

Abstract
Background
The development of abdominal aortic aneurysms (AAAs) involves a complex interplay of extracellular matrix degradation, inflammation, and apoptosis. We have previously shown that PKCδ plays a critical role in vascular smooth muscle cell (vSMC) apoptosis in the setting of oxidative stresses. Here, we show that PKCδ is also involved in the signaling that draws inflammatory cells to aneurismal tissue.
Methods
Immunostaining for monocyte chemotactic factor (MCP)-1 and PKCδ was performed on paraffin-fixed arterial sections. ELISA to detect MCP-1 produced by vSMCs was performed on media from cultured rat A10 cells after cytokine induction with or without the PKCδ specific inhibitor rottlerin. Migration of isolated lymphocytes was evaluated in response to media from activated A10 cells.
Results
Human AAAs show widespread and elevated expression of PKCδ that is not seen in normal aortic tissues. Cytokine stimulation of cultured vSMCs induced vigorous production of the key chemotactant MCP-1, the expression of which was PKCδ dependant. Stimulated vSMCs were capable of inducing the migration of leukocytes, and this effect was also dependant on PKCδ activity. Staining of human AAA tissue for MCP-1 showed an expression pattern that was identical to that of PKCδ and smooth muscle specific alpha actin (αSMA).
Conclusions
PKCδ is widely expressed in human AAA vessel walls and mediates MCP-1 expression by vSMCs, which could contribute to the inflammatory process. These findings, coupled with earlier studies of PKCδ, suggest that PKCδ plays a central role in the pathogenesis of AAAs and may be a potential target for future therapies.
Keywords: Protein kinase C-δ, monocyte chemotactic factor-1, abdominal aortic aneurysms, aneurysm formation, vascular inflammation
Introduction
Abdominal aortic aneurysm (AAA) is a progressive and lethal disorder that is the tenth leading cause of death in men over the age of 55 in the United States (1). The mainstay of treatment is either traditional open or endovascular surgical intervention and there is no medical bridging therapy that had proven to be effective to date (2, 3). The development of this devastating disorder is a dynamic and complex process that involves an intricate interplay of matrix degradation, apoptosis, and inflammation (4-6).
Central to the development of AAA is a chronic inflammatory state where both resident vSMCs as well as invading macrophages release matrix metalloproteinases (MMPs) which gradually degrade critical extracellular matrix (ECM) components (7-10). The cause of this chronic inflammatory state is incompletely understood but several stimuli have been implicated, including atherosclerosis, oxidative stress, angiotensin II, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and interferon (IFN)-γ (11). More recent work has shown that C-C motif chemokine receptor (CCR)-2, the receptor for MCP-1, is necessary to induce vascular inflammation. Inhibition of the MCP-1 signaling by the means of CCR-2 gene “knock-out” blocks the recruitment of macrophages and thus the development of aneurysms in a mouse AAA model (12).
The PKC family is a multimember group of cell membrane associated serine/threonine kinases. The kinases of the “novel” subfamily of PKCs, including PKCδ, are grouped together because their regulatory domain lacks calcium co-coordinating side chain residues (13). PKCδ has been linked to cell cycle control as well as cellular apoptosis in multiple cell types. It also has been shown that PKCδ can induce the expression of nuclear factor (NF)-κB, the inhibition of which also directly suppressed AAA development in an animal model (14, 15). NF-κB is a widely expressed transcription factor that is central in initiating and promoting an inflammatory response. Our own lab has shown that PKCδ is necessary for fibronectin synthesis as well as migration and proliferation of vSMCs which are both central to maintaining the vessel wall matrix (16).
To determine if a link existed between PKCδ and the development of AAAs we screened a number of human samples for the expression of this and other proteins. Here we report the key role that PKCδ plays in induction of MCP-1 and the effect these proteins have on the migration of inflammatory cells. A greater understanding of the cellular mechanisms responsible for the chronic inflammation of AAAs is essential if pharmacological therapies are to be targeted at this devastating disease.
Materials and Methods
General materials
Rat TNFα was obtained from R & D Systems, Minneapolis, MN. Rottlerin was obtained from Calbiochem, San Diego, CA. Dulbecco’s Modified Eagles Medium (DMEM) and cell culture reagents were from Gibco BRL Life Technologies, Carlsbad, CA. MCP-1 antibody was obtained from Santa Cruz Biotechnology, Santa Cruz, CA. Chemicals, if not specified, were purchased from Sigma Chemical Co, St. Louis, MO.
Human Tissue Procurement
Eight HAAA specimens obtained from patients undergoing surgical repair of AAA and compared them to two normal controls were obtained by members of the Pathology Department from autopsies performed on age equivalent individuals that died from non-cardiovascular diseases. No patients with known connective tissue disorders were included. The use of human samples in this study has been approved by the Institutional Review Board at Weill Cornell Medical College.
Immunohistochemistry
Immunostaining for MCP-1 and PKCδ was performed on paraffin-fixed arterial section of a diameter of 5-7 microns. MCP-1 antibody was used at 2μg/ml and PKCδ at 1 μg/ml. A negative control was performed with each stain using IgG from the matching species and the same secondary antibody to ensure that no cross-reactivity or background staining occurred. Stained sections were digitally photographed.
ELISA for MCP-1
ELISA to detect MCP-1 in SMC was performed using rat MCP-1 ELISA kit (BD Biosciences, Bedford, MA). SMC were cultured at a density of 1 × 105/ml in 1 ml of complete medium in the presence or absence of different stimuli in 12-well Costar plates (Corning Inc., Corning, NY). After incubation for various periods of time at 37°C, cell-free culture supernatants were obtained. The concentrations of MCP-1 were then measured according to the manufacturer’s instructions.
Cell Infection with Adenoviral Vectors
Adenoviral vectors expressing PKCδ were constructed as previously described(17). Cells were plated at 80,000 cells per well in a six well plate. Cells were exposed to 30,000 particles/cell for four hours and then recovered in standard high-glucose DMEM media overnight prior to treatment. Use of a GFP tagged adenovirus showed >80% infection and Western blot of cell lysate after exposure showed a strong band of expression consistent with PKCδ overexpression.
Isolation of rat bone marrow
Bone marrow from Sprague Dawley rats was isolated by flushing the long bones with Dulbecco’s PBS containing 2% bovine serum albumin (BSA), and heparin (1000 U/ml). Cells from individual animals were pooled and suspended in the above medium and filtered through a 40μm cell strainer (BD Biosciences, Bedford, MA). Viable lymphocytes were isolated from other blood components using Lympholyte-M (Cedarlane Laboratories Ltd., Hornby, Ontario) density centrifugation. The cells were then rinsed twice in PBS followed by centrifugation.
Cell migration assay
1 × 105 bone marrow derived lymphocytes or 2 × 105 Raw 264 cells were placed in the upper chamber of Costar 24-well transwell plates with 5-μm pore filters (Corning Inc., Corning, NY) and the chamber was placed in a 24-well culture dish containing cultured conditional medium. After incubating plates for 6 hours at 37 °C, migrated cells were collected from the lower chambers and counted while remaining cells in the upper chamber were discarded. Cells were then stained with Calcein AM (Invitrogen, Eugene, OR) fluorescent nuclear stain and nucleated cells were counted.
Statistics
All data are expressed as means ± s.e.m. Continuous variables including MCP-1 concentrations and migration assay results were analyzed via paired Student’s t-test. All statistical analysis was carried out using the statistical package on Microsoft Excel (Microsoft Corporation, Redmond, WA).
Results
PKCδ is highly expressed in the aortic wall of human AAAs
The link between PKCδ and proinflammatory cytokines or oxidative stresses led us evaluate a series of human abdominal aortic aneurysms obtained from surgical patients for the expression of this protein. AAA tissue samples were then compared to tissues obtained from autopsies of age similar patients without aortic aneurysms. Compared to normal aortic wall, the AAA tissues have a disordered and diminished expression of smooth muscle-specific alpha actin (αSMA) (Fig. 1B), consistent with a depletion of the vascular smooth muscle cells known to occur in AAAs. Furthermore, staining with CD68 and CD45 antibodies, markers for macrophages and monocytes, respectively, showed a diffuse signal throughout all layers of the AAA specimen consistent with previous findings of inflammatory cells in AAAs (not shown) (18). Next, we stained the tissues with a monoclonal antibody against PKCδ. The normal aortic tissues showed no detectable expression of PKCδ throughout the specimen, while displayed a pattern of organized αSMA expression in a band at the outer edges of the medial layer of the aortic wall (Fig. 1B-C). In contrast, the AAA tissues exhibited high level of PKCδ, in a staining pattern remarkable similar to that of αSMA (Fig. 1C), which suggests that much of the PKCδ expression was occurring in the remaining vascular smooth muscle cells of the aortic wall.
Figure 1. PKCδ overexpression in vSMCs of AAAs.
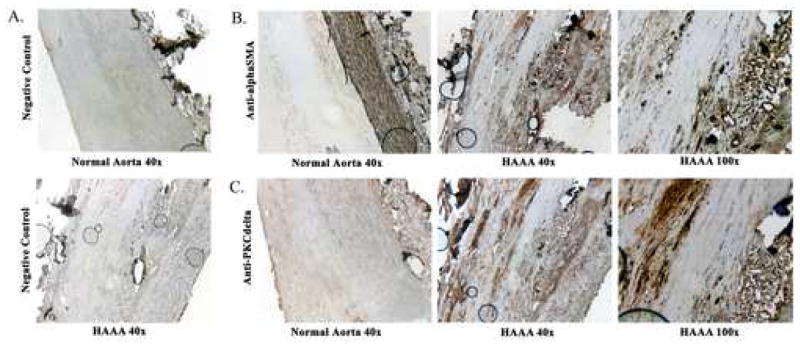
A-C) Human abdominal aortic aneurysm (HAAA) tissue harvested during surgical repair and normal aortic tissue from cadaveric specimens. A) Negative control (magnification 40x) with normal tissue above and HAAA below. B) Stain with anti-αSMA of normal (magnification 40x) and HAAA tissue (magnification 40x and 100x). C) Stain with anti-PKCδ of normal (magnification 40x) and HAAA tissue (magnification 40x and 100x). Vessel lumen is to the left aspect of each image.
PKCδ mediates expression of MCP-1 by vascular smooth muscle cells
The wide expression pattern of PKCδ in AAA tissues led us to speculate that this signaling protein may be involved in underlying activities that contribute to AAA development, such as inflammation. The recruitment of inflammatory cells into aortic wall is a critical step in development of AAAs. Mice that lack CCR-2, the receptor on inflammatory cells for monocyte chemotactic protein 1 (MCP-1), showed diminished inflammation and are resistant to the development of AAA in the setting of the angiotensin infusion model (12, 19, 20). To test whether the highly expressed PKCδ contributes to inflammation by upregulating MCP-1 production of aortic SMCs, we turned to an in vitro culture system using a rat aortic smooth muscle cell line (A10 cells). The pro-inflammatory cytokine TNFα was used to simulate an inflammatory stimulus. We found TNFα (20ng/ml) induced a 7.5 fold induction in the quantity of MCP-1 produced by A10 SMCs. To determine the role of PKCδ in MCP-1 production, we pretreated A10 SMCs with Rottlerin (2 μM), a widely used inhibitor specific for PKCδ at this dosage (21, 22). Rottlerin completely blocked the TNFα-induced MCP-1 production, indicating that PKCδ activity is necessary for MCP-1 expression (Fig. 2A) In contrast to Rottlerin, treating VSMCs with Go 6976 (10 nM), an inhibitor know to block the activity of PKCα, PKCβ1, PKCβ2, and PKCγ, did not produce any specific effect on the production of MCP-1 (Fig 2C). These results suggest that TNF-alpha induces MCP-1 expression in VSMCs through the delta type of PKC.
Figure 2. Induction of MCP-1 is PKCδ dependant.

Cultured vascular SMCs (A10 cells) were stimulated with TNFα and an ELISA was used to determine MCP-1 concentrations. Data are means ± s.e.m. A) MCP-1 production after 6 hour treatment with 20ng/mL TNFα with or without 2 μM Rottlerin (n=12). * p<0.001 when compared with controls and Rottlerin treated cells. B) MCP-1 production after TNFα induction in cells infected with AdPKCδ vs AdNull (n=4). ** p<0.001 when compared with controls and infected cells. C) MCP-1 production after TNFα induction in cells with or without Rottlerin or GO 6976 (n=4). * p<0.001 when compared with controls and Rottlerin treated cells, no difference when compared to GO 6976 treated cells.
After establishing a role of PKCδ in MCP-1 expression in normal aortic SMCs, we hypothesized that the abnormal vSMCs with high level of intracellular signaling protein PKCδ that we had observed in human AAA tissues would respond to the extracellular pro-inflammatory stimuli with even higher degree of MCP-1 production. To this end, we simulated high PKCδ expression by infecting normal A10 SMCs with an adenoviral vector that expresses PKCδ (Ad-PKCδ). As a control, a separate group of SMCs were infected with an empty viral vector (Ad-Null). Following viral infection, cells were stimulated with TNFα as before. As compared to cells infected with Ad-Null, Ad-PKCδ-infected cells showed a significantly higher expression of PKCδ. More importantly, these high PKCδ-expressing cells responded to a pro-inflammatory stimulus with a significantly higher MCP-1 induction (Fig. 2B). Together, these data suggest that PKCδ is an important signaling protein in regulating MCP-1 production within the aortic wall.
PKCδ is necessary for the recruitment of inflammatory cells
To further test a potential role of PKCδ in vascular inflammation, we next assessed the effects of inhibition of PKCδ in vSMCs on inflammatory cell chemotaxis in a transwell migration assay. Initially, lymphocytes isolated from whole rat bone marrow were used as a source of inflammatory cells. These were placed in the upper chamber that was separated from the bottom chamber by a 5-μm pore filter. Cells that migrated through the filter toward the bottom chamber were collected and stained with Casein AM, a nuclear marker. The Casein AM+ cells, presumably inflammatory cells including monocytes and macrophages, were counted. At the basal condition when the bottom chamber was filled with plain culture media, the number of migrated inflammatory cells was small. Addition of cell free media with TNFα or rottlerin to the bottom chamber had no significant effect on chemotaxis. However, conditioned media from TNFα-treated SMCs induced a nearly 10 fold increase in the chemotaxis of inflammatory cells when compared to media conditioned from control vSMCs (Fig 3A). We speculated that the chemoattractant effect of TNFα-treated SMCs is, at least in part, mediated by MCP-1. To test this hypothesis, we blocked MCP-1 production by treating vSMCs with rottlerin prior to TNFα induction. As shown in Fig. 3A, rottlerin significantly reduced the chemoattractant effect of media conditioned by TNFα-treated SMCs. In order to confirm these results the experiment was then repeated in an identical fashion using a macrophage cell line (Raw 264 cells). Similarly, we found that PKCδ is absolutely necessary for TNFα-activated vSMCs to attract macrophages (Fig. 3B).
Figure 3. Migration of inflammatory cells is blocked by PCKδ inhibition.
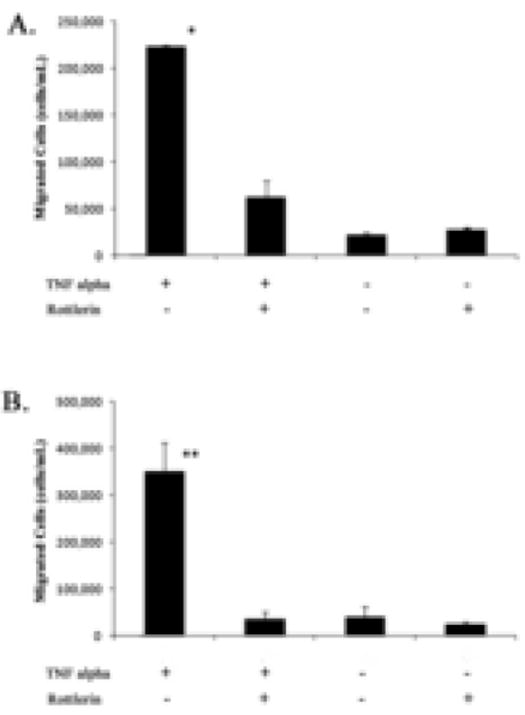
Media from TNFα treated rat aortic smooth muscle cells was used in a transwell migration assay with both bone marrow derived lymphocytes (A) and the macrophages/monocyte line Raw 264 (B). Data are means ± s.e.m. In each assay A10 vSMCs were conditionally treated with rottlerin at 2μm for 1h, followed by stimulation with TNFα fro 6h. Conditioned media was then filtered and used as a chemoattractant. * and ** p<0.001 when compared with controls and Rottlerin treated cells. n=4.
MCP-1 is also widely expressed in vascular smooth muscle cells of human AAAs
We next returned to the previously stained AAA and normal aortic tissues that we had available to us in order to determine if the correlation between PKCδ and MCP-1 was also evident in human tissues. We stained the slides for αSMA to identify the region where SMCs were most concentrated and then stained serial section again for PKCδ and now also for MCP-1 with commercially available antibodies. In each of the samples available we were able to correlate both PKCδ and MCP-1 expression to the same populations of smooth muscle (Fig. 4).
Figure 4. MCP-1 overexpression in vSMCs of AAAs correlates with PCKδ.
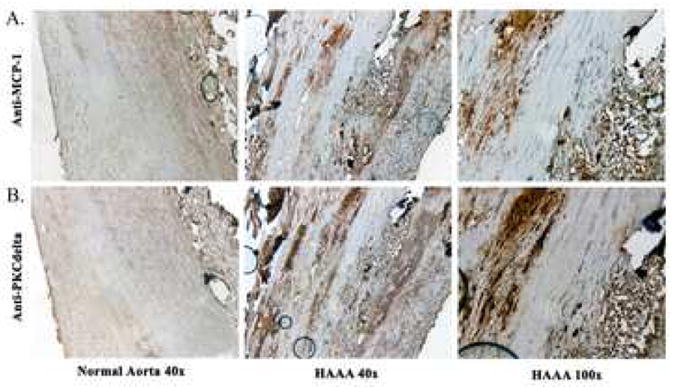
A-B) Normal aortic tissue on left (magnification 40x) with human aortic tissue harvested during surgical repair in two right panels (magnification 40x and 100x). A) Stain with anti-MCP-1. B) Stain with anti-PKCδ. Vessel lumen is to the left aspect of each image.
Discussion
Though matrix degradation and apoptosis are central to the development of AAAs, the persistent inflammation of growing aneurysms plays a significant role in the cyclical progression of this devastating disease process. In this study we have aimed to further understand the signaling systems within aortic SMCs that are responsible for recruiting inflammatory cells to the developing AAA. The PKC family mediates a number of intracellular signal transduction pathways implicated in the pathogenesis of inflammation, particularly in eicosanoid production and neutrophil migration (23). PKCδ has been implicated as an inflammatory mediator in other cell types through its ability to both directly and indirectly activate NF-κB (13). We have shown that PKCδ is widely expressed in the vSMCs of the media layer of human AAAs but not in normal aortic walls. Recent work has shown that CCR2 expression is essential for AAA development and that a murine KO model for this receptor is resistant to developing AAAs. Our data has shown that the expression of the ligand for this receptor, MCP-1, is also present in the same vSMCs of the advanced AAA as PKCδ. Additionally, we have established that PKCδ is active upstream of the cascade responsible for MCP-1 expression in this cell type and that inhibition of this kinase will lead to an inability of vSMCs to express MCP-1. Further in vitro models demonstrated that the migration of inflammatory cells which are so abundant in AAAs is readily achieved by stimulated vSMCs but that this effect is blocked by the inhibition of PKCδ.
The regulation of MCP-1 is incompletely understood at this time. Work in other cell types has shown that a promoter/enhancer region of MCP-1 contains two discrete NF-κB sites. Blocking these sites significantly reduced the production of MCP-1 in response to an inflammatory stimulus (24). Studies in TNFα induced endothelial cells showed a co-regulatory relationship between p38 mitogen-activated protein kinase (MAPK) and NFκB in the induction of MCP-1expression (25). As both MAPK and NF-κB are known downstream targets of PKCδ (26, 27) we speculate that this is the signaling pathway through which PKCδ is able to effect MCP-1 expression. Further studies will be required to fully understand the relationship between these proteins and their role in vascular inflammation.
One limiting factor of our study is the ability of rottlerin to inhibit not only PKCδ but also other PKC isotypes at various concentrations. The use of only 2 μM of rottlerin ameliorates this issue to some degree as the IC50 for the conventional PKC isotypes is at least 30 μM. Additionally, the lack of an effect by the conventional PKC inhibitor GO 6976 also leads us to believe that the effect of rottlerin on MCP-1 expression is through inhibition of PKCδ. Also, though the staining patterns for PKCδ, MCP-1, and αSMA are striking in their similarity, true co-localization with fluorescent markers would be the definitive experiment to prove expression in the same cell. Finally, though the function of MCP-1 as a chemoattractant has been well established, our data do not conclusively prove that the protein driving the migration of leukocytes in our assays is in fact MCP-1. Future studies will directly test this by treating VSMCs with MCP-1 siRNA or by blocking MCP-1 receptors on leukocytes with blocking antibodies.
The clear association of MMPs with AAA formation and the determination that the inflammatory infiltrate is spatially related to the area of ECM loss (4, 6) has led to the conclusion that mitigating the inflammation of AAAs may be the key to suppressing their growth. Recent work showing the possibility of aneurysm regression both in humans after the placement of endovascular stent grafts and in animal models with the application of recently discovered kinase inhibitors has even raised the possibility of nonsurgical therapeutic options for treated AAAs (8, 28, 29). The key initial step therefore is to determine optimal targets for such therapeutic intervention, and such targets would have to be centrally active in all aspects of aneurysm formation. By showing that PKCδ plays a central role not just in vSMC apoptosis and ECM degradation but also in recruiting inflammatory cells to AAAs we have shown that this kinase may be one of those centrally active players and warrants further study in this regard.
Acknowledgments
This work was supported by a Public Health Service Grant R01 HL-081424 (K. Kent and B. Liu) from National Heart, Lung, and Blood Institute, an American Heart Association grant-in-aid 0455859T (B. Liu), and National Institute of Health training grants T32 GM008466 (E. Ryer and S. Tsai), Society of University Surgeons Scholarship Grant (S. Tsai) and T32 HL083824 (S. Schubl). The authors like to thank Ms. Sophia Chu for the preparation of adenoviruses, and Drs. Fan Zhang and Domenick Falcone for technical assistance and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alcorn HG, Wolfson SK, Jr, Sutton-Tyrrell K, Kuller LH, O’Leary D. Risk factors for abdominal aortic aneurysms in older adults enrolled in The Cardiovascular Health Study. Arterioscler Thromb Vasc Biol. 1996;16:963–970. doi: 10.1161/01.atv.16.8.963. [DOI] [PubMed] [Google Scholar]
- 2.Nasim A, Sayers RD, Thompson MM, Healey PA, Bell PR. Trends in abdominal aortic aneurysms: a 13 year review. Eur J Vasc Endovasc Surg. 1995;9:239–243. doi: 10.1016/s1078-5884(05)80097-8. [DOI] [PubMed] [Google Scholar]
- 3.Nasim A, Thompson MM, Sayers RD, Bell PR. Elective AAA repair and rupture. Eur J Vasc Endovasc Surg. 1995;9:124–125. doi: 10.1016/s1078-5884(05)80242-4. [DOI] [PubMed] [Google Scholar]
- 4.Bobryshev YV, Lord RS, Parsson H. Immunophenotypic analysis of the aortic aneurysm wall suggests that vascular dendritic cells are involved in immune responses. Cardiovasc Surg. 1998;6:240–249. doi: 10.1016/s0967-2109(97)00168-3. [DOI] [PubMed] [Google Scholar]
- 5.Ernst CB. Abdominal aortic aneurysm. N Engl J Med. 1993;328:1167–1172. doi: 10.1056/NEJM199304223281607. [DOI] [PubMed] [Google Scholar]
- 6.Pearce WH, Koch AE. Cellular components and features of immune response in abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:175–185. doi: 10.1111/j.1749-6632.1996.tb33308.x. [DOI] [PubMed] [Google Scholar]
- 7.Bode MK, Mosorin M, Satta J, Risteli L, Juvonen T, Risteli J. Increased amount of type III pN-collagen in AAA when compared with AOD. Eur J Vasc Endovasc Surg. 2002;23:413–420. doi: 10.1053/ejvs.2002.1606. [DOI] [PubMed] [Google Scholar]
- 8.Huffman MD, Curci JA, Moore G, Kerns DB, Starcher BC, Thompson RW. Functional importance of connective tissue repair during the development of experimental abdominal aortic aneurysms. Surgery. 2000;128:429–438. doi: 10.1067/msy.2000.107379. [DOI] [PubMed] [Google Scholar]
- 9.Krettek A, Sukhova GK, Libby P. Elastogenesis in human arterial disease: a role for macrophages in disordered elastin synthesis. Arterioscler Thromb Vasc Biol. 2003;23:582–587. doi: 10.1161/01.ATV.0000064372.78561.A5. [DOI] [PubMed] [Google Scholar]
- 10.Rowe DW, McGoodwin EB, Martin GR, Grahn D. Decreased lysyl oxidase activity in the aneurysm-prone, mottled mouse. J Biol Chem. 1977;252:939–942. [PubMed] [Google Scholar]
- 11.Curci JA, Lee JK, Thompson RW. Pathogenesis of Abdominal Aortic Aneurysm. Philadelphia: Elsevier; 2001. [Google Scholar]
- 12.Ishibashi M, Egashira K, Zhao Q, Hiasa K, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K. Bone marrow-derived monocyte chemoattractant protein-1 receptor CCR2 is critical in angiotensin II-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2004;24:e174–178. doi: 10.1161/01.ATV.0000143384.69170.2d. [DOI] [PubMed] [Google Scholar]
- 13.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004;384:449–459. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB. Regulation of airway epithelial cell NF-kappaB-dependant gene expression by protein kinase C delta. J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Wang X, Evers BM. Induction of cIAP-2 in human colon cancer cells through PKCdelta/NF-kappaB. J Biol Chem. 2003;278:51091–51099. doi: 10.1074/jbc.M306541200. [DOI] [PubMed] [Google Scholar]
- 16.Ryer EJ, Hom RP, Sakakibara K, Nakayama K, Nakayama K, Faries P, Liu B, Kent KC. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26:780–786. doi: 10.1161/01.ATV.0000209517.00220.cd. [DOI] [PubMed] [Google Scholar]
- 17.Ryer EJ, Hom RP, Sakakibara K, Nakayama KI, Nakayama K, Faries PL, Liu B, Kent KC. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2006;26:780–786. doi: 10.1161/01.ATV.0000209517.00220.cd. [DOI] [PubMed] [Google Scholar]
- 18.Newman KM, Malon AM, Shin RD, Scholes JV, Ramey WG, Tilson MD. Matrix metalloproteinases in abdominal aortic aneurysm: characterization, purification, and their possible sources. Connect Tissue Res. 1994;30:265–276. doi: 10.3109/03008209409015042. [DOI] [PubMed] [Google Scholar]
- 19.Rayner K, Van Eersel S, Groot PH, Reape TJ. Localisation of mRNA for JE/MCP-1 and its receptor CCR2 in atherosclerotic lesions of the ApoE knockout mouse. Journal of vascular research. 2000;37:93–102. doi: 10.1159/000025720. [DOI] [PubMed] [Google Scholar]
- 20.Schecter AD, Berman AB, Yi L, Ma H, Daly CM, Soejima K, Rollins BJ, Charo IF, Taubman MB. MCP-1-dependent signaling in CCR2(-/-) aortic smooth muscle cells. Journal of leukocyte biology. 2004;75:1079–1085. doi: 10.1189/jlb.0903421. [DOI] [PubMed] [Google Scholar]
- 21.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. PKC-delta is an apoptotic lamin kinase. Oncogene. 2000;19:2331–2337. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 22.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochemical and biophysical research communications. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 23.Kuchera S, Barth H, Jacobson P, Metz A, Schaechtele C, Schrier D. Anti-inflammatory properties of the protein kinase C inhibitor, 3-[1-[3-(dimethylamino)propyl]-1H-indol-3-yl]-4-(1H-indol-3-yl)-1H- pyrrole-2,5-dione monohydrochloride (GF109203X) in the PMA-mouse ear edema model. Agents and actions. 1993;39:C169–173. doi: 10.1007/BF01972756. Spec No. [DOI] [PubMed] [Google Scholar]
- 24.Kutlu B, Darville MI, Cardozo AK, Eizirik DL. Molecular regulation of monocyte chemoattractant protein-1 expression in pancreatic beta-cells. Diabetes. 2003;52:348–355. doi: 10.2337/diabetes.52.2.348. [DOI] [PubMed] [Google Scholar]
- 25.Weber NC, Blumenthal SB, Hartung T, Vollmar AM, Kiemer AK. ANP inhibits TNF-alpha-induced endothelial MCP-1 expression--involvement of p38 MAPK and MKP-1. Journal of leukocyte biology. 2003;74:932–941. doi: 10.1189/jlb.0603254. [DOI] [PubMed] [Google Scholar]
- 26.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 27.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 28.Allaire E, Muscatelli-Groux B, Guinault AM, Pages C, Goussard A, Mandet C, Bruneval P, Melliere D, Becquemin JP. Vascular smooth muscle cell endovascular therapy stabilizes already developed aneurysms in a model of aortic injury elicited by inflammation and proteolysis. Ann Surg. 2004;239:417–427. doi: 10.1097/01.sla.0000114131.79899.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buth J, Harris P. Endovascular Treatment of Aortic Aneurysms. In: Rutherford RB, editor. Vascular Surgery. Philadelphia: Elsevier; 2005. pp. 1452–1475. [Google Scholar]