuri P. Differential regulation of E2F1, DP1 and the E2F1/DP1 complex by ARF. Mol Cell Biol. 2002;22:8398–8408. doi: 10.1128/MCB.22.24.8398-408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datta A, Sen J, Hagen J, Korgaonkar CK, Caffrey M, Quelle DE, et al. ARF directly binds DP1: interaction with DP1 coincides with the G1 arrest function of ARF. Mol Cell Biol. 2005;25:8024–8036. doi: 10.1128/MCB.25.18.8024-36.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat Cell Biol. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 41.Girard F, Strausfeld U, Fernandez A, Lamb NJ. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991;67:1169–1179. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- 42.Jacobs HW, Keidel E, Lehner CF. A complex degradation signal in Cyclin A required for G1 arrest, and a C-terminal region for mitosis. EMBO J. 2001;20:2376–2386. doi: 10.1093/emboj/20.10.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chibazakura T, Kamachi K, Ohara M, Tane S, Yoshikawa H, Roberts JM. Cyclin A promotes S-phase entry via interaction with the replication licensing factor Mcm7. Mol Cell Biol. 2011;31:248–255. doi: 10.1128/MCB.00630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 45.DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu K, Lin FT, Ruppert JM, Lin WC. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol. 2003;23:3287–3304. doi: 10.1128/MCB.23.9.3287-304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernardi R, Liebermann DA, Hoffman B. Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene. 2000;19:2447–2454. doi: 10.1038/sj.onc.1203564. [DOI] [PubMed] [Google Scholar]
- 48.Chin L, Pomerantz J, DePinho RA. The INK4a/ARF tumor suppressor: one gene-two products-two pathways. Trends Biochem Sci. 1998;23:291–296. doi: 10.1016/S0968-0004(98)01236-5. [DOI] [PubMed] [Google Scholar]
- 49.Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–99. doi: 10.1016/S0959-437X(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 50.Modestou M, Puig-Antich V, Korgaonkar C, Eapen A, Quelle DE. The alternative reading frame tumor suppressor inhibits growth through p21-dependent and p21-independent pathways. Cancer Res. 2001;61:3145–3150. [PubMed] [Google Scholar]
- 51.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424. doi: 10.1016/S1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 52.Cress WD, Nevins JR. A role for a bent DNA structure in E2F-mediated transcription activation. Mol Cell Biol. 1996;16:2119–2127. doi: 10.1128/mcb.16.5.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trouche D, Kouzarides T. E2F1 and E1A(12S) have a homologous activation domain regulated by RB and CBP. Proc Natl Acad Sci USA. 1996;93:1439–1442. doi: 10.1073/pnas.93.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma Y, Chen L, Wright GM, Pillai SR, Chellappan SP, Cress WD. CDKN1C negatively regulates RNA polymerase II C-terminal domain phosphorylation in an E2F1-dependent manner. J Biol Chem. 2010;285:9813–9822. doi: 10.1074/jbc.M109.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lam EWF, Bennett JS, Watson RJ. Cell cycle regulation of human B-myb transcription. Gene. 1995;160:277–281. doi: 10.1016/0378-1119(95)00184-8. [DOI] [PubMed] [Google Scholar]
- 56.Pierani A, Pouponnot C, Calothy G. Transcriptional downregulation of the retina-specific QR1 gene pp60v-src and identification of a novel v-src-responsible unit. Mol Cell Biol. 1993;13:3401–3414. doi: 10.1128/mcb.13.6.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmid I, Sakamoto KM. Analysis of DNA content and green fluorescent protein expression. In: Robinson JP, Darzynkiewicz Z, Dean P, Orfao A, Rabinovitch P, Stewart C, editors. Current Protocols in Cytometry. Vol. 7. New York: John Wiley & Sons; 2001. pp. 1–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.