

Abstract
Although caused by vastly different pathogens, the world’s three most serious infectious diseases, tuberculosis, malaria and HIV-1 infection, share the common problem of drug resistance. The pace of drug development has been very slow for tuberculosis and malaria and rapid for HIV-1. But for each disease, resistance to most drugs has appeared quickly after the introduction of the drug. Learning how to manage and prevent resistance is a major medical challenge that requires an understanding of the evolutionary dynamics of each pathogen. This review summarized the similarities and differences in the evolution of drug resistance for these three pathogens.
Introduction
Tuberculosis (Tb), malaria, and HIV are the Big Three infectious diseases worldwide. Together, they cause ~5 million deaths/year and substantially affect the lives of a billion more people. The etiologic agents (Mycobacterium tuberculosis, Plasmodium species, and HIV-1) come from vastly different parts of the tree of life (viruses do not fit on the tree) and yet have much in common. Chief on the list of similarities is drug resistance. The evolution of resistant variants has made some Tb infections virtually untreatable, threatens to make malaria untreatable, and has necessitated complex combination therapy to control HIV-1 infection. This review describes common themes and interesting differences emerging from a consideration of the resistance mechanisms, evolutionary dynamics, and persistence of these agents (Table 1). Beyond their commonalities with respect to drug resistance, these diseases are inextricably intertwined at a population level. The three infections are all widespread in the developing world, and co-infections are common. HIV-1 infection predisposes to increased severity of Tb and malaria. Interactions between the agents used to treat these diseases are complex. Considering these diseases together may yield lessons that will cross from one organism to another and will help combat all three scourges in synergistic fashion.
Table 1.
Similarties and differences in drug resistance.
| Tuberculosis | Malaria | HIV | |
|---|---|---|---|
| Causative agent | |||
| Name | Mycobacterium tuberculosis | 5 Plasmodium species | HIV-1 |
| Type of organism | mycobacterium | protozoal parasite | retrovirus |
| Number of genes | 4400 | >5000 | 9 |
| Drugs | |||
| Available agents | old and limited | old and limited | >25 drugs from 6 classes |
| Regimens | 4 drug combination | single agents or artemesinin combination therapy | 3 drug combinations |
| Required length of treatment | at least 6 months, 2 years or more in some cases | a few days | lifelong |
| Efficacy | high for sensitive strains in adherent patients, low for resistant strains | high for most regimens, influenced by Hill slope? | influenced by Hill slope, very high for combinations but not curative |
| Resistance | |||
| Extent | seen for all drugs | seen for all drugs except possibly artemisinin | seen for all drugs |
| Principal mode of acquisition | de novo and transmitted | transmitted | de novo and transmitted |
| Molecular mechanisms | target and activator mutations | target mutations, drug transporters | target mutations |
| Multidrug resistance | MDR and XDR-Tb, serious problem | MDR a problem in regional pockets | serious problem, successful salvage Rx with newer regimens |
| Evolutionary dynamics | |||
| Population size | 1010 or greater | huge at intraerythrocytic stage | large |
| Mutation rate | 10−6 –10−10/generation | 10−6–10−9/generation | 0.33/generation |
| Generation time | 18–24 hrs | doubles in 12 hrs in RBC, R0=10–16 | 2 days, R0 =20 |
| Special mechanisms | established a life-long chronic infection | genome plasticity, accelerated resistance to multiple drugs, mutagenic drugs (atovaquone) | evolution of a new enzymatic activity (excision of AZT) |
| Persistence | |||
| Persistence in treated patients | persisters | dormant parasites? | latency |
TUBERCULOSIS
A century ago, a diagnosis of pulmonary consumption was considered a death sentence. For individuals infected with the bacterial pathogen Mycobacterium tuberculosis (Mtb), the only known cure was a regimen of rest, fresh air, sunshine, and a hearty diet. This approach remained the standard of care until the mid 1900’s, when antitubercular drugs were introduced. Recently, the growing problem of drug resistant Tb has threatened to return us to a time when this diagnosis was a death sentence. Multidrug resistant Tb strains (MDR-Tb) cause 3.6% of Tb cases and require at least two years of treatment with as many as six potentially toxic drugs (World Health Organization, 2010). Extensively drug resistant strains (XDR-Tb), which may be nearly impossible to cure, have been isolated in all countries that have adequate diagnostic capability to recognize them. A concerted effort on many fronts, as outlined in the thought-provoking review by Gandhi et al. (Gandhi et al., 2010), will be necessary to confront this challenge.
The Central Problem of Tuberculosis: The Unceasing March to Resistance
Development of resistance to antibiotics is common in many bacterial infections. Resistance is often due to transmission of infrequently-arising resistant clones, generated through mutation or acquisition of resistance gene-bearing mobile genetic elements. The week or two of treatment required to clear most bacterial infections is typically insufficient to cause the de novo generation of resistance. However, cure of an uncomplicated Tb infection requires at least six months on up to four different drugs, allowing the bacteria ample opportunity to develop resistance.
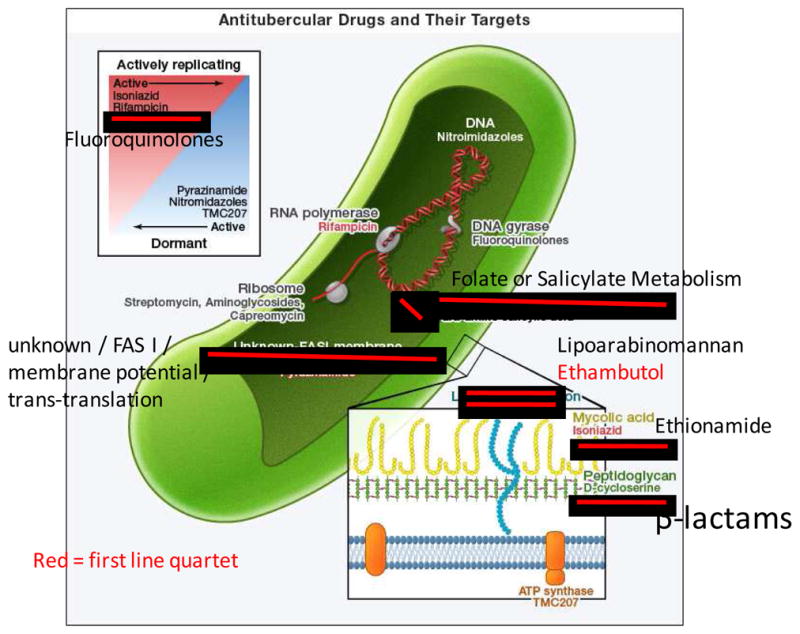
The first antitubercular drug was streptomycin (STR), shown by Schatz to have antitubercular activity in 1944 (Shatz and Waksman, 1944) and demonstrated to have substantial activity in human patients in 1946 (Hinshaw et al., 1946). Alarmingly, the evolution of resistance in over 85% of cases was reported shortly thereafter (BRMC, 1948). Streptomycin targets bacterial protein synthesis, and mutations in the 16S ribosomal DNA (rrs) and the S12 ribosomal protein gene (rpsL) can cause resistance (Figure 1). Para-aminosalicylic acid (PAS) was widely introduced in 1946 (Lehmann, 1946) and reports of resistance appeared in 1949 (Sweany, 1949; Dunner et al., 1949). Resistance mutations map to genes involved in folate and salicylate metabolism (Mathys, V. et al., 2009).
Figure 1.
Kinetics of bacterial burden in a mouse model of Tb in the alternative states of acute and chronic infection, antibiotic treatment, latency, and reactivation. In this idealized illustration, after low dose aerosol infection, the bacterial burden increases during an acute phase of infection and plateaus to a chronic infection following the onset of the adaptive immune response. Antibiotic treatment (INH + RIF) reduces burden to an apparent sterile state, which may represent true clearance or alternatively a latent infection that may be reactivated by immunosupression with the iNOS inhibitor aminoguanidine (AG). The bacterial burden during latency is comprised of persister cells that may be the substrate for the emergence of resistant strains.
In an advance that ultimately contributed to the success of HIV-1 treatment, PAS and STR were used together in combination therapy that delayed the emergence of resistance (Dunner et al., 1949; BRMC, 1950). Combination therapy became standard and was expanded with the inclusion of isoniazid (INH) in 1952 (BMRC, 1952). The target of isoniazid is InhA, which catalyzes a critical step in the synthesis of mycolic acids that comprise the Mtb cell wall (Vilchèze et al., 2006). Mutations in inhA and the gene encoding the activator of INH, katG, as well as ndh and mshB, cause resistance (Vilchèze et al., 2006; Vilchèze and Jacobs, 2007). Although this triple combination of STR+PAS+INH was successful in treating patients, in retrospect it was a decisive blow to support for Tb research, as the problem was considered “solved”. Combination therapy was further improved when Ethambutol (EMB), discovered in 1961, quickly replaced PAS (Thomas et al., 1961; Ahmad et al., 2011). It interferes with cell wall biosynthesis (Takayama and Kilburn, 1989), and mutations mapping to embB, an arabinosyl transferase, can result in resistance (Sreevatsan et al., 1997).
In 1972, two additional drugs were adopted into the standard therapy, relegating the injectable STR to second line status and reducing treatment from 18 to six months (EA/BMRC, 1972; Ahmad et al., 2011). Rifampicin (RIF), an inhibitor of transcription (resistance mutations map to the RNA polymerase subunit gene rpoB), and pyrazinamide (PZA) complete the current quartet (Yeager et al., 1952). Mutations in pncA, the gene encoding the activator of PZA, and rpsA, which encodes a ribosomal protein, result in PZA resistance (Shi et al, 2011). However, the target and mechanism of PZA remain controversial (Zimhony et al., 2000). The current first line combination of INH+RIF+PZA+EMB allowed short course treatment: two months with all four drugs followed by four months of INH+RIF (EA/BMRC, 1972). This regimen has not changed in nearly 40 years because despite substantial drawbacks of length, complexity, and toxicity, it is efficacious in the treatment of drug sensitive Tb in adherent patients. Lack of adherence with lengthy treatment is common as patients often abandon therapy when they begin to feel better, allowing the emergence of resistance.
Unfortunately, recent research has shown that the standard four-drug therapy results in drug resistance in immune-deficient mice, a finding with serious implications for the treatment of immunocompromised individuals such as those with AIDS (Zhang et al., 2011). Other problems with the regimen have been reported. Unfavorable interactions between different Tb drugs can occur (Dickinson et al, 1977; Grosset et al, 1992; Almeida et al., 2009) and for those co-infected with Tb and HIV-1, interference with the efficacy of HIV-1 antiretrovirals has been shown (MMRW, 1996). Pharmacokinetic studies suggest that combined dosing may leave patients with suboptimal plasma concentrations of individual drugs at several times during the day. Fortunately, a recent study has shown that this type of dosing may actually result in better bacterial clearance, possibly due to prevention of antagonism, and does not seem to encourage resistance (Srivastava et al., 2011). Combination therapy is clearly indispensible to treating of Tb and the preventing resistance. However, the current standard therapy may not be optimal.
Second line drugs for MDR-Tb include fluoroquinolones, which target DNA gyrase, ethionamide (ETH) which like INH targets mycolic acid biosynthesis, aminoglycosides such as kanamycin and peptides such as capreomycin, that target the ribosome, and the peptidoglycan cross-linking inhibitor D-cycloserine (Johnson et al., 2006) (Figure 1). Like the first line drugs, resistance to second line drugs is chromosomally-encoded and has not been shown to be transferred by mobile genetic elements. Initially, this gave researchers hope that multiple resistances would need to arise within a single clone and be slow to spread, but with recent reports of transmission of multidrug resistant strains, it appears the clonal structure of bacterial populations will offer little protection against the spread of drug resistance (Andrews et al., 2008).
Tolerance and Persistence: The “hunkered-down” phenotype
Apart from the problem of drug resistance, but possibly contributing to it, is the phenomenon of drug tolerance. Why does it take six months to treat drug-sensitive Tb when even the most recalcitrant of other bacterial infections can been successfully treated in a couple weeks?
The long, complex treatment regimen is necessitated by the ability of the bacteria to enter a state in which they are unaffected by or recalcitrant to drugs. Actively growing Mtb cells must undergo a developmental transition to a “hunkered-down” phenotype to survive in the face of drug and immune pressure. A typical immune response is capable of containing the infection in a chronic state, with balanced bacterial growth and killing (Ford et al., 2011; Gill et al., 2009). Drug intervention is necessary to upset this equilibrium in favor of bacterial clearance. In this war of attrition between the bacteria and the combined effects of immunologic and pharmacologic interventions, Mtb has evolved to persist by altering its metabolism and possibly slowing its replication. This is exemplified by experiments in which infected animals were treated with antibiotics for up to six months, resulting in the lack of culturable bacteria from any organ. Upon cessation of treatment, some infections spontaneously reactivated (McCune et al., 1956; McCune and Tompsett, 1956). This number increased dramatically when the animals were immunosuppressed by treatment with corticosteroids (McCune et al., 1966).
As the importance of eradicating persisters has been realized, efforts to develop drugs targeting them have increased. TMC207, a recently discovered inhibitor of the F0/F1 ATP synthase, is active against non-replicating Mtb (Andries et al., 2005; Koul et al., 2008). It is now in Phase IIb clinical trials. Recent work has shown the mycobacterial proteasome is necessary for surviving nitrooxidative stress and persistence during chronic infection. Specific inhibitors that spare the human proteasome have been identified (Darwin et al., 2003; Gandotra et al., 2007; Lin et al., 2009). Also, recent studies (Adams et al., 2011; Srivastava et al., 2010) demonstrate the importance of efflux mechanisms to drug tolerance in actively dividing cells and suggest that targeting efflux may be a useful strategy to prevent the emergence of resistance.
The Current State of Tuberculosis Chemotherapy and Future Directions
The anti-Tb arsenal of drugs is decades old and is being quickly depleted due to resistance. While it is obvious that we need new bactericidal antibiotics with novel mechanisms of action that kill replicating and non-replicating bacteria resulting in overall treatment shortening, it is imperative that future drug development address a number of other factors. Foremost amongst these is activity against MDR and XDR strains. One approach is to repurpose existing drugs approved other infections such as the MRSA drug linezolid and the antibiotic metronidazole, both currently in phase II trials. Much effort is also focused on creating novel inhibitors of the well-validated targets such as InhA (target of INH) and RpoB (target of RIF). A small number of drugs with novel targets such as TMC207, PA824, and OPC67683 are currently in Phase II clinical trials, and SQ109 and LL-3858 are both in Phase I trials (Spigelman 2007). Even for these drugs, with new modes of action, resistance develops in vitro at rates similar to the existing drugs, if not more quickly (Kaneko et al., 2011).
To design more effective drugs we need a better understanding of the basis of mycobacterial growth in vivo and the mechanism by which the bacterium achieves the developmental shift that allows persistent infection and drug tolerance. New research on genes essential for MTb survival in the host has prompted the search for inhibitors of the products of these attractive targets. Drug development processes also need a paradigm shift. Current approaches typically involve phenotypic screening of libraries against M. smegmatis or M. bovis. Auxotrophic strains of Mtb that are safe and more closely related to the target bacterium may prove more suitable for high throughput screening (Sambandamurthy, V. K. et al., 2002), Moreover, auxotrophic strains of drug-resistant Mtb could be used. New animal models that exhibit more human like-pathology would greatly improve the chances of new drugs progressing to human trials (Pichugin et al., 2009; Gil et al., 2010).
Although encouraging, these developments are still starkly minimal in the face of the enormous challenge of the explosion of drug resistant Tb and the collision of the HIV-1 and Tb epidemics. As with HIV-1, it is becoming increasingly clear that no single drug can act as a magic bullet against Tb. In this context it is essential to develop new drug combinations and consider these to be the unit of development rather than individual drugs (Ginsburg, 2011).
MALARIA
Resistance has developed to almost every drug used to treat malaria, including drugs acting at different stages in the complex life cycle of this parasite (Figure 2). The only possible exception is artemisinin. Chloroquine, once a mainstay of prophylaxis and treatment, has become obsolete in most areas where malaria is endemic. Newer treatments have also failed. As with Tb and HIV-1, resistance to some drugs has been documented almost simultaneously with drug introduction (Hyde 2005). The rate at which resistance develops depends on the mutation rate, the number of mutations needed to confer resistance, the use and abuse of the antimalarial drug, and pharmacokinetic/pharmacodynamic considerations (White, 2004). As with Tb, our current antimalarial armementarium is thin and consists of drugs that are expensive and/or toxic and that are starting to fail. We have already run out of options to protect certain subpopulations, such as women in the first trimester of pregnancy.
Figure 2.
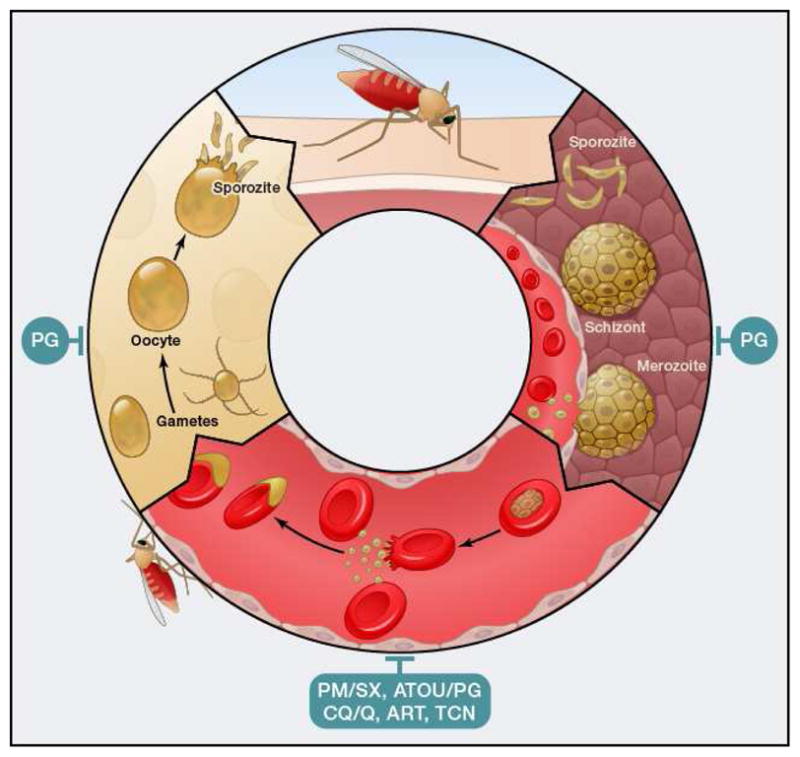
Plasmodium life cycle. Depicted are different stages. Red, intraerythrocytic stages; yellow, mosquito stages; brown, liver stages. PM/SX, pyrimethamine/sulphadoxine; ATOV/PG, atovaquone/proguanil; CQ/Q, 4-aminoquinolines like chloroquine and quinine and related compounds; ART, artemesinin and related compounds; TCN, tetracyclines; PQ, primaquine.
Artemesinin derivatives are widely used in endemic areas, and resistance is a major concern. Evidence for artemesinin resistance is not yet compelling. Certain populations are now taking longer to clear parasites from the bloodstream than was historically the case, but in culture these organisms do not have detectable resistance (Dondorp et al., 2009). Since existing combination regimens (artemesinin combination therapy or ACT) and late-stage drug development projects depend on these agents, we will have a therapeutic crisis if artemesinin resistance continues to emerge. There is an urgent need for new antimalarial agents and new strategies for overcoming resistance.
Types of Resistance
Resistance occurs via mechanisms that are well known in other organisms. Resistance to enzyme inhibitors often is due to target mutation. Usually more than a single mutation is needed to confer clinically significant resistance. Resistance to drugs that cause non-specific cellular damage (4-aminoquinolines, artemesinins) is typically through amplification and/or mutation of multidrug resistance (MDR) and MDR related protein (MRP) transporters that pump the agent out of its site of action (Koenderink et al. 2010). In the case of one 4-aminoquinoline, chloroquine, the parasite has no transporter that can efficiently eliminate the drug from its site of action, the food vacuole. The parasite has had to make one by extensive mutagenesis of a putative peptide transporter in the food vacuole, called P. falciparum chloroquine-resistance transporter or PfCRT (Bray et al. 2005). The mutations allow PfCRT to maintain its normal cellular function while now facilitating recognition and efflux of chloroquine from the food vacuole (Martin et al. 2009). Interestingly, while this mechanism of resistance has arisen independently in P. falciparum in different regions of the world, chloroquine resistance in P. vivax does not involve PvCRT (P. vivax chloroquine-resistance transporter) (Nomura et al., 2001), and its basis remains unknown.
Mechanisms for Generating Resistance
The point mutation rate in P. falciparum is 10−6 to 10−9/generation (Rathod et al., 1997; Cooper et al., 2002; Istvan et al., 2011). Given that an infected person may have 1010 to 1013 parasites in the bloodstream, and that there are an estimated 5 × 108 infections per year, there are on the order of 1020 parasites in the world each year (Rathod, 1997). It follows that any particular triple mutant parasite has a substantial chance of existing at some time somewhere in the world. Thus the sheer numbers involved represent a powerful force for the generation of resistance. Copy number variation is also readily achieved; such events occur easily in culture, and evidence for frequent amplification and deletion events is apparent in the genome (Anderson et al., 2009). Compounding genomic plasticity is a phenomenon known as ARMD (accelerated resistance to multiple drugs). Certain P. falciparum strains have up to 1000x higher frequency of resistance to selected compounds (Rathod et al. 1997). The molecular basis of ARMD is uncertain. These strains respond to drug challenge by generating multiple mutations in a random region of the genome. The concept is that if these mutations affect a gene in a way that leads to resistance, that parasite (and therefore its lineage) can survive. In this context, it is of interest that chloroquine resistant parasites have evidence of extensive mutation in and around the CRT gene (Fidock et al. 2000), some of which are important for conferring resistance. A final mechanism involves drugs whose effects are mutagenic, thereby increasing the frequency of resistance. Atovaquone is a mitochondrial cytochrome inhibitor that is potent against malaria parasites. Resistance to atovaquone as a single agent develops easily. Bockade of electron transport leads to accumulation of electrons and superoxide, which promotes mutation of the mitochondrial DNA, increasing the frequency of resistance (Vaidya and Mather, 2000).
Pharmacokinetics also play a role in the generation of resistance. Some antimalarials (many quinolines) have prolonged half-lives. New infections that arise weeks after a prophylactic dose or a treatment course will expose parasites to subtherapeutic drug concentrations that could select for resistance (White 2004). Although the parasite pool that could harbor mutation upon initial emergence from the liver is relatively low (about 100,000), the possibility of selecting a resistant mutant is significant and the probability of enriching resistant parasites from a pre-existing mixed population is high. In the case of intermittent preventive treatment in pregnancy, women treated with sulfadoxine-pyramethamine had higher overall parasitemias, suggesting a competitive facilitation of the resistant parasites (Harrington et al., 2009). Treating patients who are co-infected with HIV-1 and malaria also has the potential to engender resistant malaria parasites. Trimethoprim-sulfa, used as prophylaxis against opportunistic infections in patients with HIV-1, and HIV-1 protease inhibitors both have antimalarial activity.
Evolutionary Dynamics
Resistance to each antimalarial agent has its own evolutionary history, which is still being written. The evolution of resistance has been studied in most detail for chloroquine and the antifolates. Chloroquine was introduced in the 1940s, and resistance was first documented in the 1960s. At least two independent foci of resistance developed in Southeast Asia and South America. Resistance spread from Asia to Africa, and from east to west across the African continent (Payne 1987). There appears to be a fitness defect in resistant mutants, and in regions where chloroquine has been withdrawn, sensitive parasites have taken over (Laufer et al. 2006).
The dihydrofolate reductase (DHFR) inhibitor pyrimethamine was introduced in the 1950s, and resistance was detected almost immediately. The synergistic dihydropteroate synthetase (DHPS) inhibitor sulphadoxine was added, and the combination (SP) was successfully used for decades (Muller and Hyde, 2010). DHPS resistant parasites and SP failure were documented in the early 1990s. A double DHPS mutations combined with triple DHFR mutations confer high-level SP resistance and early treatment failure. Resistance appears to have originated in Southeast Asia and spread through Africa in a pattern similar to that of chloroquine. Interestingly, despite the ease of selecting resistance to these drugs in the laboratory, molecular epidemiology suggests a single origin and a sweep of the resistant parasite across Africa (Roper et al., 2004), although there is evidence for some independent origins of sulphadoxine resistance (Vinayak et al. 2010).
Persistence
Reinfection is common in malaria, especially in hyperendemic regions, since immunity from previous infection is partial at best. Recrudescence, the re-emergence of symptoms in an infected individual, is common with P. vivax and ovale, stemming from dormant liver forms (hypnozoites) that can activate years later. Recrudescence of P. falciparum after apparently effective drug treatment is a phenomenon seen with artemesinin. Artemesinin and its derivatives, in combination with other drugs, are rapid acting, potent antimalarials. As a single agent, artemesinin therapy fails in a significant portion of patients (de Vries 1996). Recrudescent parasites remain susceptible to artemesinin, which has given rise to the concept that there may be a population of dormant parasites that can survive treatment. In culture, a similar phenomenon can be recapitulated: a population of early stage parasites appears to be able to survive treatment and grow back (Teuscher et al. 2010). In contrast to bacterial persisters, the number of “dormant” parasites diminishes with increasing concentrations of drug. The dormancy phenomenon looks likely to be an important biological phenomenon, but one simpler explanation has not yet been clearly ruled out. If artemesinin kills with a shallow dose-response slope similar to that seen for some anti-HIV drugs (Shen et al., 2010), not all the parasites will be killed even at a dose high above the IC50 (see HIV section below, Fig. 3D).
Figure 3.
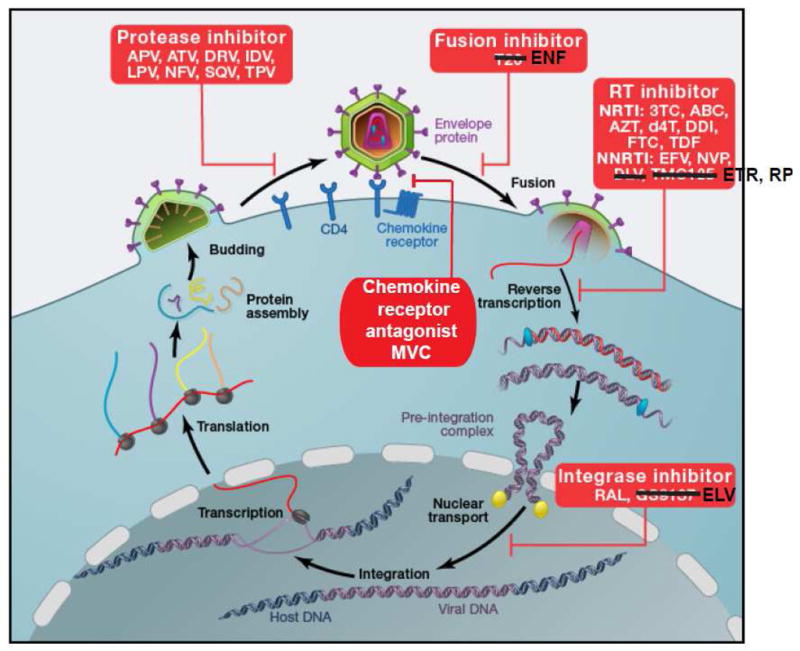
HIV-1 life cycle. HIV-1 attaches to CD4 via envelope protein spikes on the surface of the virion. The CD4 interaction triggers a conformational change in the envelope protein that allows binding to a chemokine receptor, CCR5 or CXCR4. This is followed by further rearrangements of the envelope proteins that mediate fusion of the viral envelope and the cell membrane. The genomic viral RNA is then copied by RT to give double stranded viral DNA that can be inserted into host cell DNA by integrase. Following transcription and translation, viral proteins and genomic viral RNA assemble into virions. During or shortly after release from the virus-producing cell, viral polyproteins are cleaved by HIV-1 protease into functional units, allowing infection of additional cells. Entry can be blocked by the chemokine receptor antagonist (CRA) maraviroc (MVC) or by the fusion inhibitor enfuvirtide (ENF). Reverse transcription is blocked by the NRTIs lamivudine (3TC), abacavir (ABC), zidovudine (AZT), stavudine (d4T), didanosine (ddI), emtricitabine (FTC), and tenofovir disoproxil fumarate (TDF), and by the NNRTIs efavirenz (EFV), nevirapine (NVP), etravirine (ETR), and rilpivirine (RPV). Integration is blocked by the integrase strand transfer inhibitors raltegravir (RAL) and elvitegravir (ELV). Virus maturation is blocked by the PIs amprenavir (APV), atazanavir (ATV), darunavir (DRV), indinavir (IDV), nelfinavir (NFV), saquinavir (SQV) and tipranavir (TPV).
New Strategies
The malaria parasite has managed to counter everything man has thrown at it. There is an urgent need for new approaches. One strategy is resistance reversal (Guantal and Chibale 2010). A variety of compounds, most notably calcium channel blockers such as verapamil, are able to restore chloroquine efficacy against chloroquine-resistant parasites. This is thought to be a direct effect on PfCRT (Martin et al. 2009). Further development of such agents would benefit from a better understanding of the physiology and molecular details of the PfCRT-drug interaction, which will be challenging for this large membrane protein.
Another approach similar to that used against HIV-1 reverse transcriptase is to combine two agents that work on the same enzyme. DHFR mutants that are resistant to pyrimethamine are more sensitive to the compound WR99210 and vice versa, due to conflicting requirements for accommodation in the active site (Muller and Hyde 2010). It is difficult for a parasite to become resistant to both compounds at the same time. A similar concept is operative in certain HIV-1 drug combinations for which resistance to one drug confers hypersusceptibility to another (Sarafianos et al., 2004). Of course, one could conceive of a parasite with a DHFR gene duplication in which one copy confers resistance to one drug and the second confers resistance to the other drug. Given the enormous genomic plasticity of Plasmodium, such a scenario unfortunately becomes quite feasible. Unfortunately, constructing a complex 3–4 drug regimen, such as those used for Tb and HIV-1, is currently a pipe dream for malaria, as we do not have enough new drugs to begin to construct such a regimen.
Another strategy would be to force intraerythrocytic parasites to differentiate into sexual forms en masse. This would halt parasite multiplication in the bloodstream. Patients would have to be kept away from mosquitoes until the parasites were cleared from the circulation, to prevent transmission. If resistance were to develop, it would hopefully impair differentiation, resulting in dead-end parasites. A better understanding of the physiological triggers of gametocytogenesis (Bousema and Drakely 2011) would facilitate embarking on such an approach.
A final strategy is to target parasites in the mosquito (Slavic 2011). The various life cycle forms in the insect vector range in population size from single digits (oocysts) to a few thousand (sporozoites). This is many orders of magnitude less than the number of parasites in the human bloodstream. Therefore the probability of resistance is greatly reduced. One challenge is that a drug that worked on an insect stage would need to be maintained at significant levels in the mosquito (mosquito PK!) as well as in the human, and another issue is that it would not likely be curative for the patient with malaria. This so-called transmission-blocking drug would be effective on a population level and would need to be combined with intraerythrocytic parasite-active agents for treatment.
For now, we must keep producing new agents to stay ahead of the inexorable development of resistance. People have been developing antimalarial chemotherapy for centuries. Some of the most successful efforts have stemmed from ethnobotany. The ancient Chinese herbal remedy for fever emergencies, Qinghao, was developed into the potent antimalarial artemesinin through an elegant extraction/identification project (Tu, 2011). The ancient Peruvian fever remedy from the bark of the Cinchona tree was used by the Jesuits as an antimalarial and later found to have quinine as its active principle (Rocco, 2004). Many other antimalarials have stemmed from wartime development efforts (Ockenhouse et al., 2005). Our best hope at present is the Medicines for Malaria Venture (MMV), a public-private partnership that functions as a virtual drug company. Though only a decade old, several formulations have already progressed to registration and are in clinical use. So while there is a threat that we will run out of treatments for malaria, there is a chance that we can keep ahead of this adaptable bug.
HIV-1
Twenty five antiretroviral drugs from six classes have been approved to treat HIV-1 infection (Figure 3). Typically they are used in three drug combinations (Panel on Antiretroviral Guidelines for Adults and Adolescents, 2009; Thompson et al., 2010). As with the other major human pathogens discussed here, drug resistance seriously complicates treatment (Clavel and Hance, 2004; Larder et al., 1989; Clark et al., 2007; Margeridon-Thermet and Shafer, 2011). Combination therapy can halt viral replication and reduce plasma HIV-1 levels to below the detection limit of clinical assays (Gulick et al., 1997; Hammer et al., 1997; Perelson et al., 1997). Treatment failure is typically associated with problems with adherence and does not generally increase with time in the patients who achieve an undetectable viral load (Boyd, 2009). However, resistance can occur for all six classes of antiretroviral drugs. Multidrug resistant HIV-1 can arise when mutations accumulate on the same viral genome (Palmer et al., 2005). Although multidrug treatment failure is becoming less common as regimens improve, it remains a serious problem with high mortality (Deeks et al., 2009). For each drug, a stereotypical set of mutations arise de novo in each failing patient. Resistant viruses can also be transmitted (Little et al., 2002; Wheeler et al., 2010), and among patients with newly diagnosed infection, as many as 16% have drug resistant HIV-1 in some areas (Wheeler et al., 2010). Because of the rapid rate of HIV-1 evolution, the evolutionary dynamics of drug resistance have been studied quantitatively, revealing interesting similarities and differences with the other pathogens discussed here (Table 1). Most of the principles derived from the study of HIV-1 drug resistance apply to infection with the related but less pathogenic virus, HIV-2, which is largely restricted to West Africa and declining in incidence (Ntemgwa et al., 2009).
Antiretroviral drugs target virus entry and the three virally encoded enzymes: protease, reverse transcriptase (RT), and integrase (Figure 3). With the exception of the chemokine receptor antagonists (CRAs), which bind a host protein involved in entry, antiretroviral drugs target virally encoded structures. Mutations in the viral genome appearing in response to specific drugs are well defined (summarized in Clark et al., 2007; Johnson et al., 2010), and, the molecular mechanisms by which these mutations confer resistance are understood (Acosta-Hoyos and Scott, 2010; Krishnan et al., 2010; Lobritz et al., 2010; Margeridon-Thermet and Shafer, 2011; Sarafianos et al., 2004; Wensing et al., 2010). Most resistance mutations interfere with drug binding. The protease inhibitors (PIs) and nucleoside RT inhibitors (NRTIs) are substrate analogues. Mutations conferring resistance to these drugs allow the target enzyme to better discriminate between the drug and the natural substrate. However, other types of resistance involve unexpected adaptations that permit viral replication in the presence of inhibitory drugs. Resistance to CRAs involves either mutations in the viral envelope protein that allow it to interact with the drug-bound form of the chemokine receptor or the selection of viral variants that utilize a different chemokine receptor for entry (Lobritz et al., 2010). The NRTIs are chain terminators lacking a 3′ hydroxyl group. Resistance to the thymidine analogue zidovudine (AZT) results from mutations that create a binding site on RT for ATP, the β and γ phosphates of which are positioned to function like pyrophosphate in an excision reaction that is the reverse of the reaction that incorporates the chain terminator (Acosta-Hoyos and Scott, 2010; Sarafianos et al., 2004). This remarkable adaptation can be viewed as the evolution of a new enzymatic activity that reverses drug inhibition.
When resistance develops, a change in treatment is indicated (Panel on Antiretroviral Guidelines for Adults and Adolescents, 2009; Thompson et al., 2010). However, the choice of alternative regimens is complicated by cross-resistance within drug classes, increased toxicity of second line agents, and, in many areas of the world, limited availability of additional drugs. Two types of resistance testing facilitate the choice of “salvage” regimens. Bulk sequencing of plasma virus is commonly used to detect mutations associated with resistance, and when multiple mutations are present, physicians can utilize a more expensive analysis in which the drug susceptibility of patient-derived vlral variants is directly assessed (Larder et al., 1989; Petropoulos et al., 2000). Resistance testing is less available in resource-limited settings.
Resistance was originally considered an inevitable consequence of antiretroviral therapy. However, it has become clear that resistance is largely avoidable with good adherence to modern regimens. Success in preventing resistance reflects dramatic advances in the effectiveness and tolerability of antiretroviral drugs and improved understanding of the fundamental biology underlying the evolution of resistance.
Evolutionary dynamics of HIV-1 drug resistance
The capacity for rapid evolution is a critical characteristic of HIV-1 that explains its ability to persist in the face of host immune responses and antiviral drugs. The rapid evolution of HIV-1 is generally attributed to the high error rate of RT and rapid replication rate of the virus. Following entry of the virus into a CD4+ T cell, the 9.7 kB RNA genome is reverse transcribed into double stranded DNA by RT. Base substitutions are introduced during reverse transcription, and the resulting mutant viral genomes provide the substrate for the evolution of resistance. The base substitution rate is 2.4 × 10−5 substitutions/base pair/cycle (Mansky and Temin, 1995). This mutation rate allows the virus to diversify from what is usually a single infecting genome to an increasingly complex set of related viruses (quasispecies) that coevolve over time in each infected individual (Keele et al., 2008; Shankarappa et al., 1999).
Although the RT error rate is substantially higher than the error rate of host DNA polymerases, only one in four newly infected cells should contain a substituted viral genome. It is the large size and rapid turnover rate of the infected cell population, coupled with the high error rate of RT, that give HIV-1 the capacity for rapid evolution (Bonhoeffer and Nowak, 1997; Coffin, 1995; Ho et al., 1995; Nowak et al., 1997; Wei et al., 1995). The size of the infected cell population in the average untreated patient is large enough so that at any given time every possible single point mutant in the entire viral genome is likely to preexist in some infected cell (Box 1). Preexisting resistant viruses can cause rapid treatment failure under certain conditions described below.
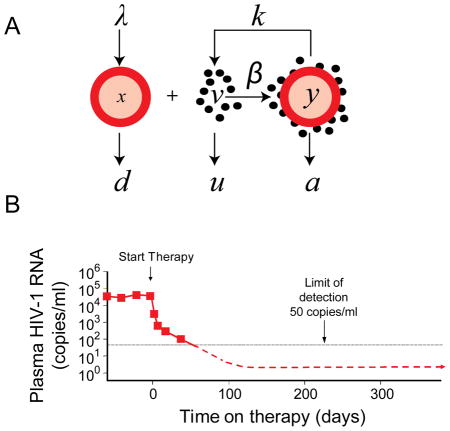
Box 1.
Insights into the evolution of HIV-1 drug resistance have come from a mathematical model of viral dynamics (Panel A, adapted from Wodarz and Nowak, 2002). This model, originally developed by Martin Nowak and Alan Perelson (Ho et al., 1995; Perelson et al., 1997; Wei et al., 1995; Wodarz and Nowak, 2002), has three components: uninfected cells (x), free virus (v), and infected cells (y). New uninfected cells are generated at a constant rate. Free virus is produced at a rate that depends on the number of infected cells (y) and a rate constant (k). Infected cells are generated through the interaction of uninfected cells and free virus at a rate that depends on concentration of each and a rate constant β. For each of these components, decay is assumed to be exponential with characteristic rate constants d, u and a, for uninfected cells, free virus, and infected cells, respectively. R0 the number of newly infected cells that arise from the virions produced by a single infected cell under conditions where target cells are not limiting is given by the expression λβk/dua (Wodarz and Nowak, 2002). Resistance mutations can reduce β. Antiretroviral drugs block new infection of susceptible cells without affecting release of virus particles from previously infected cells. Thus the drugs affect β but not k. In the case of effective HAART, they reduce β to 0 such that virus production comes only from cells infected prior to therapy Viremia decays at a rate dependent on the decay rates of free virus and various populations of virus producing cells. Analysis of the initial phase of decay has allowed measurement of the decay rates u and a Two phases of decay reflect the presence of more stable populations of infected cell (Panel B).
During the asymptomatic phase of the infection, plasma virus levels are roughly constant in the absence of therapy. In this quasi steady state, virus production equals virus clearance, as described in the fundamental equation of viral dynamics, ky=uv. This equation provides a way to estimate the total body number of productively infected cells (y) from a readily measured quantity, the level of plasma virus (v). The virus production rate k can be estimated using measurements of the burst size (N) and the decay rate of productively infected cells (a). Assuming a burst size of 2200, in the middle of published estimates, the steady state number of productively infected cells for a patient with a typical plasma HIV-1 RNA level of 30,000 copies/ml and an extracellular fluid volume of 15 liters is approximately 2 × 106. Given that average lifespan of most of the productively infected cells (1/a) is only 1 day, this is also the number of newly infected cells that arise per day. This estimate of the number of newly infected cells arising per day is lower than initial estimates of 109 that were based on erroneous assumptions about the rate of increase in CD4+ T cell counts following initiation of treatment. Nevertheless it is large enough to guarantee that most single mutants preexist in the average untreated patient. The total body number of infected cells with a particular mutation can be calculated by (μ/s)*y, where μ the mutation rate and s is the selective disadvantage conferred by the mutation. Although the precise values will depend on the nucleotide substitution involved, values >1 are obtained even for mutations with a high fitness cost because of the relationship between the mutation rate and the population size.
The evolutionary dynamics of HIV-1 drug resistance are complex (Coffin, 1995; Wei et al., 1995; Wodarz and Nowak, 2002) but are well understood in two situations. The first is the case in which a single substitution give rises to a variant that is highly resistant to a given drug but still reasonably fit. Resistance is typically evaluated as a fold change in the IC50, reflecting a shift in the drug’s dose response curve to the right (Fig. 4A). However, most resistance mutations reduce viral fitness in the absence of drug (Quiñones-Mateu and Arts, 2001), and this decrease in fitness must also be considered. Therefore, it is useful to consider the basic reproductive ratios (R0) for wild type and mutant viruses (Fig. 4B). R0 values > 1 are necessary for spreading infection. An R0 value of 20 has been measured in acute HIV-1 infection (Little et al., 1999). Most resistance mutations cause some alteration in the function of the relevant viral protein so as to reduce R0 in the absence of drug. The dose-response curves for inhibition of wild type and mutant viruses cross at a particular drug concentration (Fig. 1B, C). Below this concentration, the fitness cost of the mutation outweighs the benefit provided by resistance. Above that concentration, the mutant has a selective advantage. Whether preexisting drug resistant variants will grow out depends on where the clinical concentration range of the drug lies in relation to this crossover point. In situations where a single substitution produces a variant that has a selective advantage at clinical drug concentrations and R0 >1, resistance can emerge very rapidly in patients receiving monotherapy with that drug.
Figure 4.

Dose-response curves for antiretroviral drugs. (A) Standard dose-response curve for a hypothetical antiretroviral drug. The fraction of infection events unaffected (fu) by the drug is plotted as a function of the log of the drug concentration (log D) for wild type virus and a resistant mutant. In this example, the IC50 for wild type virus is 0.01 μM and the clinical concentration range is 1–10 μM (pink shaded box). The slope of the dose response curve is 1. The resistance mutation is assumed to increase the IC50 by 100 fold without affecting the slope. The dotted lines indicate the IC50 values for wild type and mutant virus. (B) Dose-response curve for the viruses shown in A with the reproductive ratio R0 as a measure of infection. The resistance mutation is assumed to cause a 50% decrease in fitness. The dotted lines indicate the IC50 values for wild type and mutant virus. The gray line indicates an R0 value of 1. (C) Log-log plot of the dose-response curve shown in B. The gray line indicates an R0 value of 1. (D) Effect of slope. Log-log plot of the dose-response curves for two drugs with the same IC50 (0.01 μM) and m values of 1 (blue curve) and 3.5 (green curve). Note the Y axis scale is expanded to capture the profound in inhibitory effects achieved with a high slope value. The gray line indicates an R0 value of 1. (E) Effect of a single resistance mutation on inhibition by drug with high m. The drug is assumed to have an IC50 of 0.01 uM and a slope of 3.5. A resistance mutations increases the IC50 by 3 fold, reduces the slope by 1/3rd and decreases fitness in the absence of drug by½. Nevertheless, R0 for the mutant virus remains well below 1 throughout the clinical concentration range. (F) Changes in slope can affect resistance. In this example, a drug inhibits both wild type and mutant virus with the same IC50 (0.1 μM). The mutation causes a 50% reduction in fitness but also reduces the slope from 1 to 0.5. Because there is no change in the IC50, standard methods of analysis would not classify the relevant mutation as a resistance mutation. However, in the clinical concentration range, the mutation does cause resistance due to the change in slope.
The scenario described above represents the simplest case of the evolution of resistance and is illustrated by several important clinical examples. Monotherapy with the NRTI lamivudine (Schuurman et al., 1995), the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine (Wei et al., 1995), and the fusion inhibitor enfuvirtide (Lobritz et al., 2010) all select for resistant virus in a few weeks. Plasma virus levels fall initially after the initiation of therapy but rise again as the resistant variants comes to dominate the actively replicating virus population (Wei et al., 1995). Perhaps the most dramatic example comes from studies of mother-to-child transmission of HIV-1 in resource limited settings. Transmission can be reduced by administration of a single dose of nevirapine to the mother at the time of delivery and a single dose to the infant (Guay et al., 1999). However, as a result of that single dose, the majority of the mothers develop nevirapine-resistant virus that persists for months (Flys et al., 2007).
Drug efficacy and resistance
Monotherapy does not always lead to immediate failure and resistance. Although combination therapy regimens typically produce better results in clinical trials, monotherapy with certain PIs can suppress replication in a substantial fraction of patients (Bierman et al., 2009). To understand this finding, we need to consider differences in the intrinsic antiviral activity of different drugs. Antiretroviral drug activity is typically expressed in terms of the IC50. However, the drugs are used clinically at concentrations substantially above the IC50, and inhibition at clinical concentrations can only be predicted from the IC50 if the shape of the dose-response curve is known. The shape is influenced by cooperative interactions and is described mathematically by the slope parameter or Hill coefficient (m). Recent studies have shown that certain classes of antiretroviral drugs, notably NNRTIs and the PIs, have cooperative dose-response curves with high slopes even though they target enzymes that are univalent with respect to the inhibitor (Shen et al., 2008; Jilek et al,, 2012). These high slopes may reflect a unique form of intermolecular cooperativity operative when multiple copies of a drug target participate in a given step in the virus life cycle (Shen et al., 2011).
High slopes allow for extraordinarily high-level inhibition at concentrations above IC50 because m has an exponential relationship to drug effect (Shen et al., 2008; Jilek et al., 2012). The number of logs of inhibition of single round infection events produced by clinical concentrations of a drug or regimen is called the instantaneous inhibitory potential or IIP. Fig. 4D compares the dose-response curves against wild type virus of two drugs with the same IC50 but different values of the slope parameter and hence different IIPs. High slope values are the major factor in the high IIP values of the NNRTIs and PIs and likely explain the great clinical utility of these two classes (Shen et al., 2008; Jilek et al,, 2012). Most combination therapy regimens include an NNRTI or a PI (Panel on Antiretroviral Guidelines for Adults and Adolescents, 2009; Thompson et al., 2010). The highest IIP values are observed for the PIs, and the ability of some PI monotherapy regimens to suppress viremia in a substantial fraction of patients is likely a reflection of the fact that these drugs retain sufficient inhibitory potential against the single mutants to reduce R0 to below 1 (Fig. 1E).
The other situation in which the evolutionary dynamics of HIV-1 drug resistance are straightforward is the case of effective combination antiretroviral therapy. There is strong evidence that ongoing viral replication is essentially halted in adherent patients on modern regimens. Without replication, the evolution of resistance cannot proceed. Clinically, evidence that combination therapy can halt the evolution of resistance comes from the outcomes of patients who remain adherent to current highly active antiretroviral therapy (HAART) regimens. In these patients, treatment failure and resistance is unusual (Boyd, 2009). In most clinical trials, upwards of 80% of patients achieve and maintain suppression of viremia to clinically undetectable levels.
How then does failure of combination therapy occur? Most of the current thinking points to problems with adherence (Bangsberg et al., 2006; Paterson et al., 2000; Sethi et al., 2003). Suboptimal adherence leads to drug levels that are insufficient to reduce R0 to below 1. Additional replication allow the accumulation of mutations that reduce the fitness cost of preexisting resistance mutations and confer resistance to additional drugs, ultimately leading to a situation where the resistant virus has an R0 value >1 even in the presence of optimal drug concentrations. The development of resistance and treatment failure are associated with intermediate levels of adherence that allow replication and the selection of resistant variants.
When resistance develops, choice of salvage regimens can be guided by resistance testing (Panel on Antiretroviral Guidelines for Adults and Adolescents, 2009; Thompson et al., 2010). However, a recent study suggests that the pharmacodynamics of HIV-1 drug resistance may be considerably more complex than previously appreciated (Sampah et al., 2011). Current algorithms for determining the extent of resistance caused by particular mutations are based exclusively on analysis of changes in the IC50. This approach is based on the implicit assumption that resistance mutations simple shift the dose-response curve to the right without affecting the shape or slope of the dose response curve (Fig. 1A–C). However, most resistance mutations also affect the dose-response curve slope. Thus, as is shown in Fig. 1F, the extent of resistance may be underestimated if the critical variable is ignored.
Future directions for HIV-1 drugs
The introduction of new classes of antiretroviral drugs such as entry inhibitors and integrase inhibitors has had a major impact on HIV-1 treatment by allowing the construction of effective salvage regimens for patients who develop resistance to the RT and protease inhibitors used in initial treatment regimens. Rational drug design using knowledge about resistance to existing drugs has provided second generation RT, protease, and integrase inhibitors that are effective against resistant viruses. Improvements in regimen tolerability have led to a decrease in the incidence of resistance. While new drugs in existing classes continue to be developed and new classes of drugs are being explored, there is currently a critical need to extend existing treatment in resource limited settings and improve the monitoring of therapy, which is critical to avoiding resistance.
Concluding Remarks
What we ultimately need is to develop better combinations that can treat both drug-sensitive and drug-resistant organisms. For Tb, this means using new and existing compounds to provide shorter, simpler, and less toxic regimens. For malaria, this means coming up with an arsenal of new drugs for which resistance has not yet taken hold. For HIV-1, we have drugs that can stop virus replication and prevent the evolution of resistance in adherent patients. Until a curative therapy is found, it will be important to devise regimens that can be taken for the rest of the patient’s life, with high adherence and low long term toxicity, and that can be made accessible to all infected individuals. The rapid development of numerous drugs targeting different steps in HIV-1 the life cycle, which when used together can control the replication and prevent drug resistance in this rapidly evolving pathogen, is likely to be a model for drug development for all pathogens. The relative simplicity of the organism and ease with which it can be studied in vitro undoubtedly contributed to this success. However, HIV-1 has been the subject of an intense and well funded research effort. A similar focus on Tb and malaria might accelerate the development of new agents to treat these important pathogens.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute, and by NIH Grant AI081600 to RFS. Thanks to Brian Weinrick and Dhinakaran Sandbandan for their helpful Discussions and suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta-Hoyos AJ, Scott WA. The Role of Nucleotide Excision by Reverse Transcriptase in HIV Drug Resistance. Viruses. 2010;2:372–394. doi: 10.3390/v2020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad Z, Makaya NH, Grosset J. In: Antituberculosis Chemotherapy. Donald PR, Helden PD, Bolliger CT, Karger SAG, editors. Vol. 40. 2011. pp. 1–8. [Google Scholar]
- Almeida D, et al. Paradoxical effect of isoniazid on the activity of rifampin-pyrazinamide combination in a mouse model of tuberculosis. Antimicrob Agents Chemother. 2009;53:4178–4184. doi: 10.1128/AAC.00830-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TJ, Patel J, Ferdig MT. Gene copy number and malaria biology. Trends Parasitol. 2009;25:336–343. doi: 10.1016/j.pt.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JR, et al. Exogenous reinfection as a cause of multidrug-resistant and extensively drug-resistant tuberculosis in rural South Africa. J Infect Dis. 2008;198:1582–1589. doi: 10.1086/592991. [DOI] [PubMed] [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Göhlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Bangsberg DR, Acosta EP, Gupta R, Guzman D, Riley ED, Harrigan PR, Parkin N, Deeks SG. Adherence-resistance relationships for protease and non-nucleoside reverse transcriptase inhibitors explained by virological fitness. AIDS. 2006;20:223–231. doi: 10.1097/01.aids.0000199825.34241.49. [DOI] [PubMed] [Google Scholar]
- Bierman WF, van Agtmael MA, Nijhuis M, Danner SA, Boucher CA. HIV monotherapy with ritonavir-boosted protease inhibitors: a systematic review. AIDS. 2009;23:279–291. doi: 10.1097/QAD.0b013e32831c54e5. [DOI] [PubMed] [Google Scholar]
- BMRC. Streptomycin treatment of pulmonary tuberculosis. Br Med J. 1948;2:769–782. [PMC free article] [PubMed] [Google Scholar]
- BMRC. Treatment of pulmonary tuberculosis with streptomycin and para-aminosalicylic acid; a Medical Research Council investigation. Br Med J. 1950;2:1073–1085. [PMC free article] [PubMed] [Google Scholar]
- BMRC. Treatment of pulmonary tuberculosis with isoniazid; an interim report to the Medical Research Council by their Tuberculosis Chemotherapy Trials Committee. Br Med J. 1952;2:735–746. [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S, Nowak MA. Pre-existence and emergence of drug resistance in HIV-1 infection. Proc Biol Sci. 1997;264:631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousema T, Drakeley C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev. 2011;24:377–410. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd MA. Improvements in antiretroviral therapy outcomes over calendar time. Curr Opin HIV AIDS. 2009;4:194–199. doi: 10.1097/COH.0b013e328329fc8d. [DOI] [PubMed] [Google Scholar]
- Bray PG, Martin RE, Tilley L, Ward SA, Kirk K, Fidock DA. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol Microbiol. 2005;56:323–333. doi: 10.1111/j.1365-2958.2005.04556.x. [DOI] [PubMed] [Google Scholar]
- Clark S, Calef C, Mellors J. Mutations in Retroviral Genes Associated with Drug Resistance. In: Leitner T, Foley B, Hahn B, Marx P, McCutchan F, Mellors J, Wolinsky S, Korber B, editors. HIV Sequence Compendium. Los Alamos, NM: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 2007. pp. 58–158. [Google Scholar]
- Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Gange SJ, Kitahata MM, Saag MS, Justice AC, Hogg RS, Eron JJ, Brooks JT, Rourke SB, Gill MJ, et al. Trends in multidrug treatment failure and subsequent mortality among antiretroviral therapy-experienced patients with HIV infection in North America. Clin Infect Dis. 2009;49:1582–1590. doi: 10.1086/644768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries PJ, Dien TK. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs. 1996;52:818–836. doi: 10.2165/00003495-199652060-00004. [DOI] [PubMed] [Google Scholar]
- Dickinson JM, Aber VR, Mitchison DA. Bactericidal activity of streptomycin, isoniazid, rifampin, ethambutol, and pyrazinamide alone and in combination against Mycobacterium Tuberculosis. Am Rev Respir Dis. 1977;116:627–635. doi: 10.1164/arrd.1977.116.4.627. [DOI] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunner E, Brown WB, Wallace J. The effect of streptomycin with para-amino salicylic acid on the emergence of resistant strains of tubercle bacilli. Dis Chest. 1949;16:661–666. doi: 10.1378/chest.16.6.661. [DOI] [PubMed] [Google Scholar]
- EA/BMRC. Controlled clinical trial of short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis. Lancet. 1972;1:1079–1085. [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naudé B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flys TS, Donnell D, Mwatha A, Nakabiito C, Musoke P, Mmiro F, Jackson JB, Guay LA, Eshleman SH. Persistence of K103N-containing HIV-1 variants after single-dose nevirapine for prevention of HIV-1 mother-to-child transmission. J Infect Dis. 2007;195:711–715. doi: 10.1086/511433. [DOI] [PubMed] [Google Scholar]
- Ford CB, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi NR, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med. 2007;13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill WP, et al. A replication clock for Mycobacterium tuberculosis. Nat Med. 2009;15:211–214. doi: 10.1038/nm.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil O, et al. Granuloma encapsulation is a key factor for containing tuberculosis infection in minipigs. PLoS ONE. 2010;5:e10030. doi: 10.1371/journal.pone.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg A. Research Spotlight: The TB Alliance: overcoming challenges to chart the future course of TB drug development. Future Med Chem. 2011;3:1247–1252. doi: 10.4155/fmc.11.82. [DOI] [PubMed] [Google Scholar]
- Grosset J, Truffot-Pernot C, Lacroix C, Ji B. Antagonism between isoniazid and the combination pyrazinamide-rifampin against tuberculosis infection in mice. Antimicrob Agents Chemother. 1992;36:548–551. doi: 10.1128/aac.36.3.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guantai E, Chibale K. Chloroquine resistance: proposed mechanisms and countermeasures. Curr Drug Deliv. 2010;7:312–323. doi: 10.2174/156720110793360577. [DOI] [PubMed] [Google Scholar]
- Guay LA, Musoke P, Fleming T, Bagenda D, Allen M, Nakabiito C, Sherman J, Bakaki P, Ducar C, Deseyve M, et al. Intrapartum and neonatal single-dose nevirapine compared with zidovudine for prevention of mother-to-child transmission of HIV-1 in Kampala, Uganda: HIVNET 012 randomised trial. Lancet. 1999;354:795–802. doi: 10.1016/S0140-6736(99)80008-7. [DOI] [PubMed] [Google Scholar]
- Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, Chodakewitz JA, Fischl MA. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- Harrington WE, Mutabingwa TK, Muehlenbachs A, Sorensen B, Bolla MC, Fried M, Duffy PE. Competitive facilitation of drug-resistant Plasmodium falciparum malaria parasites in pregnant women who receive preventive treatment. Proc Natl Acad Sci U S A. 2009;106:9027–9032. doi: 10.1073/pnas.0901415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw HC, Feldman WH, Pfuetze KH. Treatment of tuberculosis with streptomycin; a summary of observations on one hundred cases. J Am Med Assoc. 1946;132:778–782. doi: 10.1001/jama.1946.02870480024007. [DOI] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Hyde JE. Drug-resistant malaria. Trends Parasitol. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istvan ES, Dharia NV, Bopp SE, Gluzman I, Winzeler EA, Goldberg DE. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2011;108:1627–32. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilek BL, Zarr M, Sampah M, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med. 2012 doi: 10.1038/nm.2649. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, et al. Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol. 2006;8:97–111. [PubMed] [Google Scholar]
- Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. Update of the drug resistance mutations in HIV-1: December 2010. Top HIV Med. 2010;18:156–163. [PubMed] [Google Scholar]
- Kaneko T, Cooper C, Mdluli K. Challenges and opportunities in developing novel drugs for TB. Future Med Chem. 2011;3:1373–1400. doi: 10.4155/fmc.11.115. [DOI] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenderink JB, Kavishe RA, Rijpma SR, Russel FG. The ABCs of multidrug resistance in malaria. Trends Parasitol. 2010;26:440–446. doi: 10.1016/j.pt.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Koul A, Vranckx L, Dendouga N, Balemans W, Van den Wyngaert I, Vergauwen K, Göhlmann HW, Willebrords R, Poncelet A, Guillemont J, Bald D, Andries K. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem. 2008;283:25273–2580. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci U S A. 2010;107:15910–15915. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larder BA, Darby G, Richman DD. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science. 1989;243:1731–1734. doi: 10.1126/science.2467383. [DOI] [PubMed] [Google Scholar]
- Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–1966. doi: 10.1056/NEJMoa062032. [DOI] [PubMed] [Google Scholar]
- Lehmann J. Para-aminosalicylic acid in the treatment of tuberculosis. Lancet. 1946;1:15. doi: 10.1016/s0140-6736(46)91185-3. [DOI] [PubMed] [Google Scholar]
- Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med. 2002;347:385–394. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- Little SJ, McLean AR, Spina CA, Richman DD, Havlir DV. Viral dynamics of acute HIV-1 infection. J Exp Med. 1999;190:841–850. doi: 10.1084/jem.190.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobritz MA, Ratcliff AN, Arts EJ. HIV entry, inhibitors, and resistance. Viruses. 2010;2:1069–1105. doi: 10.3390/v2051069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margeridon-Thermet S, Shafer RW. Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C. Viruses. 2011;2:2696–2739. doi: 10.3390/v2122696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Marchetti RV, Cowan AI, Howitt SM, Bröer S, Kirk K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- Mathys V, et al. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2009;53:2100–2109. doi: 10.1128/AAC.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune RM, Jr, McDermott W, Tompsett R. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J Exp Med. 1956;104:763–802. doi: 10.1084/jem.104.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune RM, Feldmann FM, Lambert HP, McDermott W. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J Exp Med. 1966;123:445–468. doi: 10.1084/jem.123.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune RM, Tompsett R. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. J Exp Med. 1956;104:737–762. doi: 10.1084/jem.104.5.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MMWR. Clinical update: impact of HIV protease inhibitors on the treatment of HIV-infected tuberculosis patients with rifampin. Morb Mortal Wkly Rep. 1996;45:921–925. [PubMed] [Google Scholar]
- Müller IB, Hyde JE. Antimalarial drugs: modes of action and mechanisms of parasite resistance. Future Microbiol. 2010;5:1857–1873. doi: 10.2217/fmb.10.136. [DOI] [PubMed] [Google Scholar]
- Nomura T, Carlton JM, Baird JK, del Portillo HA, Fryauff DJ, Rathore D, Fidock DA, Su X, Collins WE, McCutchan TF, Wootton JC, Wellems TE. Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J Infect Dis. 2001;183:1653–1661. doi: 10.1086/320707. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Lloyd AL, Vasquez GM, Wiltrout TA, Wahl LM, Bischofberger N, Williams J, Kinter A, Fauci AS, Hirsch VM, Lifson JD. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J Virol. 1997;71:7518–7525. doi: 10.1128/jvi.71.10.7518-7525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntemgwa ML, d’Aquin Toni T, Brenner BG, Camacho RJ, Wainberg MA. Antiretroviral drug resistance in Human Immunodeficiency Virus Type 2. Antimicrob Agents Chemother. 2009;53:3611–3619. doi: 10.1128/AAC.00154-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Jr, Dewar RL, et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol. 2005;43:406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services; 2009. pp. 1–161. Available at Http://www.Aidsinfo.Nih.gov/ContentFiles/AdultandAdolescentGL.Pdf. [Google Scholar]
- Paterson DL, Swindells S, Mohr J, Brester M, Vergis EN, Squier C, Wagener MM, Singh N. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann Intern Med. 2000;133:21–30. doi: 10.7326/0003-4819-133-1-200007040-00004. [DOI] [PubMed] [Google Scholar]
- Payne D. Spread of chloroquine resistance in Plasmodium falciparum. Parasitol Today. 1987;3:241–246. doi: 10.1016/0169-4758(87)90147-5. [DOI] [PubMed] [Google Scholar]
- Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2000;44:920–928. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichugin AV, Yan BS, Sloutsky A, Kobzik L, Kramnik I. Dominant role of the sst1 locus in pathogenesis of necrotizing lung granulomas during chronic tuberculosis infection and reactivation in genetically resistant hosts. Am J Pathol. 2009;174:2190–2201. doi: 10.2353/ajpath.2009.081075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiñones-Mateu ME, Arts EJ. In: HIV-1 fitness: implications for drug resistance, disease progression, and global epidemic evolution. Kuiken C, Foley B, Hahn B, Marx P, McCutchan F, Mellors JW, Wolinsky S, Korber B, editors. Los Alamos, NM: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 2001. pp. 134–170. [Google Scholar]
- Rathod PK. Antimalarial Agents Directed at Thymidylate Synthase. J Pharmacy and Pharmacol. 1997;49:65–69. [Google Scholar]
- Rocco F. The Miraculous Fever-Tree: The Cure that Changed the World Harper Collins. San Francisco: 2004. [Google Scholar]
- Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, Sharp B. Antifolate antimalarial resistance in southeast Africa: a population–based analysis. Lancet. 2003;361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy VK, et al. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med. 2002;8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- Sampah ME, Shen L, Jilek BL, Siliciano RF. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc Natl Acad Sci U S A. 2011;108:7613–7618. doi: 10.1073/pnas.1018360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Das K, Hughes SH, Arnold E. Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr Opin Struct Biol. 2004;14:716–730. doi: 10.1016/j.sbi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Schatz ABE, Waksman S. Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Proc Soc Exp Biol Med. 1944;55:66–69. doi: 10.1097/01.blo.0000175887.98112.fe. [DOI] [PubMed] [Google Scholar]
- Schuurman R, Nijhuis M, van Leeuwen R, Schipper P, de Jong D, Collis P, Danner SA, Mulder J, Loveday C, Christopherson C. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC) J Infect Dis. 1995;171:1411–1419. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- Sethi AK, Celentano DD, Gange SJ, Moore RD, Gallant JE. Association between adherence to antiretroviral therapy and human immunodeficiency virus drug resistance. Clin Infect Dis. 2003;37:1112–1118. doi: 10.1086/378301. [DOI] [PubMed] [Google Scholar]
- Shafer RW. Genotypic testing for human immunodeficiency virus type 1 drug resistance. Clin Microbiol Rev. 2002;15:247–277. doi: 10.1128/CMR.15.2.247-277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Rabi SA, Sedaghat AR, Shan L, Lai J, Xing S, Siliciano RF. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci Transl Med. 2011;3:91ra63. doi: 10.1126/scitranslmed.3002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavic K, Delves MJ, Prudêncio M, Talman AM, Straschil U, Derbyshire ET, Xu Z, Sinden RE, Mota MM, Morin C, Tewari R, Krishna S, Staines HM. Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite’s life cycle. Antimicrob Agents Chemother. 2011;55:2824–2830. doi: 10.1128/AAC.01739-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigelman MK. New tuberculosis therapeutics: a growing pipeline. J Infect Dis. 2007;196(Suppl 1):S28–34. doi: 10.1086/518663. [DOI] [PubMed] [Google Scholar]
- Sreevatsan S, et al. Ethambutol resistance in Mycobacterium tuberculosis: critical role of embB mutations. Antimicrob Agents Chemother. 1997;41:1677–1681. doi: 10.1128/aac.41.8.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Musuka S, Sherman C, Meek C, Leff R, Gumbo T. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201:1225–1231. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Sherman C, Meek C, Leff R, Gumbo T. Pharmacokinetic mismatch does not lead to emergence of isoniazid or rifampin-resistant Mycobacterium tuberculosis, but better antimicrobial effect: a new paradigm for anti-tuberculosis drug scheduling. Antimicrob Agents Chemother. 2011;55:5085–5089. doi: 10.1128/AAC.00269-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweany HC, Turner GC, et al. A preliminary report on the use of para-amino salicylic acid in the treatment of pulmonary tuberculosis. Dis Chest. 1949;16:633–660. doi: 10.1378/chest.16.6.633. [DOI] [PubMed] [Google Scholar]
- Takayama K, Kilburn JO. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob Agents Chemother. 1989;33:1493–1499. doi: 10.1128/aac.33.9.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuscher F, Gatton ML, Chen N, Peters J, Kyle DE, Cheng Q. Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J Infect Dis. 2010;202:1362–1368. doi: 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JP, Baughn CO, Wilkinson RG, Shepherd RG. A new synthetic compound with antituberculous activity in mice: ethambutol (dextro-2,2′-(ethylenediimino)-di-l-butanol) Am Rev Respir Dis. 1961;83:891–893. doi: 10.1164/arrd.1961.83.6.891. [DOI] [PubMed] [Google Scholar]
- Thompson MA, Aberg JA, Cahn P, Montaner JS, Rizzardini G, Telenti A, Gatell JM, Gunthard HF, Hammer SM, Hirsch MS, et al. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society-USA panel. JAMA. 2010;304:321–333. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- Tu Y. The discovery of artemesinin (qinghaosu) and gifts from Chinese medicine. Nature Med. 2011;17:1217–1220. doi: 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- Vaidya AB, Mather MW. Atovaquone resistance in malaria parasites. Drug Resist Updat. 2000;3:283–287. doi: 10.1054/drup.2000.0157. [DOI] [PubMed] [Google Scholar]
- Vilchèze C, et al. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- Vilchèze C, Jacobs J, William R. The Mechanism of Isoniazid Killing: Clarity Through the Scope of Genetics. Ann Rev Micro. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- Vinayak S, Alam MT, Mixson-Hayden T, McCollum AM, Sern R, Shah NK, Lim P, Muth S, Rogers WO, Fandeur T, et al. Origin and evolution of sulfadoxine resistant Plasmodium falciparum. PLoS Pathog. 2010;6:e1000830. doi: 10.1371/journal.ppat.1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- Wensing AM, van Maarseveen NM, Nijhuis M. Fifteen years of HIV Protease Inhibitors: raising the barrier to resistance. Antiviral Res. 2010;85:59–74. doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Wheeler WH, Ziebell RA, Zabina H, Pieniazek D, Prejean J, Bodnar UR, Mahle KC, Heneine W, Johnson JA, Hall HI Variant, Atypical, and Resistant HIV Surveillance Group. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, U.S.-2006. AIDS. 2010;24:1203–1212. doi: 10.1097/QAD.0b013e3283388742. [DOI] [PubMed] [Google Scholar]
- White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Multidrug and extensively drug-resistant tuberculosis: 2010 global report on surveillance and response. World Health Organization; Geneva, Switzerland: 2010. [Google Scholar]
- Wodarz D, Nowak MA. Mathematical models of HIV pathogenesis and treatment. Bioessays. 2002;24:1178–1187. doi: 10.1002/bies.10196. [DOI] [PubMed] [Google Scholar]
- Yeager RL, Munroe WG, Dessau FI. Pyrazinamide (aldinamide) in the treatment of pulmonary tuberculosis. Am Rev Tuberc. 1952;65:523–546. [PubMed] [Google Scholar]
- Zhang M, et al. Treatment of Tuberculosis with Rifamycin-containing Regimens in Immune-deficient Mice. Am J Respir Crit Care Med. 2011;183:1254–1261. doi: 10.1164/rccm.201012-1949OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimhony O, Cox JS, Welch JT, Vilchèze C, Jacobs WR., Jr Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med. 2000;6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]