

Abstract
The members of the Suppressor of Cytokine Signaling (SOCS) protein family mainly modulate the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway. SOCS-1 and SOCS-3 have already been shown to influence growth and apoptosis of pancreatic β-cells. We hypothesized that SOCS-2, which is expressed in pancreatic islets, also contributes to β-cell physiology. We tested this hypothesis in vivo in SOCS-2−/− knockout mice and in vitro in Ins-1E rat insulinoma cells. We found that SOCS-2−/− mice have normal islet insulin secretion and unchanged glucose and insulin tolerance compared to wildtype controls. SOCS-2−/− are bigger than wildtype mice but body weight-corrected β-cell mass and islet morphology were normal. Growth hormone-induced proliferation of Ins-1E cells was not affected by either siRNA-mediated SOCS-2 knockdown or stable SOCS-2 overexpression. Interleukin-1β mediated cell death in vitro was unchanged after SOCS-2 knockdown. Similarly, autoimmune destruction of β-cells in vivo after multiple low-dose injections of streptozotocin (STZ) was not altered in SOCS-2−/− mice.
In summary, SOCS-2−/− knockout mice have a normal function of insulin-producing pancreatic β-cells, a fully adapted β-cell mass and a normal morphology of the endocrine islets. Based on in vitro evidence, the increased β-cell mass in the mutants is likely due to indirect adaptive mechanisms and not the result of altered growth hormone signaling within the β-cells. Immune mediated β-cell destruction is also not affected by SOCS-2 ablation in vitro and in vivo.
Keywords: SOCS1, SOCS2, SOCS3, C57Bl/6, α-cell, δ-cell, glucagon, somatostatin, pancreatic polypeptide
Introduction
Many cytokines such as growth hormone or interleukins signal through cell surface receptors that activate cytoplasmatic tyrosine kinases, particularly members of the Janus kinase (JAK) family. Those subsequently phosphorylate signal transducer and activator of transcription (STAT) proteins, which dimerize and translocate to the nucleus for transcriptional activation of target genes. The intracellular cytokine signal is negatively regulated by a number of proteins including the suppressors of cytokine signaling (SOCS). The members of this protein family are upregulated by the JAK/STAT pathway and inhibit it by a negative feedback loop. Eight SOCS proteins have been described so far.1
Members of the SOCS family became of interest to diabetes researchers because of their ability to antagonize signals of pro-apoptotic cytokines. SOCS-1 as well as SOCS-3 can block the toxic effects of interleukin-1β and interferon-γ on insulin-producing β-cells in vitro.2,3 For SOCS-1 a protective effect against immune-mediated diabetes was also demonstrated in vivo.4 SOCS family members can also inhibit growth hormone (GH) and prolactin (PRL) signaling.1 Both hormones are important β-cell growth factors and transgenic mice overexpressing SOCS-3 in a β-cell-specific manner indeed have a reduced β-cell mass.5 Taken together, at least some SOCS proteins can modulate β-cell proliferation and death. JAK/STAT signaling might also be involved in insulin gene transcription,6 although possibly only in rodents,7 and insulin secretion.8 SOCS family members could therefore potentially also influence β-cell function.
SOCS-2 has mainly been implicated in GH and PRL signaling but it can also act downstream of other cytokines.1 The main phenotype of SOCS-2−/− knockout mice is gigantism due to excessive growth hormone and IGF-1 signaling.9 SOCS-2 function has not yet been examined in pancreatic islets or β-cells but a recent study performed in a Japanese population linked a polymorphism in the SOCS-2 gene to type 2 diabetes.10 SOCS-2 expression has also been demonstrated in human pancreatic islets.11
Based on the functions of SOCS-1 and SOCS-3 in pancreatic islet β-cells and the detection of SOCS-2 in human islets we hypothesized that SOCS-2 also contributes to β-cell physiology. We tested this hypothesis in vivo and in vitro.
Results and Discussion
SOCS-2 is expressed in mouse pancreatic islets and in Ins-1E rat insulinoma cells.
SOCS-2 expression in human pancreatic islets has already been demonstrated.11 We tested the expression of the gene in rodent islets and found SOCS-2 mRNA expressed in mouse pancreatic islets by rtPCR (Fig. 1A). Attempts at immunostaining pancreatic tissue were unsuccessful. To confirm expression of SOCS-2 in β-cells we therefore analyzed Ins-1E rat insulinoma cells.12 These cells were found to express SOCS-2 mRNA (Fig. 1A) and protein (Fig. 1B and C).
Figure 1.
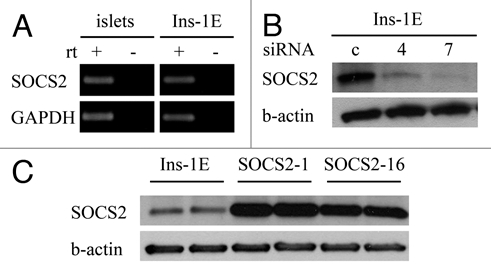
SOCS-2 expression in mouse pancreatic islets and Ins-1E rat insulinoma cells; suppression by siRNA and overexpression in stably transfected cell clones. (A) SOCS-2 mRNA is detectable in isolated mouse islets and in Ins-1E rat insulinoma cells by rtPCR. (B and C) Western blots of whole cell lysates. (B) SOCS-2 protein is present in Ins-1E cells and is effectively suppressed by two different gene-specific siRNAs (numbers 4 and 7) 72 hours after transient transfection. (C) Two Ins-1E clones stably overexpressing SOCS-2 were studied (numbers 1 and 16).
Glucose metabolism is normal in SOCS-2−/− knockout mice.
After demonstrating that SOCS-2 is expressed in pancreatic islets and also in a well established insulin-producing β-cell line we next tested glucose metabolism in adult SOCS-2−/− knockout mice in comparison to wildtype (wt) controls. In intraperitoneal glucose tolerance tests (ipGTT) the area under the glucose curve (AUC blood glucose) was unchanged in SOCS-2−/− mice of both sexes compared to wt controls (Fig. 2A). Thus, overall glucose tolerance is not altered by SOCS-2 ablation. This finding is in line with a previous report on the metabolic changes in male SOCS-2−/− mice.13 Data on female mice was not reported in this study. We observed a statistically significant difference in the peak glycemia at 15 minutes between SOCS-2−/− and wt female mice (p < 0.01). Insulin tolerance was again equal between SOCS-2−/− and wt animals, both male and female (Fig. 2B). We therefore hypothesized that a difference in first phase insulin secretion was responsible for the more pronounced rise in blood glucose in SOCS-2−/− female mice. To test this hypothesis we performed intravenous glucose tolerance tests (ivGTT) but found no difference in first phase insulin secretion between SOCS-2−/− and wt female mice (Fig. 2C). Hence the higher peak glucose level in SOCS-2−/− females is likely due to different absorption kinetics of glucose in the ipGTT and not to alterations in β-cell function. Along this line, glucose-induced insulin secretion from SOCS-2 knockout islets in vitro was also normal (Fig. 2D).
Figure 2.
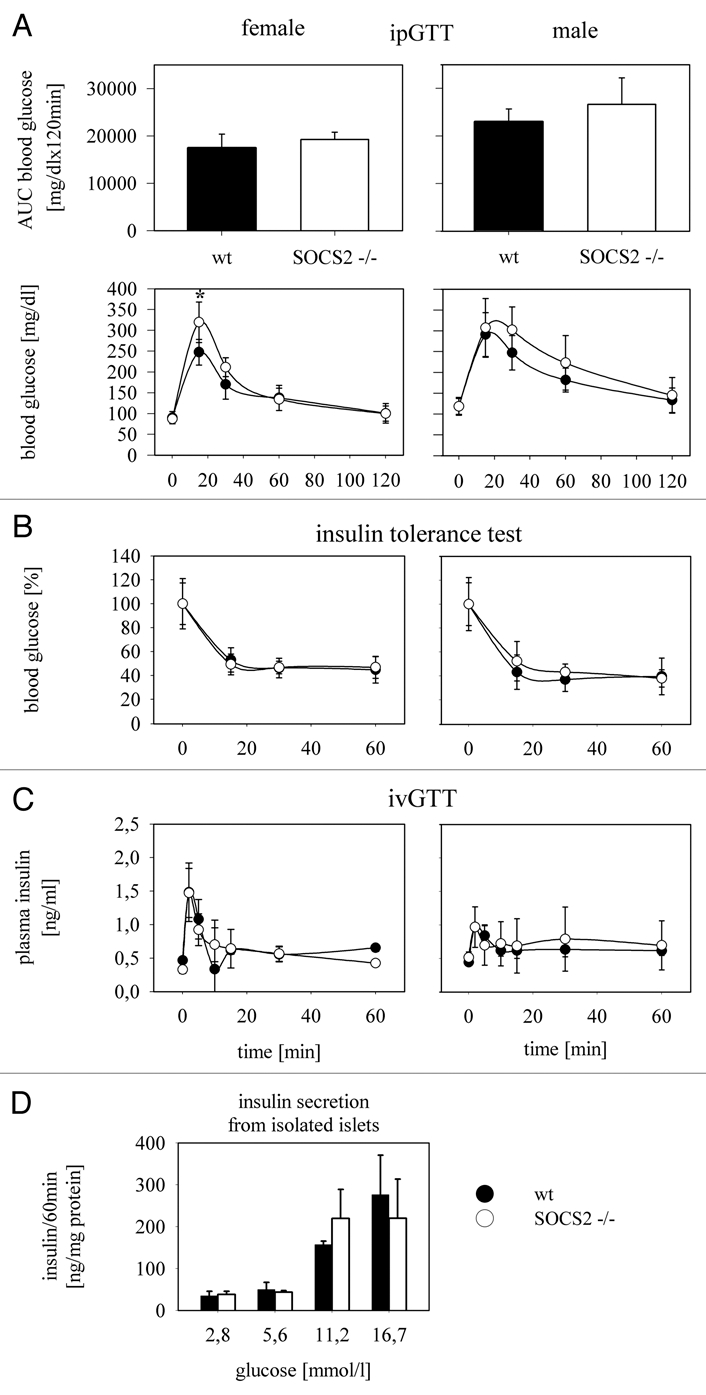
Glucose metabolism in SOCS-2−/− knockout mice. (A) Intraperitoneal glucose tolerance tests were done with 2 g glucose/kg body weight after 6 hrs of fasting in SOCS-2−/− mice and matched wt controls. Female (n = 7) and male (n = 8) animals were analyzed separately. The area under the glucose curve as a measure of overall glucose tolerance is not different between wt and SOCS-2 null mice in both sexes. A difference in blood glucose is only seen in female mice 15 min after glucose injection when SOCS-2−/− animals show significantly higher values (p < 0.01; student's t-test). (B) Intraperitoneal insulin tolerance tests were done with 0.75 IU/kg body weight (n = 6 for female, n = 7 for male mice). Insulin sensitivity is comparable in SOCS-2−/− and wt animals of both sexes. (C) To test whether the higher early glucose values in the ipGTT in female SOCS-2−/− mice are the result of a reduced first phase insulin secretion, intravenous glucose tolerance tests with 1 g glucose/kg body weight were done (n = 2 for female and male mice, respectively). First phase insulin secretion is detected at a comparable magnitude in SOCS-2 null and wt mice, ruling out a defect in this β-cell function as a reason for the more extensive early rise in blood glucose in SOCS-2−/− female mice. (D) Glucose-induced insulin secretion from isolated SOCS2−/− and wt islets is also not significantly different.
SOCS-2−/− knockout mice have a normal islet structure and an adequate β-cell mass.
We next examined the pancreas of SOCS-2 null mice histologically. Immunostaining with antibodies against insulin, glucagon, somatostatin and pancreatic polypeptide revealed normal proportions and distributions of β, α, δ and PP cells, respectively (Fig. 3A). SOCS-2−/− mice show increased growth compared to their wt counterparts and many organs including the pancreas are enlarged proportionally.9 We confirmed the findings of increased body and pancreatic weight in 11 to 17 weeks old male and female SOCS-2−/− mice (data not shown). Pancreatic β-cell mass was also increased in the knockouts but body weight-corrected β-cell mass, which is the physiologically relevant parameter, was unchanged compared to wt controls (Fig. 3B). Together with the finding of normal glucose tolerance and insulin secretion this result indicates full adaptation of the pancreatic β-cell “organ” to the overall enlargement of the SOCS-2 null mice. β-cell mass is regulated by a number of factors to adapt to changes in insulin requirements, probably including glucose and insulin signaling itself.14 Thus the adequately increased β-cell mass in SOCS-2−/− mice could be the result of these adaptive mechanisms. On the other hand, growth hormone, whose signaling is probably the central target of SOCS-2,9 can directly act as a β-cell growth factor.5 Therefore the higher β-cell mass in the SOCS-2−/− mice could also be the result of altered intracellular signaling in β-cells. To address this question we carried out in vitro experiments using Ins-1E insulinoma cells with siRNA mediated knockdown (Fig. 1B) or stable overexpression of SOCS-2 (Fig. 1C). Growth hormone induced proliferation was examined in SOCS-2 knockdown and SOCS-2 overexpressing cells and compared to the respective controls (Fig. 3C and D). No significant differences were seen with either alteration of SOCS-2 expression. This suggests no non-redundant function of SOCS-2 in growth hormone signaling in insulin producing β-cells. One likely explanation is that other members of the SOCS protein family blunt the effects of SOCS-2 modulation in this cell type. The gain of β-cell mass in the SOCS-2 knockout mice therefore is probably the result of general adaptive mechanisms and not of altered growth hormone signaling within the β-cells themselves. However, in vivo evidence derived from a β-cell specific knockout of SOCS-2 or islet transplantation experiments will be necessary to definitively answer this question.
Figure 3.
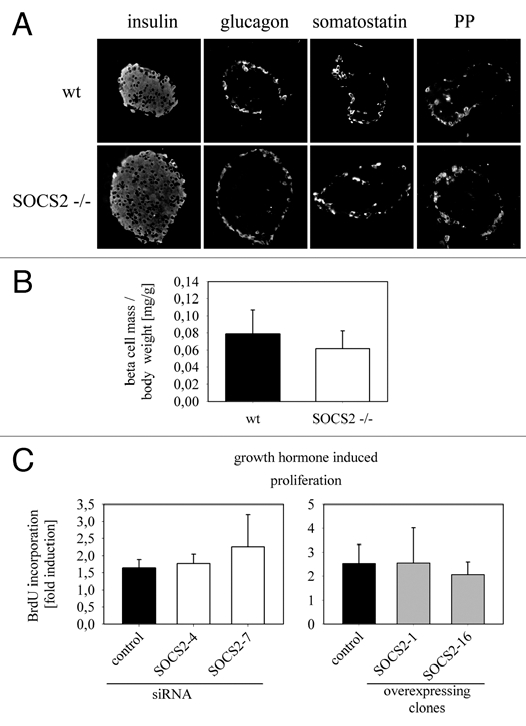
Morphology of the SOCS-2−/− mouse pancreas, β-cell mass and growth hormone-induced proliferation of Ins-1E insulinoma cells in vitro. (A) Immunostainings with antibodies against the four major islet hormones were performed on paraffin sections from SOCS-2−/− and wt pancreas. The proportions of the different cell types and the distribution of the cells within the islet are comparable in knockout and wt tissue. (B) pancreatic β-cell mass corrected for body weight was determined in 11–17 week old SOCS-2−/− mice and wt controls (n = 6; male and female combined). No significant difference is seen between the two groups indicating a fully adapted β-cell mass in the knockout mice, which are generally bigger than their wt counterparts. (C) To determine whether SOCS-2 has a direct effect on growth hormone signaling in insulin-producing β-cells, Ins-1E cells were incubated for 24 hours with and without growth hormone (50 ng/ml; 2.2 nmol/l) in serum-free media. BrdU incorporation during that time period was quantified by immunostaining. Induction of proliferation by growth hormone is not significantly different in cells transfected with control or SOCS-2 specific siRNA (left) and also not in control and SOCS-2 overexpressing cells (right).
SOCS-2 does not influence autoimmune mediated cell death of pancreatic β-cells.
SOCS-1 and SOCS-3 have been shown to protect pancreatic β-cells from cytokine induced cell death in vitro and from autoimmune destruction in vivo (SOCS-1).2–4 Since SOCS-2 also has anti-inflammatory properties in other tissues1 we tested whether it affects cytokine mediated cell death in vitro and autoimmune destruction of β-cells after multiple low-dose injections of streptozotocin (STZ) in vivo.15 In Ins-1E insulinoma cells the reduction of viability after incubation with interleukin-1β was not affected by siRNA-mediated SOCS-2 knockdown (Fig. 4A). We also incubated isolated wt and SOCS-2−/− islets for 24 hours with interleukin-1β (1 ng/ml), IFNγ (1,000 U/ml) and TNFα (1,000 U/ml) in serum-free media. After intravital staining with Hoechst 33342 and propidium iodide no difference in cell death was observed between the two types of islets by visual inspection (data not shown). SOCS-2−/− mice were also indistinguishable from wt controls in the in vivo multiple low-dose STZ model. Male mice developed hyperglycemia with the same time course as controls while female mice remained normoglycemic as expected for the C57Bl/6 strain15 (Fig. 4B). Taken together these data indicate that, in contrast to other members of the SOCS protein family, SOCS-2 does not influence the autoimmune destruction of pancreatic β-cells.
Figure 4.

Interleukin-1β induced cell death in vitro and autoimmune mediated β-cell destruction in vivo. (A) To analyze whether SOCS-2 influences cytokine-mediated β-cell death, Ins-1E cells were incubated with and without 100 pg/ml recombinant mouse interleukin 1-β for 24 hours in serum-free media. Seventy-two hours earlier the cells were transfected with control or SOCS-2 specific siRNA. No effect of SOCS-2 knockdown on the interleukin-induced reduction of viability is seen (MTT assays). (B) The multiple low dose STZ regimen was used as a model of autoimmune diabetes and β-cell destruction in vivo. SOCS-2 and wt mice received five consecutive daily i.p. injections of STZ (40 mg/kg body weight; n = 3 for males; n = 2 for females). Fed blood glucose was recorded daily (except for days 12 and 13), starting on day 7 after the first injection. Male mice became diabetic with the same kinetics in both groups. Female mice, as expected for wt C57Bl/6, remained normoglycemic.
In summary, we were able to show that SOCS-2−/− mice have a normal function of pancreatic β-cells, a fully adapted β-cell mass and a normal morphology of the endocrine islets. Based on in vitro evidence, the increased β-cell mass in the mutants is likely due to indirect adaptive mechanisms and not the result of altered growth hormone signaling within the β-cells. Immune mediated β-cell destruction is also not influenced significantly by SOCS-2 in vitro and in vivo. One likely explanation for these findings is redundancy within the SOCS protein family.
Further studies examining the effects of combined modulation of different SOCS proteins in islet β-cells will be necessary to address this issue.
Material and Methods
Mice.
C57Bl/6 SOCS-2−/− mice9 were provided by W. S. Alexander and bred as homozygotes. C57Bl/6 controls were obtained from Charles River laboratories. All animal studies were carried out in accordance with the principles of laboratory animal care and institutional and governmental regulations.
Reagents and antibodies.
Recombinant human growth hormone was purchased from R&D-Systems, recombinant mouse interleukin-1β from Calbiochem. The following antibodies were used in this study: Rabbit anti-SOCS-2 (1:500; Abcam), Mouse anti-β-actin (1:10,000, Sigma), Mouse anti-BrdU (1:1,000, Sigma), Guinea pig anti-insulin (1:1,000, Dako), Rabbit anti-glucagon (1:500, Chemicon); Rabbit anti-somatostatin (1:100, Chemicon); Rabbit anti-pancreatic polypeptide (1:30, Chemicon). Secondary antibodies for immunofluorescence were purchased from Jackson Immunoresearch.
Cell culture.
Ins-1E cells were maintained in RPMI1640 medium (PAA), supplemented with 5% fetal calf serum, 100 IU/ml penicillin, 100 µg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate and 50 µM β-mercaptoethanol and cultured in a humidified incubator with 5% CO2 at 37°C. To analyze the growth hormone induced proliferation cells were seeded at a density of 150,000 per well in 6-well plates. Each well contained a gelatin-coated cover slip for later immunostaining and microscopic analysis. For all other experiments Ins-1E cells were seeded at the same density per well in 24-well plates.
Stable overexpression of SOCS-2 and siRNA mediated SOCS-2 knockdown.
The mouse SOCS-2 ORF was cloned into pcDNA3. The resulting overexpression vector pcDNA3-SOCS2 was sequenced and stably transfected into Ins-1E cells. Stable clones were selected by incubation with G418 (Roth). Expression of the correct protein was confirmed by western blot. Transient transfections with SOCS-2 specific siRNAs (Invitrogen) were used to knock down endogenous gene expression in wildtype cells (Lipofectamin RNAi MAX, Invitrogen). Effective gene knockdown after 72 hours was confirmed by rtPCR and western blot. The used siRNA sequences were as follows: CCC ACU CAG ACU ACC UAU U (siSOCS2–4) and CCA ACA AGA CUG AAA GAU U (siSOCS2–7).
RNA isolation and semiquantitative rtPCR.
Total RNA was extracted using RNeasy and DNA removal columns from Qiagen. Oligo(dT) primed cDNA was prepared from total RNA (ImProm-II Reverse Transcription System; Promega) and semiquantitative rtPCR performed by using the Taq PCR Core Kit (Qiagen). GAPDH was used for normalization and the minimal cycle number required was applied for each gene product. The primer sequences were as follows: SOCS2 (mouse): 5′-TGG CTG CTC AAG ATC AAA TG-3′ and 5′-TGT CCT CCT GGA AAT GGA AG-3; SOCS2 (rat): 5′-CGG GAA TTT GGA GAG AAA CA-3′ and 5′-CAG GGT CAT GGG AGA TGA GT-3′; GAPDH (mouse): 5′-AAC TTT GGC ATT GTG GAA GG-3′ and 5′-ACA CAT TGG GGG TAG GAA CA-3′; GAPDH (rat): 5′-GGC ATT GCT CTC AAT GAC A-3′ and 5′-TGT GAG GGA GAT GCT CAG T-3′.
Protein extraction and western blot analysis.
Cells were harvested in lysis buffer (50 mM HEPES, 0.1% Triton X-100, 1 mM DTT, 1x protease inhibitor cocktail (Roche)), and maintained under constant agitation for 30 min at 4°C. After a centrifugal step at 10,000 rpm for 20 min at 4°C the supernatant was mixed in Laemmli loading buffer and boiled for 5 min. Proteins were separated by SDS-PAGE and transferred onto an Immobilon-P membrane (PVDF; Millopore). Membranes were blocked with blocking buffer containing 5% dry milk before overnight incubation at 4°C with primary rabbit or mouse antibodies. Signals were detected using horseradish peroxidase-conjugated antirabbit or anti-mouse IgG antibodies and ECL detection reagent (Thermo scientific).
Analysis of growth hormone induced proliferation in Ins-1E insulinoma cells.
Cells were seeded at low density on gelatine-coated glass cover-slips and grown for 24 hours in serum-free media +0.25% BSA+/− 50 ng/ml recombinant human growth hormone in the presence of BrdU (3.33 nM). The cells were then fixed with 4% PFA and stained with anti-BrdU and DAPI. For each condition at least 200 cells were counted.
Analysis of viability after incubation with interleukin-1β.
Cells were cultured in the presence and absence of 100 pg/ml recombinant mouse interleukin-1β under serumfree conditions for 24 hours. At the end of the assay cellular viability was determined by an MTT [3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay.
Pancreatic islet isolation and static insulin secretion assay.
Mice were killed by cervical dislocation after isoflurane inhalation. Islets were isolated by intraductal collagenase digestion. For this method, the common bile duct is exposed and a clamp is placed directly proximal of the junction of the bile duct with the small intestine. A collagenase solution [Collagenase V (Sigma-Aldrich) 1 mg/ml in HBSS with 10 mM Hepes and 0.01% BSA] is injected into the pancreatic duct system via the common bile duct. The inflated pancreas is removed and digestion is completed at 37°C. After several washes with HBSS/Hepes/10% FCS islets are purified through three rounds of hand picking under a dissection microscope.
For static insulin secretion assays freshly isolated islets were pre-incubated in Krebs-Ringer Buffer containing 2.8 mmol/l Glucose for one hour, washed with fresh buffer and hand-picked into eppendorf tubes (10 islets per tube) containing 500 µl of Krebs-Ringer buffer supplemented with different glucose concentrations and 0.1% BSA. After one hour of incubation 300 µl of buffer were removed for analysis. Secreted insulin was quantified using a mouse and rat insulin specific ELISA (Linco Research). Insulin secretion was normalized for total protein content of the islets in each individual tube as determined by fluorometry. Three samples/tubes per glucose concentration were analyzed in each assay.
Glucose and insulin tolerance tests.
At the age of 12 weeks an intraperitoneal glucose tolerance test (ipGTT) was performed in SOCS2−/− mice and age-matched C57BL/6 controls after a fasting period of 6 h. Blood glucose was measured using an Ascensia CONTOUR glucometer (Bayer) at the basal state (0 min) and at 15, 30, 60, 90 and 120 min after glucose injection (2 g/kg body weight). Insulin plasma levels were determined by an ultrasensitive ELISA (CrystalChem). ivGTT was done by injecting 1g glucose/kg body weight into a tail vein. Blood glucose and insulin serum levels were measured at the basal state (0 min) and at 2, 5, 10, 15, 30 and 60 min after glucose injection. Insulin tolerance testing was carried using human insulin at 0.75 IU/kg body weight i.p.
Quantification of β-cell mass.
β-cell mass was quantified by measuring β-cell area on immunostained pancreatic sections using a 11 × 11 dot grid. The number of grid points overlapping insulin-positive cells tissue was determined in non-overlapping fields. A minimum of 150 fields per animal were counted. The total β-cell fraction was then multiplied with the pancreatic weight.
Multiple low dose STZ model.
Wild type and SOCS-2 null mice received i.p. injections of streptozotocin (STZ; 40 mg/kg body weight; Sigma) dissolved in 20 mM citrate puffer (pH 4.5) on 5 consecutive days. A fed blood glucose was determined daily beginning on day 7 after the first injection.
Tissue preparation and immunostaining.
Tissues were fixed overnight in 4% buffered formalin, and embedded in paraffin. Sections were cut at 4 µm. After dewaxing slides were washed in PBS and microwave antigen retrieval was performed. For immunostaining, slides were blocked with 1% normal donkey serum in PBS containing 0.1% Triton X-100. Visualization was achieved using the VectastainABC kit and the VectorNovaRED substrate kit (Vector Laboratories) for quantification of β-cell mass and Cy2/Cy3 labeled secondary antibodies for all other stainings. Images were captured with a CCD camera attached to a Zeiss Axioscope.
Statistical analysis.
Unpaired Student's t-tests were used in all analyses except for the experiments depicted in Figure 3C, which required a one-way ANOVA.
Acknowledgements
We are grateful to Dr. W.S. Alexander for the provision of the SOCS-2 knockout mice. This study was supported by grants from the Deutsche Forschungsgemeinschaft (LE 1302/3-1) and the Münchner Medizinische Wochenschrift Foundation to A.L.
Footnotes
Previously published online: www.landesbioscience.com/journals/islets/article/12556
References
- 1.Rico-Bautista E, Flores-Morales A, Fernandez-Perez L. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 2006;17:431–439. doi: 10.1016/j.cytogfr.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Cottet S, Dupraz P, Hamburger F, Dolci W, Jaquet M, Thorens B. SOCS-1 protein prevents Janus Kinase/STAT-dependent inhibition of beta cell insulin gene transcription and secretion in response to interferongamma. J Biol Chem. 2001;276:25862–25870. doi: 10.1074/jbc.M103235200. [DOI] [PubMed] [Google Scholar]
- 3.Karlsen AE, Heding PE, Frobose H, Ronn SG, Kruhoffer M, Orntoft TF, et al. Suppressor of cytokine signalling (SOCS)-3 protects beta cells against IL-1beta-mediated toxicity through inhibition of multiple nuclear factor-kappaB-regulated proapoptotic pathways. Diabetologia. 2004;47:1998–2011. doi: 10.1007/s00125-004-1568-3. [DOI] [PubMed] [Google Scholar]
- 4.Chong MM, Chen Y, Darwiche R, Dudek NL, Irawaty W, Santamaria P, et al. Suppressor of cytokine signaling-1 overexpression protects pancreatic beta cells from CD8+ T cell-mediated autoimmune destruction. J Immunol. 2004;172:5714–5721. doi: 10.4049/jimmunol.172.9.5714. [DOI] [PubMed] [Google Scholar]
- 5.Lindberg K, Ronn SG, Tornehave D, Richter H, Hansen JA, Romer J, et al. Regulation of pancreatic beta-cell mass and proliferation by SOCS-3. J Mol Endocrinol. 2005;35:231–243. doi: 10.1677/jme.1.01840. [DOI] [PubMed] [Google Scholar]
- 6.Galsgaard ED, Gouilleux F, Groner B, Serup P, Nielsen JH, Billestrup N. Identification of a growth hormoneresponsive STAT5-binding element in the rat insulin 1 gene. Mol Endocrinol. 1996;10:652–660. doi: 10.1210/mend.10.6.8776725. [DOI] [PubMed] [Google Scholar]
- 7.Hay CW, Docherty K. Comparative analysis of insulin gene promoters: implications for diabetes research. Diabetes. 2006;55:3201–3213. doi: 10.2337/db06-0788. [DOI] [PubMed] [Google Scholar]
- 8.Weinhaus AJ, Stout LE, Bhagroo NV, Brelje TC, Sorenson RL. Regulation of glucokinase in pancreatic islets by prolactin: a mechanism for increasing glucose-stimulated insulin secretion during pregnancy. J Endocrinol. 2007;193:367–381. doi: 10.1677/JOE-07-0043. [DOI] [PubMed] [Google Scholar]
- 9.Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405:1069–1073. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- 10.Kato H, Nomura K, Osabe D, Shinohara S, Mizumori O, Katashima R, et al. Association of single-nucleotide polymorphisms in the suppressor of cytokine signaling 2 (SOCS2) gene with type 2 diabetes in the Japanese. Genomics. 2006;87:446–458. doi: 10.1016/j.ygeno.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Santangelo C, Scipioni A, Marselli L, Marchetti P, Dotta F. Suppressor of cytokine signaling gene expression in human pancreatic islets: modulation by cytokines. Eur J Endocrinol. 2005;152:485–489. doi: 10.1530/eje.1.01856. [DOI] [PubMed] [Google Scholar]
- 12.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 13.Rico-Bautista E, Greenhalgh CJ, Tollet-Egnell P, Hilton DJ, Alexander WS, Norstedt G, et al. Suppressor of cytokine signaling-2 deficiency induces molecular and metabolic changes that partially overlap with growth hormone-dependent effects. Mol Endocrinol. 2005;19:781–793. doi: 10.1210/me.2004-0040. [DOI] [PubMed] [Google Scholar]
- 14.Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev. 2005;85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 15.O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Beta-cell apoptosis is responsible for the development of IDDM in the multiple low-dose streptozotocin model. J Pathol. 1996;178:176–181. doi: 10.1002/(SICI)1096-9896(199602)178:2<176::AID-PATH433>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]