

Abstract
The HIV-1 Rev protein plays a key role in the late phase of virus replication. It binds to the Rev Response Element found in underspliced HIV mRNAs, and drives their nuclear export by the CRM1 receptor pathway. Moreover, mounting evidence suggests that Rev has additional functions in viral replication. Here we employed proteomics and statistical analysis to identify candidate host cell factors that interact with Rev. For this we studied Rev complexes assembled in vitro with nuclear or cytosolic extracts under conditions emulating various intracellular environments of Rev. We ranked the protein-protein interactions by combining several statistical features derived from pairwise comparison of conditions in which the abundance of the binding partners changed. As a validation set, we selected the eight DEAD/H box proteins of the RNA helicase family from the top-ranking 5% of the proteins. These proteins all associate with ectopically expressed Rev in immunoprecipitates of cultured cells. From gene knockdown approaches, our work in combination with previous studies indicates that six of the eight DEAD/H proteins are linked to HIV production in our cell model. In a more detailed analysis of infected cells where either DDX3X, DDX5, DDX17, or DDX21 was silenced, we observed distinctive phenotypes for multiple replication features, variously involving virus particle release, the levels of unspliced and spliced HIV mRNAs, and the nuclear and cytoplasmic concentrations of these transcripts. Altogether the work indicates that our top-scoring data set is enriched in Rev-interacting proteins relevant to HIV replication. Our more detailed analysis of several Rev-interacting DEAD proteins suggests a complex set of functions for the helicases in regulation of HIV mRNAs. The strategy used here for identifying Rev interaction partners should prove effective for analyzing other viral and cellular proteins.
HIV-1 utilizes many host cell factors for its replication (1–3), similar to other viruses. There is strong interest in identifying and understanding these components to shed light on the molecular mechanisms of virus replication. Moreover, this can provide the potential for developing new therapeutics. The HIV Rev protein is a key regulator of viral replication that is critical for the late stages of virus replication (4, 5). The best-characterized function of Rev involves its potent stimulation of the nuclear export of unspliced and singly spliced (“underspliced”) HIV transcripts that encode the viral structural proteins and accessory factors (5). In the absence of Rev, these transcripts are retained in the nucleus because of their incomplete splicing. At the molecular level, Rev binds and oligomerizes along the 351-nt Rev Response Element (RRE)1 (6) in the env gene that is present in all underspliced HIV transcripts. Rev contains a classical leucine-rich nuclear export sequence that recruits CRM1, a transport receptor of the karyopherin family (7, 8). CRM1 commonly is used for nuclear export of cellular proteins, and only infrequently is involved in cellular mRNA export (9, 10). Upon binding to the RRE together with the GTP-bound form of Ran, CRM1 forms the core of an export complex that directs the transport of underspliced transcripts through the nuclear pore complex to the cytoplasm (5). Rev has been reported to promote additional functions in HIV infection besides mRNA export, including translation of underspliced HIV mRNAs (11–13) and genome encapsidation (14). The molecular mechanisms of these other functions and the identity of relevant host cell proteins remain unknown.
A number of potential Rev cofactors in addition to CRM1 and Ran have been reported, including certain members of the DEAD/H box RNA helicase family (15, 16). DEAD/H box proteins are members of a major subgroup of RNA helicases in eukaryotes (17, 18). They are modular, multidomain proteins that contain a conserved central RecA-like domain involved in ligand recognition and ATP hydrolysis, and nonconserved N- and C-terminal domains involved in helicase targeting and regulation. They have been linked to virtually all steps of gene expression, from the initial transcription to mRNA processing, turnover, translation, and intracellular trafficking. They can have many functions in addition to duplex RNA unwinding, including protein displacement, RNA folding and ribonucleoprotein remodeling (17, 18). DEAD/H helicases commonly carry out their functions in concert with additional cofactors that promote helicase targeting and activity.
To contribute to a more comprehensive understanding of Rev regulation and functions, we have carried out a proteomics screen to identify host cell proteins that physically associate with Rev. Proteins identified by the proteomics approach were analyzed by statistical methods to generate a ranked list of binding proteins, which reflected the abundance and binding specificity of hits. We chose the eight DEAD/H box proteins present in the top 5% of the scores as a validation set. From RNAi analysis in cultured cells, our work in combination with previous studies established that six of the eight DEAD/H proteins in the validation set are linked to HIV production. In a more detailed phenotypic analysis of four of the validated helicases, we found that the knockdown of each protein influences multiple aspects of HIV production in distinctive ways. This suggests considerable functional complexity for these helicases in viral production. Based on the results with our validation set, we conclude that our top-scoring data set is enriched in Rev-interacting proteins relevant to HIV replication. This information is expected to be a valuable resource for directing future studies of Rev functions.
EXPERIMENTAL PROCEDURES
Isolation of Rev-binding Proteins from Cell Extracts
HIV-1 Rev (accession code P04620) was cloned into the pMAL-c2X bacterial expression vector (New England Biolabs, Ipswich, MA), yielding a fusion protein with Rev attached to the C terminus of maltose binding protein (MBP) separated by a 24-amino acid linker. The resulting MBP-Rev was expressed in E. coli BL21 cells by inducing with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. Cells were collected and resuspended in 20 mm Tris pH 8.0, 500 mm NaCl, 1 mm MgCl2, 1 mm dithiotreitol (DTT), 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, aprotinin, and pepstatin, 1 mg/ml lysozyme and 10 μg/ml DNase I and frozen at −20 °C. All subsequent steps were performed at 4 °C. Thawed cells were centrifuged at 30,000 × g and the supernatant was incubated with 10 ml amylose resin (New England Biolabs) per liter of original culture, and equilibrated with Column Buffer (20 mm Tris pH 8.0, 500 mm NaCl, 1 mm MgCl2, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, aprotinin, and pepstatin). The resin was then packed into a column, washed with Column Buffer, and the MBP-Rev was eluted with Column Buffer containing 10 mm maltose. Next, the MBP-Rev solution was adjusted to 1 m NaCl using a stock, and the nucleic acids were removed by precipitation with 0.5% poly(ethylenimine) and centrifugation. MBP-Rev in the supernatant was precipitated by adding ammonium sulfate to 75% saturation. The resulting precipitate was collected by centrifugation and dissolved in SP-Sepharose Buffer (50 mm Hepes pH 7.4, 400 mm NaCl, 1 mm DTT) and applied to an SP-Sepharose column equilibrated with SP-Sepharose Buffer. MBP-Rev was eluted with a salt gradient from 0.4 to 2 m NaCl. Finally, MBP-Rev was dialyzed against 50 mm Hepes pH 7.4, 1 m NaCl, 1 mm DTT, concentrated to 100 μm and stored at −80 °C. The resulting protein was greater than 99% pure as judged by SDS-PAGE and Coomassie Blue staining (supplemental Fig. S1), and its absorbance values at 254 nm versus 280 nm indicated no nucleic acid contamination. RanQ69L was expressed, purified, and loaded with GTP as described (19) and stored as a 125 μm stock at −80 °C. Purified MBP was purchased from New England Biolabs. The 351 nucleotide long RRE was transcribed in vitro and purified as described earlier (20), and stored at −20 °C at 50 μm concentration. RRE was annealed directly before use as described earlier (20). Cytosolic extracts of HeLa cells, grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, were prepared as described (21). Nuclear HeLa extracts were prepared from 3 × 109 cells as described (22), yielding 11 ml of extract containing ∼11 mg/ml protein and ∼1 mg/ml RNA.
The general scheme for identification of Rev-binding proteins involved incubation of cytosolic or nuclear extracts with either MBP-Rev or MBP (control) in the presence or absence of RRE, and with or without RanQ69L-GTP. Where indicated, cytosolic extracts were pretreated with 0.1 mg/ml RNase A at RT for 1 h. Binding experiments were done in 1 ml final volume in Transport Buffer (20 mm HEPES, 110 mm KOAc, 2 mm Mg(OAc)2, 2 mm DTT, 1 μg/ml leupeptin, aprotinin, and pepstatin; (21)) containing either 0.8 mg total protein from cytosolic extracts or 1.2 mg protein from nuclear extracts. MBP or MBP-Rev was added to 1 μm final concentration, and 0.1 μm RRE was added to specified samples together with 100 units of RNaseIN (Promega, Madison, WI). RanQ69L-GTP was added to the designated samples at 10 μm final concentration. Samples were incubated with 20 μl of amylose resin pre-equilibrated in Transport Buffer for 2 h at 4 °C. The resin was washed 3 times with 1 ml Transport Buffer, and specifically bound complexes were eluted with three consecutive 3-min incubations in 0.1 ml Transport Buffer containing 10 mm maltose. Pooled eluted samples were centrifuged to remove residual resin, and were analyzed by MudPIT (Multidimensional Protein Identification Technology) mass spectrometry.
MudPIT Proteomic Analysis
The method was that described (23) with the modifications noted below. Proteins eluted from amylose resin were precipitated with 20% trichloroacetic acid overnight on ice followed by centrifugation. Pellets were washed two times with acetone, placed in an evaporator for 5 min, and resuspended in 30 μl of 100 mm Tris-HCl (pH 8.5). Solid urea was added to 8 m. After reduction (5 mm Tris(2-carboxyethyl)phosphine hydrochloride, Roche) and alkylation (20 mm iodoacetamide, Sigma), Endoproteinase Lys-C (Roche) was added to a 1:100 enzyme/substrate ratio, and samples were incubated overnight at 37 °C. After 4 × dilution with 100 mm Tris-HCl (pH 8.5) and addition of CaCl2 to 2 mm, proteins were further digested using modified Trypsin (Roche) at a 1:100 enzyme/substrate ratio and incubation at 37 °C overnight. Digested peptide mixtures were pressure loaded onto a 100-μm inner diameter, fused-silica column packed first with 8–9 cm of 5-μm C18 reverse phase particles (Polaris 2000, Metachem Technologies) followed by 4–5 cm of 5 μm-strong cation exchange material (Partisphere SCX, Whatman). Loaded microcapillary columns were installed in-line with a Quaternary Agilent 1100 series HPLC pump. An overflow tubing was used to decrease the flow rate to <200–300 nl/min. The application of a 2.4-V distal voltage electrosprayed the eluting peptides directly into an LCQ-Deca ion trap mass spectrometer equipped with a nano-LC electrospray ionization source (ThermoFinnigan). Three different elution buffers were used as follows: 5% acetonitrile, 0.1% formic acid (Buffer A); 80% acetonitrile, 0.1% formic acid (Buffer B); and 500 mm ammonium acetate, 5% acetonitrile, 0.1% formic acid (Buffer C). Fully automated 6-step chromatography runs were carried out. In such sequences of chromatographic events, peptides were sequentially eluted from the strong cation exchange resin to the reverse phase resin by increasing salt steps (increase in Buffer C concentration), followed by organic gradients (increase in Buffer B concentration). The last chromatography step consisted of a high-salt wash with 100% Buffer C, followed by the acetonitrile gradient. Full MS spectra were recorded on the peptides over a 400–1600 m/z range, followed by three tandem mass (MS/MS) events sequentially generated in a data-dependent manner on the first, second, and third most intense ions selected from the full MS spectrum (at 35% collision energy).
Raw data were extracted to the MS2 file format using an in house algorithm (Xtract_RAW version 1.0) that does not alter raw mass spectra (24). PEP_PROBE (contained within SEQUEST version 3.0) (25) was used to match MS/MS spectra to peptides in the RefSeq NCBI human database (downloaded 8/14/06) plus common contaminants (such as trypsin, keratins, and IgG sequences) and the HIV-1 Rev protein sequence. A reverse version of the entire database was concatenated with the forward sequences for a total of 78,983 sequences searched against by PEP_PROBE. PEP_ PROBE is a modified version of SEQUEST (26) and uses a hypergeometric probability model to calculate the confidence for a match to be nonrandom. No enzyme specificity was used for the database search. Carbamidomethyl cysteine was considered as a fixed modification and no variable modifications were considered. A ± 1.5 Da mass tolerance was used for parent ion matches and a 1 Da mass tolerance was used for fragment ion matches in the search algorithm. The validity of peptide/spectrum matches was therefore assessed using the SEQUEST-defined parameters, cross-correlation score (XCorr), and normalized difference in cross-correlation scores (DeltaCn), as well as the PEP_PROBE-defined parameters, probability, and confidence for a match to be nonrandom. Spectra/peptide matches were only retained if they had a DeltaCn of at least 0.08 and minimum XCorr of 1.8 for +1, 2.5 for +2, and 3.5 for +3 spectra. An 85% confidence for the peptide/spectrum matches not to be random (as defined by PEP_PROBE) was used as cut-off. Peptides that were not fully tryptic were removed. In addition, the minimum sequence length was seven amino acid residues. False positives rates were estimated by the number of peptide/spectrum matches to reverse protein sequences. The false positive rate for proteins identified in all analyses was below 5%. DTASelect (27) was used to select and sort peptide/spectrum matches passing this criteria set and to group peptides identified by proteins from which they were derived. DTASelect was run such that peptides mapping to multiple proteins will be listed for all proteins containing that sequence, however these peptides were denoted as not unique in the DTASelect output. Peptide hits from multiple runs were compared using CONTRAST (27). Proteins were considered detected in the samples if they were identified by at least two peptides passing all of the selection criteria.
Scoring System and Computational Methods
Our strategy to identify biologically relevant interaction partners of Rev is based on the notion that many relevant partners will have increased or decreased binding to Rev in a comparison of conditions that emulate different environments of Rev in cells (i.e. without or with the RRE or RanGTP). Four parameters entered into our scoring system to assess the significance of the protein “hits” (proteins identified in the proteomics). The first parameter was the hit abundance in each sample. This was described by spectral counts, which is related to the quantity of protein in label-free “shotgun” mass spectrometry (28). To allow sample-to-sample comparisons, we normalized the spectral counts of each hit to the number of spectral counts derived from MBP in the same sample. The second feature of our scoring system assessed background significance, and was obtained by dividing the sum of hit abundances in samples involving MBP-Rev by the sum of abundances in the MBP control samples. The zero-division problem was avoided by setting the control hit abundance to the value corresponding to an empirically determined background level. The third feature, the fold-change, was defined as the ratio of hit abundance in pairs of samples involving the addition of RanGTP or RRE. The zero-division problem was avoided as above. The fourth feature, the enrichment significance, measured the statistical significance of the fold-change induced by RRE or RanGTP. It was defined as a negative logarithm of the two-tailed p value estimated by the χ2-test (29). Next, to combine the four features into a single composite score, we used the first principal component scores from standard principal component analysis (PCA), as implemented in the MDP toolkit (30). PCA is a mathematical procedure, frequently used in dimension reduction tasks, that transforms the feature space and identifies the first principal component of the different features (e.g. abundance, background significance, etc.) that maximizes the variance of the data (i.e. accounts for as much of the variability in the data as possible). The new scores (the first principal component scores) thus become a linear combination of original features, each one multiplied by a weight that corresponds to the loadings of the first principal component. In our case, the weights of the four features were 0.18, 0.24, 0.26, and 0.32 for abundance, background significance, fold-change, and enrichment significance, respectively. Finally, 250 of the highest-ranking hits from the experiments comparing the effects of RanGTP and RRE were chosen for further analyses. GO-term enrichments for top ranking bait-prey pairs were calculated using AmiGO Term Enrichment tool version 1.8 (31). The graphic presentation was generated with Cytoscape (32).
Association of DEAD Box Proteins with Rev by Immunoprecipitation
HeLa cells were seeded ∼16 h prior to cDNA transfection at a density of 106 cells/10 cm dish. The Rev expression plasmid (pRev-V5) was cotransfected with pGagRRE or pGagCTE using Lipofectamine 2000 (Invitrogen) and cells were grown for 24 h. After washing with phosphate-buffered saline, transfected cells (2–3 × 106) were lysed in 750 μl of IP Buffer [50 mm Tris (pH 7.4), 75 mm NaCl, 1 mm MgCl2, 1% Nonidet P-40, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride and protease inhibitor mixture (Roche Diagnostics)]. Where indicated, cell lysates were treated with 67 units/ml RNase A (Affymetrix) for 15 min at room temperature. Lysates were centrifuged at 10,000 × g at 4 °C, and an aliquot of clarified supernatant was reserved as “lysates.” The remainder was incubated for 1 h at 4 °C with either anti-V5 antibodies (Invitrogen, Carlsbad, CA) or control mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Samples were then incubated with protein G-agarose Dynabeads (Invitrogen) for 30 min, the beads were washed, and bound proteins were eluted in SDS sample buffer. Samples were analyzed by SDS-PAGE and Western blotting (see below section for details). DEAD box proteins were detected with mouse anti-DDX3 (Abcam), rabbit anti-DDX5 (Aviva Systems Biology), rabbit anti-DDX17 (Novus Biologicals, Littleton, CO), rabbit anti-DDX21 (Aviva Systems Biology), rabbit anti-DHX36 (Bethyl Laboratories, Montgomery, TX), rabbit anti-DDX47 (Aviva Systems Biology), rabbit anti-hnRNP D (Aviva Systems Biology), and rabbit anti-hnRNP A0 (Abcam, Cambridge, MA).
HIV-1 Production and Infectivity Assays
For the HIV production assay, HeLa CD4+ TZM-bl cells (Catalog # 8129 AIDS Reference Reagent Program, NIH) were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. Silencing of helicases was accomplished by two sequential transfections with siRNA to enhance knockdown efficiency. First, TZM-bl cells were plated in six-well plates (1.7 × 105 cells/well) with 20 nm siRNA and RNAiMAX (Invitrogen). After 48 h, the cells were transfected with 20 nm siRNA with Lipofectamine 2000 (Invitrogen). After another 24 h, the cells were challenged with 50 ng HIV-1 using an NL4.3 derivative clone R9 (kind gift P. Gallay TSRI La Jolla) (33). After 18 h, cells were washed with PBS and fresh medium was added. The complete removal of free virus particles was verified by immediate testing for the presence of HIV-1 capsid (p24) in the medium by ELISA (Alliance HIV-1 p24 Antigen ELISA kit, Perkin Elmer). At 48 h postinfection, the culture medium and cells were harvested. Virus production was determined by measuring p24 levels in the medium by ELISA. The p24 production shown represents triplicate samples involving three separate experiments. Aliquots of the virus-containing medium were stored at −80 °C prior to evaluation of infectivity (below). The infected cells were trypsinized, divided into two halves and pelleted. One set of the cells was resuspended with 50 μl SDS sample buffer and used for Western blot analysis, and the other was used for subcellular fractionation (below). We tested 3–5 different Stealth siRNA oligonucleotides (Invitrogen) for each DEAD box target, and used GC content-matched control siRNAs.
The knockdown efficiency for each DEAD/H protein was determined by Western blot analysis of the cell sample taken at the end of the infection assay. Western blotting involved use of HRP-conjugated secondary antibodies. Chemiluminescent detection involved use of SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and exposure of x-ray film, followed by quantification with ImageG software. We made a standard curve for the signal obtained with decreasing amounts of the si-Ctrl sample (100%, 50%, 25%, 12.5%, 6.25%), and evaluated the degree of silencing by quantitatively comparing the Western blot signal of each si-DDX sample to the si-Ctrl curve. For determining Gag and Env expression (Fig. 4) we used mouse anti-p24 (Chemicon, Temecula, CA) and anti-gp120 (kind gift of Dennis Burton, TSRI, La Jolla, CA). Gag and Env protein expression (Fig. 3) and the selectivity of DDX silencing by the siRNAs (supplemental Fig. S4) were estimated by densitometric scanning of Western blots, with internal normalization of samples to α-tubulin.
Fig. 4.

Effects of DDX silencing on HIV-1 production and infectivity. TZM-bl cells were treated with control siRNA (Ctrl), or with each of two separate siRNAs for each target (labeled A and B). In the case of DDX3X, treatment involved a siRNA pool. Samples then were infected with HIV-1 clone R9 (NL4.3). At 48 h post-infection, samples were harvested and analyzed. Data shown is derived from 3 independent experiments, with triplicate samples for each siRNA. Error bars represent standard error. A, Virus production measured by p24 release into the culture medium. The p24 production was normalized to cell numbers and is scored as a percentage relative to cells treated with control siRNA. B, Gag and Env levels in infected cells. Lysates of infected cells were analyzed by Western blotting to detect Gag, Env and α-tubulin. Values of Gag and Env were normalized to α-tubulin, and are expressed as the percentage of expression relative to control siRNA treated cells. C, Infectivity of virus released into the medium of DDX-silenced cells. Samples of culture supernatant from cells treated with control siRNAs were normalized for p24 content and used to infect TZM-bl cells. Virus infectivity was measured by a luciferase assay 24 h after infection, and is scored as a percentage relative to control siRNA treated cells.
Fig. 3.
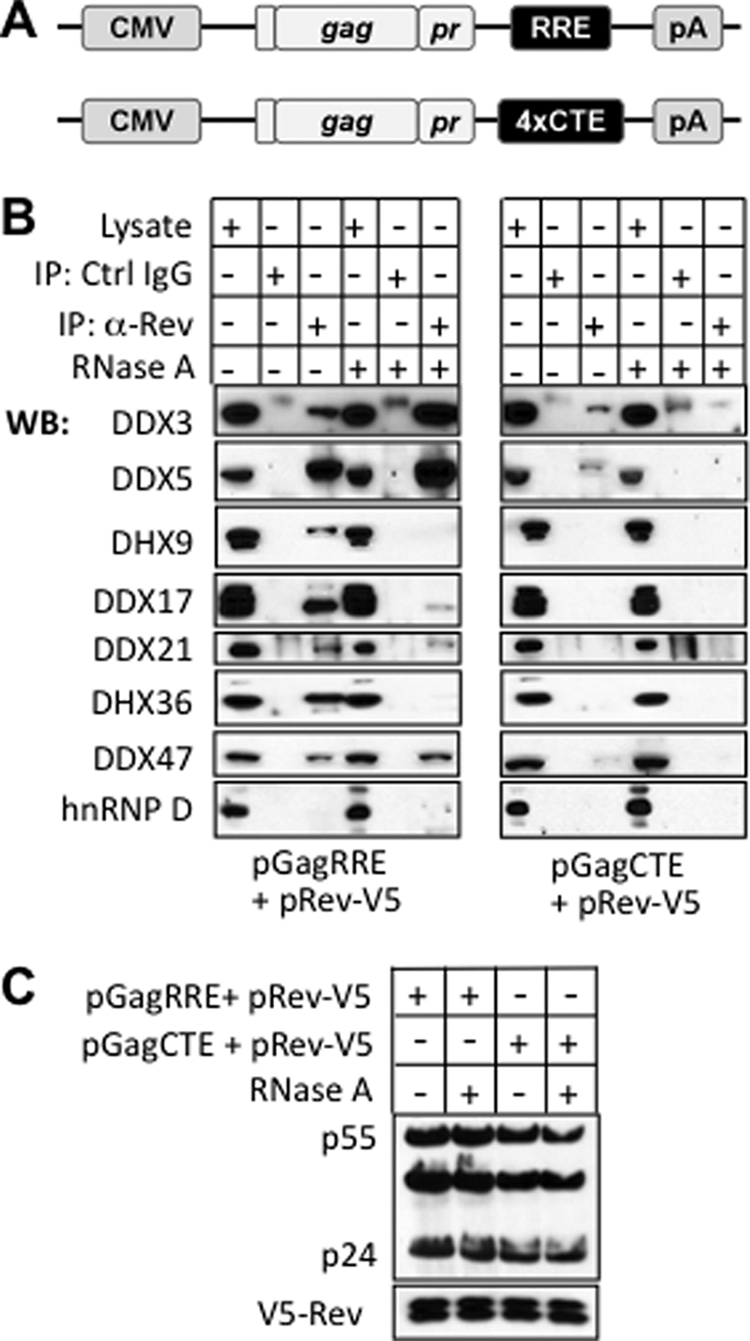
Association of DEAD/H proteins with Rev by co-immunoprecipitation. A, Schematic representation of the pGagRRE and pGagCTE constructs used in the analysis. pGagRRE contains a fragment of the the env gene with the 351 nt RRE, whereas pGagCTE instead contains four copies of the constitutive transport element (CTE). B, Western blot analysis to detect the indicated DEAD/H helicases in whole cell lysates of transfected cells and in immunoprecipitates derived from these with anti-V5 antibody or with control IgG. Cells were transfected with pRev-V5 and one of the two plasmids described in panel A. Where indicated, cell lysates were pretreated with RNase A. Fifteen sample equivalents were loaded on the immunoprecipitate lanes, as compared with one equivalent for the input lanes (C) Western blot analysis of whole cell lysate with anti-p24 and anti-V5.
To evaluate the infectivity of culture supernatants obtained from siRNA treated cells, single-round infections were carried out with CD4+ TZM-bl cells, by seeding 100,000 cells/well onto 24-well plates 24 h prior to infection. Cells were challenged in quadruplicate with culture supernatants from virus production assays (above). Equivalent viral challenges were achieved by normalization of p24 content. Infectivity was measured 24 h after challenge using an enzymatic β galactosidase activity assay with Galacton-Star substrate (Applied Biosystems) for luminometric detection by a microplate reader. Infectivity shown represents triplicate samples from three independent experiments.
Subcellular Fractionation and qRT-PCR
Preparation of nuclear and cytosolic fractions was performed as described (34) with the following modifications. Washed cells (∼ 106) were lysed with 10 up and down excursions in a micropipette in 100 μl lysis buffer [10 mm Tris (pH 8.4), 140 mm NaCl, 1.5 mm MgCl2, 0.5% Nonidet P-40, 1 mm dithiothreitol, and RNasIN (100 units/ml)]. The lysate was centrifuged at 1000 × g for 10 min, yielding the cytosolic (supernatant) and the nuclear (pellet) fractions Total mRNA in the nuclear and cytosolic fractions were isolated, reverse-transcribed to cDNA and quantified using the Power SYBR Green Cells-to-Ct Kit (Applied Biosystems). qPCR was performed to quantitatively analyze the levels of HIV-1 unspliced and spliced mRNA, as well as the levels of cellular HPRT and pre-GAPDH mRNA, using the primers denoted in supplemental Table S5. The qPCR involved incubation at 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, 55 °C for 15 s and 60 °C for 15 s using the 7900 HT Fast Real-Time PCR System (Applied Biosystems). The specificity of PCR product was examined by a dissociation curve, and results were analyzed by the 2−ΔΔCT method (35). Values were normalized to pre-GAPDH.
RESULTS
Identification of Rev Interacting Host Cell Proteins by Comparative Proteomics and Statistical Analysis
Our strategy for identifying candidate host cell factors involved in Rev functions is depicted in Fig. 1A. We assembled Rev complexes in vitro by incubating nuclear or cytosolic extracts of HeLa cells with recombinant maltose binding protein fused to Rev (MBP-Rev; supplemental Fig. S1). This was done with a range of conditions emulating different intracellular environments of Rev, where the association of functionally pertinent binding partners was predicted to be different. The purified complexes then were analyzed by MudPIT proteomics (23). For nonspecific background determination, we carried out a parallel analysis of samples containing MBP instead of MBP-Rev. We then ranked the statistical significance of bound proteins by computational methods.
Fig. 1.

Proteomic analysis of Rev/RRE interacting host cell proteins. A, Flow diagram depicting the proteomic and statistical analyses. Cytosolic or nuclear extracts of HeLa cells were incubated with either MBP-Rev or MBP (control) in the presence or absence of RRE and/or RanQ69LGTP (denoted RanGTP hereafter). Certain experiments (not depicted) involved pretreatment of cytosolic extract without or with RNase prior to incubation with MBP-Rev or MBP. The complexes were adsorbed to amylose resin and eluted with maltose. Eluates were analyzed by MudPIT proteomics. Hits were scored by PCA and the list of the 250 top-scoring proteins was assembled. B, Western blot analysis of samples bound to MBP-Rev from cytosolic extracts. Samples were incubated with or without RanQ69L-GTP or RRE as indicated. C, D, GO term enrichments for proteins in the top 250 hits where the interactions with Rev were altered by the addition of RanGTP (C) or RRE (D). Only GO terms with an increased relative enrichment over the proteome are displayed. For simplicity, actins, tubulins, ribosomal proteins and histones were omitted from the graphs. Proteins lacking GO terms (such as predicted proteins) also were omitted. The size of a node reflects the number of proteins with that specific GO term (also indicated in parentheses) and the width of the lines connecting the various nodes reflects the number of proteins annotated with both respective GO terms at both ends of the lines, as designated by the scale.
As physiologically relevant conditions to modulate protein binding to Rev, we tested the effects of the 351 nucleotide long RRE (6) transcribed in vitro (∼0.1 mol RRE/mol MBP-Rev), and a recombinant GTP hydrolysis resistant form of the small GTPase Ran (RanQ69L) (7) loaded with GTP (hereafter termed RanGTP; Fig. 1A). We also tested the effect of RNase pretreatment of cytosolic extracts. Because the RRE serves as a platform for Rev oligomerization (5, 36), proteins that preferentially bind to the Rev/RRE complex as compared with Rev alone might sense features of Rev that occur in its oligomeric RNA-bound form and/or specific structural features of the RRE. RanGTP binds to karyopherins in the nucleus to dissociate cargo from import receptors and to promote cargo binding to export receptors (7). Thus, proteins whose binding to Rev is affected by the addition of hydrolysis-resistant RanGTP might be components of nuclear import or export complexes involving Rev or Rev binding partners. To validate the activity of exogenous MBP-Rev, RanGTP, and RRE, we examined the effects of RanGTP and RRE on the binding of major nuclear transport receptors to MBP-Rev in cytosolic extracts (Fig. 1B). As expected, the addition of RanGTP promoted the binding of CRM1 to Rev in the absence or presence of RRE. Moreover both RanGTP and RRE caused dissociation of Rev from the import receptor importin β, consistent with the functions of RanGTP and with the overlap of the nuclear localization signal of Rev with the arginine rich RRE-binding motif of the latter (37). Finally, we found that recombinant MBP-Rev was active as a nuclear import substrate in vitro (supplemental Fig. S1).
Our proteomic analysis revealed 3161 unique proteins (supplemental Table S1). Following normalization of the spectral counts in individual samples to the MBP peptide counts of the bait, we compared each of the Rev-associating proteins in six sample pairs where the only variable between the two sample conditions was the absence or presence of either RRE or RanGTP, and one sample pair where the only variable was preincubation without or with RNase. We used four metrics to statistically rank the hits: (1) relative protein abundance (normalized spectral counts), (2) fold difference of spectral counts in the MBP-Rev samples as compared with the MBP samples, (3) fold change of spectral counts of a specific protein in response to Ran or RRE addition, or to RNase pretreatment, and (4) the statistical significance (two-tailed p value) of metric 3 as determined by the χ2 test. The score of an interacting pair is a weighted sum of the normalized four metrics as determined by PCA. Thus the proteins with the highest scores are ones that are abundant in the samples, have a specific interaction with MBP-Rev as compared with MBP, and have a Rev association that dramatically changes in the presence of RRE and/or RanGTP, or with RNase pretreatment.
Most of the Rev-associating hits from pairwise comparisons yielded low scores that were not deemed to be significant by our metric (supplemental Fig. S2). We assembled a list of the highest scoring 250 Rev interacting partners for more detailed consideration (hereafter termed “top-ranking list”; supplemental Table S2). These were above the PCA cutoff of 0.0368 and represented 5% of the total pairwise comparisons. The 250 proteins in supplemental Table S2 result in 435 entries because some proteins occur as multiple isoforms, and because certain binding partners have a high PCA score for more than one pairwise comparison. After excluding the minor number of predicted but unverified proteins from the data set, we functionally annotated the remainder using the Gene Ontology database (supplemental Table S3), and determined the enrichment of GO terms (31) among these proteins (Figs. 1C and 1D). As expected, the 110 hits whose interaction with Rev was strongly affected by RanGTP (Fig. 1C) included a set of proteins with nucleocytoplasmic transport functions. Compared with the distribution of GO terms across whole human proteome, nucleocytoplasmic transport was statistically significantly enriched in this group (p value of 7.8 × 10−15). However, the largest group of proteins affected by RanGTP was linked to RNA binding, and this was statistically very enriched (p value of 3.0 × 10−18). This could reflect the fact that many RNA-binding proteins shuttle between the nucleus and the cytoplasm, and are part of RanGTP-sensitive import or export complexes. Proteins related to translation and with GTPase activity also were affected by RanGTP more than proteins with other GO functions (p values of 3.1 × 10−8 and 5.1 × 10−11, respectively).
Among the 150 proteins whose binding to the Rev complex was strongly influenced by the addition of RRE, RNA binding proteins were by far the largest group (p value of 1.4 × 10−69) (Fig. 1D), although proteins involved in transcription, RNA splicing and translation also were included (p values of 3.8 × 10−2, 1.2 × 10−57, and 1.3 × 10−7, respectively). Many of these interactions undoubtedly were mediated directly through the RRE. Also, some of the interactions could be because of nonspecific RRE binding despite the 10-fold excess of endogenous RNA over RRE in the samples, which was expected to strongly diminish nonspecific interactions. Smaller groups of proteins that were strongly enriched in the presence of RRE were proteins with ATPase and helicase activity (p values of 2.2 × 10−4 and 3.8 × 10−7, respectively), as were proteins linked to chromosome organization, cell cycle, and nucleocytoplasmic transport (p values of 6.2 × 10−9 6.0 × 10−2, and 1.6 × 10−6, respectively). The RRE related GO term enrichments are generally consistent with previously described RRE-related functions of Rev complexes, and with the possibility that Rev is associated with RRE-containing HIV transcripts in multiple locations in the cell. Approximately 11% of the proteins in our top 250 hits were encoded by genes linked to HIV-1 production by recent genome-wide RNAi sceens (38–41), and a largely nonoverlapping ∼11% were present in the NIAID HIV-1 Rev Human Protein Interaction Database (supplemental Table S3).
A detailed Cytoscape representation (32) of the individual proteins from the top ranking list whose binding to Rev was affected by the addition of either RRE or RanGTP highlighted the complex web formed by Rev-interacting proteins (Fig. 2 and supplemental Fig. S3). Most of the Rev interacting proteins whose levels were affected by the addition of RRE were identified in nuclear extracts rather than cytosol. However, a small subset of the cytosolic proteins was found to dissociate from Rev upon RRE binding, including some nuclear import receptors (KPNA2, IPO7). Certain nuclear proteins that associated with Rev are apparently recruited by the RRE more readily in the presence of RanGTP than in the absence (e.g. DDX1, PRPF6, PRPF19), others did this in the absence of RanGTP (e.g. CUL4A, SK2L2, DHX36). Preferred association with Rev upon RRE binding in the presence of RanGTP might include proteins that are involved in forming the Rev/RRE nuclear export complex. Another group of proteins associated strongly with Rev upon RRE binding in a manner independent of RanGTP (e.g. DDX3X, DDX17, HNRNPD). Some members of this group potentially could have roles at multiple stages of the Rev/RRE complex lifecycle.
Fig. 2.

Cytoscape representation of Rev interacting proteins whose abundance was altered by the addition of RRE. Yellow and tan colored nodes and emanating lines each represent the proteomic analysis of a pair of experimental samples where the only difference between the samples was the addition or omission of RRE. The proteins come from the 250 top- ranking hits, with actins, tubulins, ribosomal proteins and histones omitted. The three nodes come from experiments involving cytosolic (top) or nuclear (bottom) extracts and without or with RanGTP, as indicated. Proteins identified in the respective experiments are connected to the nodes and their gene symbols written in gray circles, where the size of the circles represents their relative PCA scores. Proteins where the association with Rev is increased by RRE are connected with blue lines, and those where it is decreased by RRE are denoted in red.
As with any affinity tag/purification assay, many interacting partners of Rev identified in our top ranking list may associate indirectly with the Rev bait, because of their binding to the RRE or their presence in multi-protein complexes that contain only one or a few proteins directly involved in Rev binding. Moreover, because a single Rev-binding protein might coexist in different multiprotein complexes, pull-downs could include functionally irrelevant complexes containing this protein as well as relevant ones. Finally, some of the proteins detected in our analysis might nonspecifically interact with Rev even though they show strong changes upon addition of RRE or RanGTP, because these components could affect nonspecific as well as specific binding sites. Direct biochemical and functional analysis of individual members of the top hit list from the proteomics data set is required to establish their relevance to Rev and to HIV replication.
DEAD/H Proteins as a Validation Set for the Top Hit List
We selected the DEAD/H helicase family members found in the top ranking protein-protein interaction list as a validation set. There were 8 proteins in this group (Table I), out of a total of 30 DEAD/H proteins that appeared in our proteomics data set as MBP-Rev interactors (supplemental Table S1). In addition to the eight proteins in the validation set from the top-ranking list, we also analyzed DDX21. Although the highest PCA score of DDX21 (0.0287) was below our cutoff, this target was of interest because the mRNA for DDX21 was reported to be up-regulated upon activation of HIV production in latently infected cells (42), and its PCA score was still in the top 10%. Notably, four of the eight proteins in the validation set already have been linked to HIV production by RNAi approaches: DDX1 (15), DDX3X (16), DHX9 (43), and DDX24 (44). Furthermore, three of these proteins were reported to associate with Rev: DDX1 (15), DDX3X (16), and DDX24 (44).
Table I. Top-Ranking HIV-1 Rev Binding DEAD/H Box Proteins.
| Rank | Gene name | PCA score | Comparisona |
|---|---|---|---|
| 1 | DHX36 | 0.097 | NX vs NX+RRE |
| 2 | DDX24 | 0.092 | NX vs NX+RanGTP |
| DDX24 | 0.068 | NX vs NX+RRE | |
| 3 | DDX3X | 0.070 | NX vs NX+RRE |
| DDX3X | 0.050 | NX+RanGTP vs NX+RanGTP+RRE | |
| 4 | DDX1 | 0.052 | NX+RanGTP vs NX+RanGTP+RRE |
| DDX1 | 0.038 | NX+RRE vs NX+RRE+RanGTP | |
| 5 | DDX17 | 0.049 | NX vs NX+RRE |
| DDX17 | 0.040 | NX+RanGTP vs NX+RanGTP+RRE | |
| 6 | DHX9 | 0.048 | NX vs NX+RRE |
| 7 | DDX47 | 0.043 | NX+RanGTP vs NX+RanGTP+RRE |
| DDX47 | 0.040 | NX vs NX+RRE | |
| 8 | DDX5 | 0.040 | NX vs NX+RRE |
a Abundance of DEAD/H Box proteins is increased in second condition. Some proteins have PCA scores above the cutoff for two different comparisons (as shown). NX: nuclear extract.
Because our identification of Rev-binding proteins was based on in vitro reconstitution, we investigated whether the proteins in our validation set that have not previously been shown to bind Rev (DDX5, DHX9, DDX17, DHX36, and DDX47) have the capacity for interaction with Rev by coimmunoprecipitation from cell lysates (Fig. 3). We included DDX3X as a positive control, and also examined DDX21. Our strategy involved cotransfecting cells with a plasmid encoding epitope-tagged Rev, together with either pGagRRE or pGagCTE (45). Both of the latter plasmids encode the Gag protein, but pGagRRE contains the RRE and expresses Gag in a Rev-dependent manner, whereas pGagCTE contains the constitutive transport element (CTE) and expresses Gag in a Rev independent manner (Figs. 3A). Both samples expressed equivalent levels of Rev and Gag (Fig. 3C).
These experiments indicated that all of the DEAD/H proteins analyzed specifically associated with Rev when pGagRRE was cotransfected, since they were absent in samples involving control IgG beads (Fig. 3B). By contrast, either much less or no association of the DEAD/H box proteins with Rev was seen when the pGagCTE plasmid was cotransfected. This indicates that the binding of the DEAD/H proteins to Rev in cultured cells is substantially promoted by the RRE. These results are consistent with our in vitro binding analysis, where the high scores of the DEAD/H proteins in the top ranking list in most cases derived from a comparison of −/+ RRE samples (Table I). All of the DEAD box protein interactions with Rev were sensitive to RNase pretreatment of cell extracts except for DDX3X and DDX47 (Fig. 3B), suggesting that in the other cases Rev association was at least partly dependent on RRE integrity, and thus may involve recognition of an RNA sequence/structure or a multimeric array of Rev on the RRE. We did not detect co-immunoprecipitation with Rev of the abundant RNA binding protein, HNRNP D (heterogenous nuclear ribonucleoprotein D), which was in the top ranking list (Fig. 3B). This indicates that HNRNP D does not have a stable Rev interaction with our cell lysis conditions.
We next used siRNA-mediated knockdown to determine whether the previously uncharacterized DEAD/H proteins in our validation set (DDX5, DDX17, DHX36, and DDX47) and DDX21 have a role in HIV-1 production in HeLa cells. As a positive control we included DDX3X, a member of the validation set that was previously implicated in Rev functions in HIV production (16). We examined two separate siRNAs for each target (supplemental Table S4) that each gave strong knockdown, showed no cytotoxicity in HeLa cells by trypan blue staining, and yielded similar molecular phenotypes (Figs. 4 and 5). However, since all of the individual siRNAs for DDX3X that we tested yielded inefficient knockdown, we used a pool of three different siRNAs to silence this protein. By Western blot analysis of siRNA-treated cells, we determined that we achieved > 75–90% knockdown of all the targets with the siRNAs selected (Materials and Methods). It should be noted that silencing of each individual DEAD/H protein did not substantially alter the level of the other DEAD/H proteins analyzed (supplemental Fig. S4), showing that our siRNA treatments were selective.
Fig. 5.

Effects of DDX silencing on HIV-1 mRNA levels and localization. TZM-bl cells were treated with siRNAs as in Fig. 4, and were harvested 48 h postinfection. Nuclear and cytosolic fractions were prepared, and RNA was extracted from each and analyzed by qRT-PCR with primers to detect the unspliced and the fully spliced HIV mRNAs, together with primers for pre-GAPDH and HPRT. Data shown is derived from three independent experiments, with triplicate samples for each siRNA. Error bars represent standard errors. A, Total unspliced and spliced HIV mRNA levels in siRNA-treated cells. The mRNA from cytosolic and nuclear fractions was normalized to total preGAPDH mRNA and these values were combined to represent total mRNA. Values are scored as a fold-change relative to that of siRNA control cells. B, C/N ratio of unspliced and spliced HIV mRNA. The C/N ratio of HIV mRNAs is scored as a percentage relative to that for siRNA control cells.
After two sequential treatments with siRNA, cells were infected with HIV-1. At 48 h after virus challenge, the effects of silencing on virus production were assessed by three criteria: the release of p24 into the medium, the expression of Gag and Env proteins in cells, and in the cases where substantial p24 was released, the infectivity of the viral particles (Fig. 4). We were unable to monitor Rev expression because of the lack of antibodies capable of detecting the low levels of Rev in the infected cells.
The siRNAs targeting the positive control DDX3X diminished p24 release to ∼25% of the control siRNA, although there was no significant change in the levels of cellular Gag and Env (Figs. 4A and 4B). Each of the siRNAs targeting DDX17 and DDX21 reduced p24 release to < 25% of control levels. The levels of cellular Gag and Env were strongly reduced (to 30–40% of controls) by silencing DDX17 but were not significantly diminished by silencing DDX21. Conversely, siRNAs targeting DDX5 moderately increased the cellular level of Gag (to 125–150% of control) and elevated p24 release to ∼200% of control levels. Furthermore, DDX5 silencing strongly increased viral infectivity to ∼400% of controls (Fig. 4C). It is known that the DDX5 and DDX17, which are paralogs with ∼70% sequence identity, form both homodimers and heterodimers in cells (46). Accordingly, the silencing of DDX5 or DDX17 is predicted to increase the concentration of the DDX17 or DDX5 homodimers, respectively. This suggests that a DDX17 homodimer is the entity involved in HIV production. Consistent with this model, silencing of both DDX5 and DDX17 together, similar to the silencing of DDX17 alone, led to a strong reduction in p24 release (supplemental Fig. S5).
There was no significant change in Gag and Env levels, in p24 release, or in infectivity by siRNAs targeting DHX36 (Figs. 4A–4C). This suggests that DHX36 is not involved in HIV replication in HeLa cells. However, a role for this protein cannot be eliminated because low levels of DHX36 persist after silencing. Finally, silencing of DDX47 resulted in no significant change in Gag and Env levels, and a modest reduction in p24 release (to 35–55% of control levels), even though the level of DDX47 protein was reduced by > 90% with both siRNAs (supplemental Fig. S4). Because the weak phenotype with DDX47 silencing might reflect an indirect effect, we did not analyze this target further. In summary, our analysis of the four previously uncharacterized DEAD/H proteins in our validation set has robustly linked two of these proteins to HIV production: DDX5 and DDX17. Thus, the studies here combined with previous work indicate that six out of eight DEAD/H proteins in our validation set are related to HIV production. In addition, we implicated DDX21 in HIV production, although it had a somewhat lower score than the cutoff value for the top-ranking set.
Functions of Novel DEAD Proteins in the Levels and Localization of HIV mRNAs
The previously characterized DEAD/H box helicases in our validation set are suggested to function at discrete steps of HIV replication, involving nuclear export (DDX1 and DDX3) (15, 16), translation (DHX9) (43) and encapsidation of unspliced HIV mRNA (DDX24) (44). We analyzed the three novel DEAD box proteins that we implicated in HIV production to get further insight into the molecular basis for our phenotypes. For this we carried out qRT-PCR of material from siRNA treated, infected HeLa cells to determine the levels of fully spliced and unspliced HIV mRNAs, and the nuclear (N) versus cytosolic (C) distribution of these transcripts. The total transcript level is a measure of the rate of synthesis and turnover of the mRNAs, and the C/N ratio reflects the nuclear export rate in concert with the stability of the mRNAs. We analyzed HPRT as an endogenous mRNA for comparison, and evaluated DDX3X as a control (Fig. 5). The primer binding sites for the unspliced mRNA occur in the ORF of the gag gene, and those for the fully spliced mRNA occur in the overlapping coding regions of the tat and rev genes (supplemental Table S5). Nuclear integrity was retained with our fractionation protocol and siRNA treatments, as > 95% of the pre-GAPDH mRNA occurred in the nuclear fraction of all samples (supplemental Fig. S6). As indicated below, our phenotypic analysis of siRNA-treated cells often revealed changes for spliced HIV mRNA as well as for unspliced transcripts, suggesting that the helicases are involved in both Rev-independent and Rev-dependent pathways. This is consistent with finding that helicases typically are multifunctional (17, 18).
From our analysis we found that silencing of DDX3X resulted in an ∼threefold increase in the level of spliced HIV mRNA, but had no effect on the level of unspliced mRNA (Fig. 5A). Correspondingly, there was a substantial (∼threefold) increase in the C/N ratio of both unspliced and spliced mRNAs (Fig. 5B). Our results showing an increased level of cytoplasmic unspliced mRNA with DDX3 silencing differ from those of a previous study, which reported a strong decrease in cytoplasmic unspliced mRNA with DDX3X knockdown (16). This discrepancy could be explained if more efficient knockdown of DDX3X was obtained in the previous study, and if this condition altered steps of unspliced mRNA metabolism upstream of the changes we have observed.
Silencing of DDX5 led to ∼two- fourfold increases in the levels of total unspliced and spliced HIV mRNAs, with the largest increase seen for unspliced mRNA (Fig. 5A). The silencing caused a smaller (< ∼1.5-fold) elevation in the C/N ratio of both transcripts (Fig. 5B). In comparison, silencing of DDX17 led to ∼two- fivefold decreases in the levels of total unspliced and spliced mRNAs, with the largest decrease seen for unspliced mRNA (Fig. 5A). Furthermore, there was an ∼two- fivefold decrease in the C/N ratio of both transcripts (Fig. 5B). The mRNA phenotypes obtained with DDX5 and DDX17 support the possibility considered above (Fig. 4) that the DDX17 homodimer is the entity that functions in HIV production, and that DDX5 antagonizes this species by heterodimer formation.
Finally, silencing of DDX21 resulted in two- threefold greater levels of both unspliced and spliced HIV mRNAs (Fig. 5A). By contrast, there was an ∼ twofold decrease in the C/N ratio of unspliced mRNA, and no significant change in the level of spliced mRNA. The silencing of certain members of the group of DEAD proteins that we analyzed (most notably, DDX5 and DDX21) modestly changed the levels or C/N ratio of the HPRT transcript relative to the internal standard pre-GAPDH, although these changes were substantially less than those seen for HIV transcripts.
Altogether, our analysis revealed distinctive mRNA phenotypes for each of the four DEAD box proteins analyzed. Since each of two separate siRNAs for DDX5, DDX17, and DDX21 yielded similar phenotypes for all of the parameters measured, it is very unlikely that these results are due to off-target effects. This indicates that these helicases directly or indirectly affect multiple distinct features in the late stages of HIV production, not simply one aspect.
DISCUSSION
Identification of Rev-Interacting Proteins Using Comparative Proteomics
Here we used comparative proteomics to identify candidate host cell factors that associate with HIV Rev, and statistical analysis to judge the likelihood of binding specificity. Our strategy involved analysis of Rev complexes formed in vitro with a range of conditions resembling those to which Rev is exposed in cells, which could change the association of binding partners. To evaluate the significance of Rev association, we assigned each binding partner a statistical score from sample-to-sample comparisons. The score was a composite measure of the binding partner abundance, its specificity and the changes in its Rev association with different conditions.
Examination of the top-scoring 250 hits (5% of the comparisons) revealed a strong enrichment in GO designations that were consistent with a role in Rev or Rev/RRE functions. We selected the eight DEAD/H proteins in the top-ranking list as a validation set for further analysis. We verified that each of the novel helicases not previously characterized as Rev binders has the capacity to associate specifically with Rev in vivo when RRE-containing mRNA is co-expressed. Our RNAi analysis combined with previous work indicates that six of the eight DEAD/H proteins in the validation set are linked to HIV production. Altogether, these results indicate that our top- ranking list is enriched in HIV-relevant host proteins that associate with Rev or Rev/RRE. It should be noted that some of the proteins revealed in our analysis may associate only indirectly with Rev due to their presence in multi-component complexes or because they recognize structural features of the RRE. In this regard, three of the DEAD/H proteins on our top-ranking list, DDX1, DDX21 and DHX36, have been reported to co-immunoprecipitate from lysates of lymphoid cells (47). This could account for our finding that DHX36 was in our top-ranking list, but had no detectable role in HIV production in cultured cells, unlike DDX21 (this study) and DDX1 (15). Although the HeLa cell model we used in this study is commonly employed for analyzing the molecular virology of HIV, it remains to be determined whether the helicases we have examined are involved in HIV replication in CD4+ T cells and macrophages, the predominant targets of infection in humans (1–3).
The finding that two of the eight DEAD/H proteins in our top-ranking list do not show a robust phenotype in HIV production suggests that these proteins either are not rate-limiting for HIV production in HeLa cells, or that they are false positives as HIV cofactors. We do not expect our analysis to detect all physiologically relevant interaction partners of Rev or assign them a high PCA score because of intrinsic limitations of in vitro Rev complex formation. For example, although DDX21 had PCA scores below the cutoff, we found that siRNAs targeting DDX21 strongly inhibited HIV production. Key specificity parameters for our proteomics analysis were defined by incubation of samples in the absence or presence of RanGTP and/or RRE, which modulate the formation of nuclear transport complexes and Rev-RRE RNP complexes, respectively. The pairwise comparisons that resulted in a high PCA score for most of the DEAD/H proteins in our validation set involved incubation −/+ RRE. For at least some of these helicases, and for a substantial number of the other top PCA hits identified by −/+ RRE sample comparisons, it is possible that the proteins recognize structural features of the RRE RNA that are not enriched in the bulk competitor RNA in the extract, either separate from or in combination with structural features of the Rev oligomer.
Only a small number of proteins in the top hit list have well-established functional connections to Rev. It is possible to imagine many potential functions for the uncharacterized candidates. Some of these proteins could be additional components of RNA helicase complexes, or of other multi-protein complexes, involved in various aspects of HIV transcript function. Other proteins could be linked to more subtle aspects of Rev biology, including its regulation. For example, Rev undergoes posttranslational modification by phosphorylation (48), and its level in host cells could be regulated by protein turnover as well as by translation. Moreover, since Rev has a high net positive charge due to the long stretch of basic amino acids in its RNA binding domain/nuclear localization sequence (37), hypothetical Rev chaperones also could be represented in the top-ranking list to help promote its intracellular “solubility” when it is not bound to HIV transcripts. Clearly, focused analysis of individual proteins in the data set will be needed to characterize the nature of their Rev interaction, as well as to determine whether they have an essential role in HIV replication.
Role of Rev and DEAD Box Proteins in Multiple Facets of HIV Production
Our in vitro and in vivo binding analyses, combined with previous work on DDX1 (15), DDX3X (16) and DDX24 (44), show that Rev has the capacity to directly or indirectly interact with all of the helicases in our top-ranking list. Moreover, at least six of these binding partners are linked to HIV replication. The association of Rev with underspliced HIV transcripts could help to specify targeting of the helicases to these mRNAs, and also could help to locally modulate the helicase functions. Rev is known to initially associate with underspliced HIV mRNAs in the nucleus, where it functions in nuclear export (5). Although Rev is not detectably associated with mature budded virions (49), it nonetheless may be associated with underspliced HIV transcripts following their transport to the cytoplasm, either through the RRE (5), or via a secondary binding site in the 5′ UTR that has been proposed (50). Whether Rev is associated with cytosolic HIV transcripts in the context of a native virus infection has not yet been resolved, although there is evidence to support this possibility from several studies of Rev in ectopic contexts. A cytoplasmic association of Rev with HIV transcripts could help explain the reported role of Rev in stimulating infectious virion budding (44) and in translation of structural genes for the virus (11, 13).
Our more detailed characterization of the phenotypes of HIV-infected cells where DDX3X, DDX5, DDX17, and DDX21 were silenced suggests considerable functional complexity for these proteins in HIV regulation. In each case, we obtained distinctive effects involving multiple facets of HIV mRNA/protein regulation. These variously involved the cellular levels of spliced and unspliced HIV mRNAs, the C/N ratio of these transcripts, the levels of Gag and Env proteins, and the release of p24 particles. Silencing of two of the helicases, DDX3X and DDX21, resulted in strong inhibition of p24 release even though the cellular levels of Gag and Env were close to the control, implying defects in Gag/Env intracellular localization or in virus release. Moreover, with DDX3X the level of Gag was similar to the control, even though the level of the cytoplasmic unspliced mRNA was substantially increased, implying an effect on translation or conceivably, on protein stability. Silencing of DDX5, the heterodimerization partner of DDX17 (46), resulted in increased levels and C/N ratios of unspliced and spliced mRNAs, together with elevated virus production. Silencing of DDX17 gave phenotypes that were generally the converse of those with DDX5 silencing. The changes in HIV transcript levels and localization we obtained with the silencing of DDX5 and DDX17 as well as with the other helicases could result from effects on many processes, including transcription, nuclear export and stability of the transcripts. In the future, it will be important to implement mutational approaches to help assign and distinguish the different phenotypes.
The complexity of phenotypes caused by helicase silencing raises the question of whether the helicases directly or indirectly affect the processes that are perturbed. It is well established that individual RNA helicases can affect multiple processes in the life cycle of cellular transcripts (17, 18). For example, Dbp5 is involved in mRNA export through the nuclear pore complex as well as in translational initiation and termination (51). Thus it is reasonable to propose that the helicases we have analyzed have direct effects on multiple aspects of the behavior of HIV transcripts. Some of the functions implied for DDX3X and DDX5/17 in HIV replication by our studies are consistent with previous studies of these proteins in the context of cellular mRNAs. DDX3X is known to be important for translational initiation and turnover of some mRNAs, and DDX5/17 have been implicated in transcription, splicing and turnover of certain mRNAs (18, 52). Nonetheless, it is conceivable that some of the phenotypes we observed are indirect consequences of helicase silencing. Notwithstanding this caveat, the effects of helicase knockdown clearly are selective, since silencing of each of the 6 helicases analyzed did not detectably affect the levels of the other 5 proteins. In addition to the question of direct versus indirect effects of helicases on different aspects of the HIV phenotypes, it is possible that only a subset of the phenotypes result from interaction with Rev, and that others are independent of Rev. Resolving these complex issues will require structural, biochemical, and functional studies.
It is well established that separate steps of mRNA metabolism can be closely linked. For example, proteins of exon junction complex, which are deposited on transcripts in the nucleus during splicing, have effects on these transcripts after their transport to the cytoplasm related to nonsense mediated decay and translational regulation (9). Thus, it is possible that some of the effects of helicases related to the metabolism and functions of cytoplasmic HIV transcripts could be specified by helicase-mediated mRNP remodeling in the nucleus that persists as “memory” on the particles after their nuclear export, analogous to the exon junction complex. This concept is consistent with the finding that the nuclear export receptor pathway used by Gag-encoding transcripts in murine cells substantially influences the subsequent translational fate of Gag mRNA as well as Gag localization (53). For example, although DDX5/17 (18, 52) and DDX21 (54) are highly concentrated in the nucleus and hypothetically might act on HIV mRNP particles at this location, the consequences of this action might only be manifested in the cytoplasm. Moreover, it is possible that some of the helicases become associated with underspliced HIV transcripts in the nucleus, possibly for one set of functions, and accompany the transcripts to the cytoplasm for a separate set of functions. In this regard, it is noteworthy that DDX1 (15), DDX3 (16), DDX5 (55), and potentially DHX9 (56) are nucleocytoplasmic shuttling proteins. Understanding the dynamics of association of specific helicases with HIV transcripts will be important for addressing these questions.
In summary, our proteomics and statistical analysis has identified a set of Rev-interacting proteins that is enriched in functionally relevant Rev binding partners, based on the results with a validation set of DEAD box proteins. Our more detailed analysis of some of these helicases by RNAi indicates that they each have distinctive effects on multiple features of HIV replication in HeLa cells. Finally, the general methodology we describe here, involving comparative proteomics and statistical analysis of protein complexes assembled in vitro, should be effective for the identification of interaction partners of other proteins, particularly RNA-binding nucleoplasmic shuttling proteins. This can complement the more conventional strategy for identification of protein interaction partners involving proteomic analysis of pull-downs from cell lysates.
Footnotes
* This work was funded by National Institutes of Health grants P50-GM082545 to LG, P41 RR011823 to JRY, and P50 GM082250, P01 AI090935 to NJK. SN and GA were supported by postdoctoral fellowships from the California HIV/AIDS Research Program. PV was supported by fellowships from the Austrian Science Fund and the American Heart Association. NJK is a Searle Scholar and a Keck Young Investigator.
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S5.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S5.
1 The abbreviations used are:
- HIV-1
- human immunodeficiency virus 1
- RRE
- Rev Reponse Element
- CRM1
- chromosome region maintenance 1
- PCA
- principal component analysis.
REFERENCES
- 1. Freed E. O. (2004) HIV-1 and the host cell: an intimate association. Trends Microbiol. 12, 170–177 [DOI] [PubMed] [Google Scholar]
- 2. Lever A. M., Jeang K. T. (2011) Insights into cellular factors that regulate HIV-1 replication in human cells. Biochemistry 50, 920–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin-Serrano J., Neil S. J. (2011) Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 9, 519–531 [DOI] [PubMed] [Google Scholar]
- 4. Frankel A. D., Young J. A. (1998) HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67, 1–25 [DOI] [PubMed] [Google Scholar]
- 5. Pollard V. W., Malim M. H. (1998) The HIV-1 Rev protein. Annu. Rev. Microbiol. 52, 491–532 [DOI] [PubMed] [Google Scholar]
- 6. Mann D. A., Mikaelian I., Zemmel R. W., Green S. M., Lowe A. D., Kimura T., Singh M., Butler P. J., Gait M. J., Karn J. (1994) A molecular rheostat. Co-operative rev binding to stem I of the rev-response element modulates human immunodeficiency virus type-1 late gene expression. J. Mol. Biol. 241, 193–207 [DOI] [PubMed] [Google Scholar]
- 7. Cook A., Bono F., Jinek M., Conti E. (2007) Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 76, 647–671 [DOI] [PubMed] [Google Scholar]
- 8. Xu D., Farmer A., Chook Y. M. (2010) Recognition of nuclear targeting signals by Karyopherin-beta proteins. Curr. Opin. Struct. Biol. 20, 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carmody S. R., Wente S. R. (2009) mRNA nuclear export at a glance. J. Cell Sci. 122, 1933–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cullen B. R. (2003) Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28, 419–424 [DOI] [PubMed] [Google Scholar]
- 11. D'Agostino D. M., Felber B. K., Harrison J. E., Pavlakis G. N. (1992) The Rev protein of human immunodeficiency virus type 1 promotes polysomal association and translation of gag/pol and vpu/env mRNAs. Mol. Cell Biol. 12, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Groom H. C., Anderson E. C., Dangerfield J. A., Lever A. M. (2009) Rev regulates translation of human immunodeficiency virus type 1 RNAs. J. Gen. Virol. 90, 1141–1147 [DOI] [PubMed] [Google Scholar]
- 13. Perales C., Carrasco L., Gonzalez M. E. (2005) Regulation of HIV-1 env mRNA translation by Rev protein. Biochim. Biophys. Acta 1743, 169–175 [DOI] [PubMed] [Google Scholar]
- 14. Brandt S., Blissenbach M., Grewe B., Konietzny R., Grunwald T., Uberla K. (2007) Rev proteins of human and simian immunodeficiency virus enhance RNA encapsidation. PLoS Pathog. 3, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fang J., Kubota S., Yang B., Zhou N., Zhang H., Godbout R., Pomerantz R. J. (2004) A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology 330, 471–480 [DOI] [PubMed] [Google Scholar]
- 16. Yedavalli V. S., Neuveut C., Chi Y. H., Kleiman L., Jeang K. T. (2004) Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119, 381–392 [DOI] [PubMed] [Google Scholar]
- 17. Bleichert F., Baserga S. J. (2007) The long unwinding road of RNA helicases. Mol. Cell 27, 339–352 [DOI] [PubMed] [Google Scholar]
- 18. Jankowsky E. (2010) RNA helicases at work: binding and rearranging. Trends Biochem. Sci. 36, 19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hintersteiner M., Ambrus G., Bednenko J., Schmied M., Knox A. J., Meisner N. C., Gstach H., Seifert J. M., Singer E. L., Gerace L., Auer M. (2010) Identification of a small molecule inhibitor of importin beta mediated nuclear import by confocal on-bead screening of tagged one-bead one-compound libraries. ACS Chem. Biol. 5, 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edgcomb S. P., Aschrafi A., Kompfner E., Williamson J. R., Gerace L., Hennig M. (2008) Protein structure and oligomerization are important for the formation of export-competent HIV-1 Rev-RRE complexes. Protein Sci. 17, 420–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cassany A., Gerace L. (2009) Reconstitution of nuclear import in permeabilized cells. Methods Mol. Biol. 464, 181–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res.11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 24. McDonald W. H., Tabb D. L., Sadygov R. G., MacCoss M. J., Venable J., Graumann J., Johnson J. R., Cociorva D., Yates J. R., 3rd (2004) MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 18, 2162–2168 [DOI] [PubMed] [Google Scholar]
- 25. Sadygov R. G., Yates J. R., 3rd (2003) A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 75, 3792–3798 [DOI] [PubMed] [Google Scholar]
- 26. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 27. Tabb D. L., McDonald W. H., Yates J. R., 3rd (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 29. Zhang B., VerBerkmoes N. C., Langston M. A., Uberbacher E., Hettich R. L., Samatova N. F. (2006) Detecting differential and correlated protein expression in label-free shotgun proteomics. J. Proteome Res. 5, 2909–2918 [DOI] [PubMed] [Google Scholar]
- 30. Zito T., Wilbert N., Wiskott L., Berkes P. (2008) Modular Toolkit for Data Processing (MDP): A Python Data Processing Framework. Frontiers Neuroinformatics 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carbon S., Ireland A., Mungall C. J., Shu S., Marshall B., Lewis S. (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cline M. S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., Hanspers K., Isserlin R., Kelley R., Killcoyne S., Lotia S., Maere S., Morris J., Ono K., Pavlovic V., Pico A. R., Vailaya A., Wang P. L., Adler A., Conklin B. R., Hood L., Kuiper M., Sander C., Schmulevich I., Schwikowski B., Warner G. J., Ideker T., Bader G. D. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gallay P., Hope T., Chin D., Trono D. (1997) HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. U. S. A. 94, 9825–9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rousseau D., Kaspar R., Rosenwald I., Gehrke L., Sonenberg N. (1996) Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc. Natl. Acad. Sci. U. S. A. 93, 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 36. Daugherty M. D., D'Orso I., Frankel A. D. (2008) A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol. Cell 31, 824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Truant R., Cullen B. R. (1999) The arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin beta-dependent nuclear localization signals. Mol. Cell Biol. 19, 1210–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brass A. L., Dykxhoorn D. M., Benita Y., Yan N., Engelman A., Xavier R. J., Lieberman J., Elledge S. J. (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 [DOI] [PubMed] [Google Scholar]
- 39. Yeung M. L., Houzet L., Yedavalli V. S., Jeang K. T. (2009) A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284, 19463–19473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou H., Xu M., Huang Q., Gates A. T., Zhang X. D., Castle J. C., Stec E., Ferrer M., Strulovici B., Hazuda D. J., Espeseth A. S. (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell. Host Microbe 4, 495–504 [DOI] [PubMed] [Google Scholar]
- 41. Konig R., Zhou Y., Elleder D., Diamond T. L., Bonamy G. M., Irelan J. T., Chiang C. Y., Tu B. P., De Jesus P. D., Lilley C. E., Seidel S., Opaluch A. M., Caldwell J. S., Weitzman M. D., Kuhen K. L., Bandyopadhyay S., Ideker T., Orth A. P., Miraglia L. J., Bushman F. D., Young J. A., Chanda S. K. (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krishnan V., Zeichner S. L. (2004) Alterations in the expression of DEAD-box and other RNA binding proteins during HIV-1 replication. Retrovirology 1, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bolinger C., Sharma A., Singh D., Yu L., Boris-Lawrie K. (2010) RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 38, 1686–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ma J., Rong L., Zhou Y., Roy B. B., Lu J., Abrahamyan L., Mouland A. J., Pan Q., Liang C. (2008) The requirement of the DEAD-box protein DDX24 for the packaging of human immunodeficiency virus type 1 RNA. Virology 375, 253–264 [DOI] [PubMed] [Google Scholar]
- 45. Wodrich H., Bohne J., Gumz E., Welker R., Krausslich H. G. (2001) A new RNA element located in the coding region of a murine endogenous retrovirus can functionally replace the Rev/Rev-responsive element system in human immunodeficiency virus type 1 Gag expression. J. Virol. 75, 10670–10682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ogilvie V. C., Wilson B. J., Nicol S. M., Morrice N. A., Saunders L. R., Barber G. N., Fuller-Pace F. V. (2003) The highly related DEAD box RNA helicases p68 and p72 exist as heterodimers in cells. Nucleic Acids Res. 31, 1470–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Z., Kim T., Bao M., Facchinetti V., Jung S. Y., Ghaffari A. A., Qin J., Cheng G., Liu Y. J. (2011) DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34, 866–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fouts D. E., True H. L., Cengel K. A., Celander D. W. (1997) Site-specific phosphorylation of the human immunodeficiency virus type-1 Rev protein accelerates formation of an efficient RNA-binding conformation. Biochemistry 36, 13256–13262 [DOI] [PubMed] [Google Scholar]
- 49. Chertova E., Chertov O., Coren L. V., Roser J. D., Trubey C. M., Bess J. W., Jr., Sowder R. C., 2nd, Barsov E., Hood B. L., Fisher R. J., Nagashima K., Conrads T. P., Veenstra T. D., Lifson J. D., Ott D. E. (2006) Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 80, 9039–9052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gallego J., Greatorex J., Zhang H., Yang B., Arunachalam S., Fang J., Seamons J., Lea S., Pomerantz R. J., Lever A. M. (2003) Rev binds specifically to a purine loop in the SL1 region of the HIV-1 leader RNA. J. Biol. Chem. 278, 40385–40391 [DOI] [PubMed] [Google Scholar]
- 51. Kutay U., Panse V. G. (2008) Gle1 does double duty. Cell 134, 564–566 [DOI] [PubMed] [Google Scholar]
- 52. Fuller-Pace F. V. (2006) DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34, 4206–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sherer N. M., Swanson C. M., Papaioannou S., Malim M. H. (2009) Matrix mediates the functional link between human immunodeficiency virus type 1 RNA nuclear export elements and the assembly competency of Gag in murine cells. J. Virol. 83, 8525–8535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Holmstrom T. H., Mialon A., Kallio M., Nymalm Y., Mannermaa L., Holm T., Johansson H., Black E., Gillespie D., Salminen T. A., Langel U., Valdez B. C., Westermarck J. (2008) c-Jun supports ribosomal RNA processing and nucleolar localization of RNA helicase DDX21. J. Biol. Chem. 283, 7046–7053 [DOI] [PubMed] [Google Scholar]
- 55. Wang H., Gao X., Huang Y., Yang J., Liu Z. R. (2009) P68 RNA helicase is a nucleocytoplasmic shuttling protein. Cell Res. 19, 1388–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tang H., McDonald D., Middlesworth T., Hope T. J., Wong-Staal F. (1999) The carboxyl terminus of RNA helicase A contains a bidirectional nuclear transport domain. Mol. Cell. Biol. 19, 3540–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]