

Abstract

We present a flexible, modular route to GlcNAc-MurNAc-oligosaccharides that can be readily converted into peptidoglycan (PG) fragments to serve as reagents for the study of bacterial enzymes that are targets for antibiotics. Demonstrating the utility of these synthetic PG substrates, we show that the tetrasaccharide substrate lipid IV (3), but not the disaccharide substrate lipid II (2), significantly increases the concentration of moenomycin A required to inhibit a prototypical PG-glycosyltransferase (PGT). These results imply that lipid IV and moenomycin A bind to the same site on the enzyme. We also show the moenomycin A inhibits the formation of elongated polysaccharide product but does not affect length distribution. We conclude that moenomycin A blocks PG-strand initiation rather than elongation or chain termination. Synthetic access to diphospholipid oligosaccharides will enable further studies of bacterial cell wall synthesis with the long-term goal of identifying novel antibiotics.
Keywords: Lipid IV, Moenomycin, Peptidoglycan, Transglycosylation, Carbohydrate synthesis
1. Introduction
Bacteria have evolved a cell wall that protects the cell membrane from hostile environments and allows the cells to withstand varying osmotic pressures.1 The supportive structure of the cell wall is peptidoglycan (PG), consisting of polysaccharide threads of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) units.2 These polysaccharides are cross-linked through peptide side chains appended to the MurNAc units to form a stable polymeric mesh. Many highly effective antibacterial drugs disrupt the formation of the cell wall, which ultimately results in cell death. It is important to better understand cell wall synthesis in order to develop new therapies for bacterial infections.3
Work over the past several decades has shed light on the biosynthesis of PG. In the final stages of biosynthesis lipid II (2, Figure 1) is polymerized by peptidoglycosyl transferases (PGT) and the resulting polysaccharide strands are then linked by transpeptidases (TPs).2 Many different classes of natural product antibiotics that inhibit strand polymerization and crosslinking have been identified (e. g. Scheme 1). These antibiotics function by three major type of mechanisms: 1) They bind to PGT substrates such as 2, sequestering them so they are unavailable for PG-strand synthesis and cross-linking (e.g. vancomycin4 and ramoplanin5); 2) They acylate TP-domains, inactivating them so that they are incapable of strand cross-linking (e.g. the beta-lactams);6 or 3) they bind to PGT domains, preventing PG strand formation (e.g., moenomycin A (1), Figure 1).7 Antibiotics having the first or second mechanism of action have been highly effective in the clinic but there are no approved drugs with the third mechanism.8
Figure 1.

Molecular structures of moenomycin A (1), lipid II (2), and lipid IV (3).
Scheme 1.

The final stages of peptidoglycan biosynthesis involve polymerization of lipid II to make oligosaccharide chains followed by crosslinking via transpeptidation. This sequence is often performed by bifunctional enzymes containing distinct glycosly transferase (GT) and transpepdidase (TP) domaines (depicted in gray). Peptidoglycan synthesis can be disrupted at different steps by various antibiotics.
The natural product moenomycin A (1), which binds to PGTs, has potent antibacterial activity against Gram-positive pathogens and has been used as a growth promoter in animals for decades without engendering resistance.9 Its use in humans has been prevented by unfavorable pharmacokinetic properties (e.g. a long half-life and low oral bioavailability)10 but it can serve as a blueprint for the development of other PGT inhibitors. Hence, a detailed understanding of how 1 inhibits PGTs is important for exploiting these crucial enzymes as antibiotic targets.
A major impediment to study antibiotics that inhibit PGTs has been the lack of access to substrates used by these enzymes. PGTs catalyze the transformation of lipid II (2) into a polymer. In the first round of synthesis, two units of lipid II are coupled. In subsequent rounds of synthesis, lipid II adds in a processive manner to the elongating glycan strand. In order to dissect the reaction, it is necessary to have both disaccharide and oligosaccharide substrates available to discriminate between the glycosyl acceptor and the glycosyl donor sites of the enzyme. For over a decade we, and others, have been developing methods to obtain PGT substrates as well as assays using these substrates to probe the mechanisms and inhibition of PG biosynthetic enzymes.11 Here we report a modular synthesis of polysaccharides to obtain differentiated diphospholipid oligosacharide substrates that serve as specific mimics of the glycosyl donor strand. Furthermore, we used these substrates to learn more about the mode of action of moenomycin A.
2. Results and Discussion
An efficient synthesis of lipid IV (3) and its higher homologues, i. e. lipid VI, VIII etc., must address a number of challenges associated with these intriguing, structurally complex, primary metabolites. First and foremost we had to fundamentally change our previously described synthetic strategy (Scheme 2).11n,o Whereas the previous route was specifically tailored to obtain 3, we now required a strategy that would easily allow access to a variety of polysaccharides of different chain lengths, comprising n × GlcNAc-MurNAc-units (cf. 10). We envisioned the use of a disaccharide building block 4 that could be converted into both a disaccharide donor (5) and acceptor (6), which would then be coupled to obtain tetrasaccharide 7. A similar sequence of differentiation and coupling would give access to octasaccharide 10 and higher polysaccharides at will. Additionally, we required a large amount of Z,Z,Z,Z,E,E-heptaprenyl phosphate 12 for use as an acceptor substrate. We thus sought a scalable, stereoselective route to the heptaprenyl alcohol starting from the readily available E,E-farnesol. Since current methods for the preparation of pyrophosphates (13) are inefficient, an effective protocol for the formation of pyrophosphates by condensation of two monophosphates needed to be established.
Scheme 2.

Disaccharide 4, equipped with a suitable C4-OH group (red dot) and an anomeric protecting group (black dot), gets converted into both a disaccharide donor and acceptor (5 and 6). Coupling of 5 and 6 yields tetrasaccharide 7 that can be converted into octasaccharide 10 in a similar sequence via donor 8 and acceptor 9. Repetition of this sequence followed by coupling to 12 allows access to diphospholipid oligosaccharides 13 with different length of the oligosaccharide.
2.1. Synthesis of Z,Z,Z,Z,E,E-heptaprenyl alcohol
For the synthesis of heptaprenyl alcohol 21 we devised a synthetic route that relied on the finding of Casey and coworkers that β-keto esters can be stereoselectively converted into their diethylphosphoroxy enol ethers.12 These, in turn, react stereospecifically in a substitution reaction with methylcuprates.13 Thus, E,E-farnesol (14) was converted into β-keto ester 15 in 84% yield (over 2 steps). Execution of Casey’s and Weiler’s protocols followed by reduction of the ester with DIBAL-H provided E,E,Z-tetraprenyl alcohol 17 as the only stereoisomer observed and yields ranged from 30 to 50% over 5 steps.12,13
Upon iteration of this procedure with 17 we noted, however, that the Z/E ratio eroded in the third cycle. The desired hexaprenyl alcohol 19 was obtained in a 7/3 mixture along with its double bond isomer. Purification by chromatography proved unsuccessful and we were forced to optimize the reaction conditions with regard to stereospecificity of the substitution of the phosphate by a methyl group. To this end we switched from a diethyl phosphate to a diphenyl phosphate leaving group and were able to derive enol ether 18 in 60% yield over 3 steps. Gratifyingly, we found that 18 participated in a highly efficient and stereospecific cross coupling reaction with MeMgCl using Fe(acac)3 as catalyst.14 After DIBAL-H reduction we obtained 19 in 75% (over 2 steps). Moreover, we found that the iron-catalyzed cross coupling retained its high stereospecificity upon iteration of the sequence to provide the desired heptaprenyl alcohol 21 in an average yield of 7% from farnesol. With this procedure in hand we obtained more than 2 g of 21, and we next turned our attention to the development of an efficient coupling procedure for the formation of pyrophosphates.
2.2. N-Methylimidazolium chloride as an effective catalyst for pyrophosphate formation
We recently reported the identification of N-methyl imidazolium chloride as a catalyst for pyrophosphate formation by condensation of two phosphates (Scheme 4).15 This catalyst is superior to the commonly employed tetrazole with respect to activity, cost, and safety. Using this catalyst we established a synthesis of lipid I. Sugar phosphate 22 was first converted to phophorimidazolide 23, which was coupled with heptaprenyl phosphate 21 in 12 h. After removal of the protecting groups, pyrophosphate 24 was obtained in acceptable yields (54%) and was subsequently transformed into Lipid I, showcasing the utility of this protocol for the synthesis of lipid pyrophosphate saccharides.
Scheme 4.
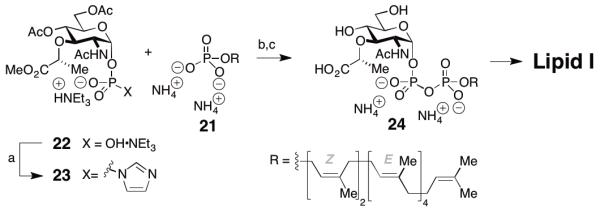
Reagents and conditions: a) CDI, DMF then MeOH; b) N-Methylimidazole·HCl (4 equiv.), DMF/THF 1/1, rt, 12 h; c) 1 M LiOH·H2O, THF-MeOH-H2O (3:1:1), 54%.
2.3. Modular synthesis of polysaccharides
The modular synthesis of the polysaccharide part of the lipid pyrophosphate sugars started with readily available glucosamine derivate 25 and took advantage of the bifunctionality of its thiophenol group, the phthalimide group, as well as a benzylic acetal at C4 and C6 (Scheme 5).16 Thiophenol is a powerful protecting group of the anomeric position but can easily be converted into its sulfoxide,17 which in turn is activated with Tf2O under mild conditions (-78 °C) to provide a superb glycosyl donor.18 Phthalimides at C2 are known to sterically shield the C3 position, obviating an additional protecting group for the C3 hydroxy group.19 Phthalimide groups provide anchimeric assistance to allow glycosylation to occur with high ß-selectivity. Finally, a number of procedures have been put forth for the reduction of a C4/C6 acetal, which allow selective liberation of the C4 group.20
Scheme 5.

Reagents and conditions: a) BnBr (1.1 equiv.), NaH (1.2 equiv.), Bu4NI (0.1 equiv.), DMF, rt, 70%; b) mCPBA (1 equiv.), CH2Cl2, −78 °C to rt, 88%; c) HSiEt3 (5 equiv.), TFA (5 equiv.), CH2Cl2, 0 °C, 93%; d) 27 (1.5 equiv.), ADMB (5 equiv.), DTBMP (5 equiv.), Tf2O (1 equiv.), CH2Cl2, MS 4Å, 58 %; e) mCPBA (1.1 equiv.), CH2Cl2, 77%; f) HSiEt3 (12 equiv.), BF3·OEt2 (2 equiv.), CH2Cl2, 70%; g) 29 (1.25 equiv.), ADMB (6.25 equiv.), DTBMP (3.75 equiv.), Tf2O (1.25 equiv.), CH2Cl2, MS 4Å, 55%. Abbreviations: ADMB = 4-allyl-1,2-dimethoxybenzene, DTBMP = 2,6-di-tert-butyl-4-methylpyridine.
Glucosamine 25 was thus converted into both glycosyl donor 26, by protection of C3 and oxidation of the thioether, and glycosyl acceptor 27 by reduction of the acetal using triethylsilane and TFA. These two building blocks were then coupled under conditions previously developed in our group to give β-1,4-disaccharide 28 in 57% yield.18 To suppress formation of a trisaccharide (at C3), it proved beneficial to employ an excess of the glycosyl acceptor, which could be reisolated after the reaction. Disaccharide 28 was then subjected to a similar sequence to obtain glycosyl acceptor 30 (HSiEt3, BF3·OEt2, 70%) and glycosyl donor 29 (77%). These disaccharides were again subjected to Tf2O-induced glycosylation and tetrasaccharide 31 was obtained in 55% yield. In this case excess disaccharide donor proved beneficial and residual acceptor 30 could be reisolated (20%). The regioselectivity of the reaction to give the C4-glycosylated product was verified by acylation of the C3-OH group, which leads to a significant 1H-NMR downfield shift of the proton at C3 (ca. 5.7 ppm).21 Furthermore, we found that the carbon atom that bears the glycosyl substituent experiences a 13C-NMR downfield shift, which allows facile structure determination (C3 from ca. 70 ppm to 80 ppm; C4 from ca. 75 ppm to ca. 85 ppm, as determined by HSQC). We obtained precursors to Lipid II (28) and Lipid IV (31) as required for the studies reported below but this synthetic sequence could be further extended to obtain similarly substituted hexa-, octa-, deca-, etc. saccharides.
To show the utility of this approach we converted 31 into heptaprenyl-lipid IV (3b). To this end we installed the N-acetates by aminolysis of the phthalimide with ethylenediamine, followed by acylation of the free amine. The C3-lactic acid substituent was subsequently installed by SN2-reaction with S-(-)-2-bromopropionic acid followed by methylation of the carboxylate with TMS-diazomethane to obtain 33 in 53% yield from 31. Substrate 33 intercepted a late stage in our previously published synthesis and can be converted into Lipid IV.11o
2.4. Use of synthetic PG substrates to study the mechanism of moenomycin A inhibition
We have previously reported that inhibition of the prototypical PGT, E. coli PBP 1a, by moenomycin A cannot be overcome by increasing the concentration of Lipid II.11i The complexity of the enzymatic reaction, which involves substrate differentiation and processive elongation, makes it challenging to interpret this result. Therefore, we have revisited the question of how moenomycin A inhibits PGT using a different approach that employs both lipid II and a lipid IV substrate containing a blocked non-reducing end (Gal-Lipid IV).22 The concentration of moenomycin A required to inhibit by 50% (IC50) the polymerization of lipid II by E. coli PBP 1a (20 nM) was found to be 9.2 nM (Figure 2); or approximately 1/2 the enzyme concentration. This IC50 reflects the high affinity of moenomycin A for the enzyme. The addition of 1.4 μM Gal-Lipid IV, which functions only as a glycosyl donor, shifts the IC50 of moenomycin A 5-fold.23 The shift implies that Gal-Lipid IV and moenomycin A compete for binding to the same site on the enzyme. It has previously been suggested that moenomycin A is a mimetic of the elongating glycan strand but the results reported here provide the first kinetic evidence that the inhibitor and the elongating substrate occupy the same site24.
Figure 2.

Dose-response curves of the moenomycin A inhibition of peptidoglycan formation. Solutions of E. coli PBP1a (20 nM) were incubated with moenomycin A in different concentrations and then treated with 14C-Lipid II (4 μM). Distribution of Lipid II/polysaccharide was determined by scintilation count after paperchromatography of the product solution. Blue curve: no Gal-Lipid IV added; red curve: PBP1a was pretreated with Gal-Lipid IV (1.36 μM).
We next examined whether moenomycin A has any effect on the length distribution of products formed by the PGT catalyzed polymerization of lipid II. E. coli PBP1a was incubated with lipid II in the presence of increasing concentrations of moenomycin A and the products were analyzed by SDS-PAGE (Figure 3).11i, 25 PG-polymers could not be detected at moenomycin concentrations equal to or exceeding the enzyme concentration even at extended reaction times. At moenomycin concentrations below the enzyme concentration, products were detected but the length distribution appeared similar regardless of the inhibitor concentration. Since the resolution of the page assay is limited for very long products, we repeated the experiments with a different PGT that was previously shown to produce much shorter products.11n,o As before, products were not detected at equimolar enzyme:moenomycin concentrations. Products formed at lower moenomycin concentrations, but the length distribution did not vary significantly as a function of concentration. These results imply a mechanism for inhibition in which moenomycin A blocks chain initiation rather than elongation or termination since effects on the latter stages of the enzymatic reaction would alter product length (Scheme 7).
Figure 3.

SDS-PAGE of enzymatic 14C-lipid II-polymerization reactions (8 μM) with varying amounts of moenomycin A (MmA) present. Red lines mark equimolar ratio of enzyme to moenomycin A. In reactions with c(MmA) ≥ c(PBP) no polymerization of Lipid II is observed. If c(MmA) < c(PBP), polysaccharide is produced. Note that the length distribution of polysaccharides formed varies with the enzyme employed but is not influenced by moenomycin A.
Scheme 7.

Model for Lipid II polymerization by peptidoglycan glycosyltransferases (PGTs) and their inhibition by moenomycin A (MmA). Moenomycin A blocks initation of the polymerization of lipid II by binding to the donor site (D) of the PGT.
3. Conclusion
We report a modular synthesis of diphospholipid oligosaccharides using a bifunctional disaccharide building block. Along with our improved synthesis of pyrophosphates and of heptaprenyl alcohol, this synthetic sequence provides access to higher-order oligosaccharide cell wall fragments. We have used these PG fragments, containing diphosphate activated reducing termini, to evaluate the mechanism of action of moenomycin A. We have provided the first kinetic evidence that moenomycin A occupies the same binding site as the elongating PG strand. Since we have previously shown that PG strands elongate by addition of disaccharide units to their reducing ends,11n the kinetic data is consistent with the proposal by Welzel and coworkers that moenomycin A mimics the growing glycan strand.7 Since PGTs can exist in both open and closed conformations,24 however, moenomycin A and the growing chain may not bind to the same conformation. Efficient access to PG fragments that bind in the donor site may make it possible to obtain structures of PGTs bound to substrates.11r,t
In addition to providing substrates to study PGTs, the reported synthetic route enables access to oligosaccharide fragments of PG to study the transpeptidation reaction. (i.e., peptide cross-linking). The transpeptidases are the lethal targets of the β-lactams but an inability to obtain PG oligosaccharides has precluded detailed mechanistic studies of these enzymes. The methods reported here will provide the tools necessary to understand the TPs and other penicillin-binding proteins in detail.
4. Experimental section
4.1. General Methods
All reactions were carried out under an argon atmosphere with solvents dried by passage over activated alumina. Reagents were used as obtained with the following exceptions: BF3OEt2 was distilled prior to use. Ethylenediamine was stirred over MS 4 Å for 24 h, then stored over solid KOH and distilled prior to use. TLC was carried out using SiO2 coated glass plates (Merck, 200×200×0.25 mm3) and spots were visualized by fluorescence quench at 254 nm or by staining with cerium ammonium sulfate or anisaldehyde solution. Flash column chromatography was performed on silica gel obtained from Sorbent Technologies (60 Å, 32-63 μm). Chromatography paper was obtained from Whatman (3 MM CHR). Analytical HPLC/MS was carried out on a Agilent Series 1100 instrument, using a Phenomenex Luna C18 3 μm column and ESI ionization. NMR spectra were recorded on a Varian I500, Varian M400, or Varian M300 spectrometer. Chemical shifts are reported in ppm as d and referenced to the residual internal solvent signal (CDCl3: 7.26 (1H) and 77.16 (13C); DMSO-D6 1: 2.50 (H) and 39.52 (13C)). High resolution mass spectrometry was performed on a Bruker microTOF instrument using ESI. Enzymes were obtained and handled as previously reported.11o 14C-Heptaprenyl-Lipid II was obtained as previously described.11c Moenomycin A was isolated from flavomycin as reported.26 Autoradiography was carried out using storage phosphor screens by GE Healthcare and a GE Healthcare Typhoon 9400 scanner.
4.2. General procedure for the Z-prenylation of allylic alcohols 14, 17, 19 and 20
The allylic alcohol was dissolved in Et2O (1 M) and cooled to −30 °C before PBr3 (0.37 equiv.) was added. MeOH and H2O were added as TLC analysis indicated completion of the reaction. The mixture was extracted with hexane and the organic phases were washed with water, NaHCO3 (sat.), and brine and dried over MgSO4. Evaporation of the solvent in vacuo provided the allylic bromide, which was used as such in the next step.
NaH (2.2 equiv.) was suspended in THF (1.1 M) cooled to 0 °C before ethyl acetoacetate (2.0 equiv.) were added. After 15 min nBuLi (2.5 M, 2.1 equiv.) was added and the mixture was stirred for 15 min before a solution of the allylic bromide (1 equiv., 2.0 M in THF) was added. After 1.5 h, Et2O (1 x reaction volume) and 3 M HCl (1/2 x reaction volume) were added. The phases were separated and the organic layer was washed with H2O and brine and dried over MgSO4. Evaporation of the solvent in vacuo and column chromatography provided the desired ketoester.
The ketoester previously obtained (1 equiv.), NEt3 (2.0 equiv.), and DMAP (0.2 equiv.) were dissolved in DMPU (0.3 M in ketoester) and cooled to −30 °C. Diphenyl chlorophosphate (2.0 equiv.) was added and the solution was brought to rt over 3 h. NH4Cl (sat.) and Et2O was added, the phases were parted and the organic layer was washed with H2O and brine and dried over MgSO4. Removal of the solvent in vacuum and column chromatography provided the desired enol ether.
The above mentioned enol phosphate was dissolved in THF (0.06 M), NMP (9.0 equiv.) and Fe(acac)3 (0.05 equiv.) were added, and the mixture was cooled to −40 °C. MeMgCl in THF (3.0 equiv.) was added and the solution was stirred for 30 min before the NH4Cl (sat.) was added. The aqueous layer was extracted with Et2O and the combined organic phases were washed with 1 M HCl, NaHCO3 (sat.) and brine and dried over MgSO4. The solvent was evaporated in vacuo and the residue was dissolved in PhMe (0.5 M), cooled to −78 °C and treated with DIBAL-H (1.0 M in PhMe, 2.7 equiv.). After 1 h MeOH was added and the mixture was brought to rt. NH4Cl (sat.), 1 M HCl, and Et2O were added, the suspension was stirred for 1.5 h and then extracted with Et2O. The organic phases were washed with water and brine and dried over MgSO4. Evaporation of the solvent and column chromatography provided the desired prenylated allylic alcohol. The analytical data obtained matched the published data.27
4.3. Phenyl 6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (27)
Acetal 25 (2.17 g, 4.44 mmol) was dissolved in CH2Cl2 (11.1 mL) and cooled to 0 °C. Triethylsilane (3.54 mL, 22.2 mmol) and TFA (1.70 mL, 22.2 mmol) were added and the mixture was stirred for 4 h at 0 °C. Volatile components were removed in vacuo and the residue was purified by column chromatography on SiO2, eluting with hexane/EtOAc 1/1 to obtain the title compound 27 as colorless solid (2.04 g, 4.15 mmol, 93%).16 Rf (hexane/EtOAc 1/1) 0.33; dH (300 MHz CDC13) 7.83 (br, 2H), 7.73-7.70 (m, 2H), 7.39-7.26 (m, 6H), 7.22-7.19 (m, 3H), 5.60 (d, J 10.2 Hz, 1H), 4.60 (d, J 11.7 Hz, 1H), 4.56 (d, J 11.7 Hz, 1H), 4.36-4.30 (m, 1H), 4.23-4.16 (m, 1H), 3.85-3.75 (m, 2H), 3.70-3.55 (m, 2H), 3.35 (br, 1H), 3.00 (br, 1H); HRMS (ESI): M+Na+, found 514.1293; C27H25NO6SNa requires 514.1295.
4.4. Phenyl 4,6-O-benzylidene-3-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside S-oxide(26)
Acetal 25 (3.60 g, 7.40 mmol) was dissolved in DMF (37 mL) and cooled 0 °C before NaH (356 mg, 60% in mineral oil) was added. After gas evolution had ceased nBu4NI (260 mg, 704 mmol) and BnBr (0.96 mL, 8.1 mmol) were added. After stirring the reaction at RT for 20 h, MeOH (5 mL) was added dropwise and the resulting mixture was partitioned between H2O and CH2Cl2. The phases were separated and the aqueous phase was extracted three times with CH2Cl2. The combined organic phases were washed with 10% LiCl in water and dried over Na2SO4. After removal of the volatiles in vacuo the residue was purified by column chromatography on SiO2 (hexane/EtOAc 9/1 → 8/2 → 7/3) to obtain 3-benzyloxy-25 as colorless solid (3.01 g, 5.18 mmol, 70%).28 Rf (hexane/EtOAc 1/1) 0.81; dH (300 MHz CDC13) 7.90-7.85 (m, 1H), 7.73-7.64 (m, 4H), 7.53-7.50 (m, 2H), 7.42-7.38 (m, 6H), 7.26-7.24 (m, 2H), 6.99-6.86 (m, 5H), 5.62 (s, 1H), 5.61(d, J 10.5 Hz, 1H), 4.77 (d, J 12.3 Hz), 4.49 (d, J 12.3 Hz, 1H), 4.47-4.40 (m, 2H), 4.33-4.25 (m, 1H), 3.88-3.70 (m, 3H); HRMS (ESI): M+Na+, found 602.1617; C34H29NO6SNa requires 602.1608.
A solution of this sulfide (3.01 g, 5.19 mmol) in CH2Cl2 (173 mL) was cooled to −78 °C and a solution of mCPBA (77% pure, as commercially available, 896 mg) in CH2Cl2 (104 mL) was added over 1 h. The cloudy reaction mixture was then brought to rt over 45 min and then quenched with Na2SO3 (10% aqueous solution). The phases were separated and the organic phase was washed with NaHCO3 (sat.) and brine and dried over Na2SO4. Removal of the solvent in vacuo and column chromatographic purification of the residue (SiO2, hexane/EtOAc 6/4) gave the title compound as colorless solid and as a 1/1 mixture of diastereomers (2.72 g, 4.57 mmol, 88%). Rf (hexane/EtOAc 6/4) 0.25; Due to formation of two sulfoxide diastereomers NMR data is not given; HRMS (ESI): M-HOSPh+Na+, found 492.1422; C27H24NO7S requires 492.1418.
4.5. Phenyl 4,6-O-benzylidene-3-O-benzyl-2-deoxy-2-phthalimido-b-D-glucopyranosyl-(1→4)-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (28)
Glycosyl acceptor 27 (1.29 g, 2.62 mmol), 4-allyl-1,2-dimethoxybenzene (1.50 mL, 8.75 mmol), and 2,6-di-tert-butyl-4-methylpyridine (1.08 g, 5.25 mmol) were dissolved in benzene and concentrated in vacuo to remove traces of water. The residue was dissolved in CH2Cl2 (65.5 mL), MS 4Å (400 mg) was added and the suspension was stirred at rt for 45 min before it was cooled to −78 °C. After addition of Tf2O (0.29 mL, 1.8 mmol) a solution of glycosyl donor 26 (1.04 g, 1.75 mmol) in CH2Cl2 (44 mL) was added over 2 h by syringe pump. After these 2 h, the solution was stirred at −60 °C for 1 h, followed by 1 h at −40 °C. The reaction was washed with NaHCO3 (sat.) and the organic phase was dried over Na2SO4. Removal of the solvent in vacuo, followed by column chromatography on SiO2 (hexane/EtOAc 7/3) yielded disaccharide 28 as colorless solid (970 mg, 1.01 mmol, 58%). Rf (hexane/EtOAc 7/3) 0.44; dH (500 MHz CDC13) 7.89-7.82 (m, 2H), 7.74-7.68 (m, 4H), 7.51–7.48 (m, 1H), 7.49-7.39 (m, 3H), 7.32-7.26 (m, 4H), 7.19-7.15 (m, 1H), 7.12-7.08 (m, 1H), 7.06 (d, J 6.0 Hz, 1H), 6.96 (d, J 6.5 Hz, 1H), 6.00-6.84 (m, 2H), 5.58 (s, 1H), 5.47 (d, J 10.0 Hz, 1H), 5.31 (d, J 8.0 Hz, 1H), 4.75 (d, J 12.5 Hz, 1H), 4.48-4.44 (m, 2H), 4.40-4.34 (m, 2H), 4.23-4.16 (m, 2H), 4.08-4.00 (m, 2H), 3.79-3.64 (m, 4H), 3.50-3.47 (m, 1H), 3.26-3.20 (m, 2H); dC (125 MHz, CDCl3) 168.4, 167.8, 138.3, 137.9, 137.2, 134.3, 134.2, 133.1, 132.0, 131.7, 129.4, 129.0, 128.6, 128.4, 128.3, 128.3, 128.1, 127.7, 127.6, 127.6, 126.3, 124.1, 123.7, 123.5, 101.7, 100.0, 83.27, 82.7, 81.6, 78.2, 74.4, 73.1, 71.2, 68.5, 68.2, 66.3, 56.0, 55.2, 30.4; HRMS (ESI): M+Na+, found 983.2772; C55H48N2O12SNa+ requires 983.2820.
4.6. Phenyl 6-O-benzyl-3-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-glucopyranoside (30)
Disaccharide 28 (1.35 g, 1.40 mmol) was dissolved in CH2Cl2 (28.1 mL) and cooled to 0 °C. Triethylsilane (2.68 mL, 16.8 mmol) and BF3·OEt2 (0.36 mL, 2.8 mmol) were added and the mixture was stirred for 2 h at 0 °C. NaHCO3 (sat.) was added to the reaction mixture and the phases were parted. The organic phase was concentrated in vacuo and the residue was purified by column chromatography on SiO2 (hexane/EtOAc 6/4→1/1 to obtain the title compound 30 as colorless solid (950 mg, 0.986 mmol, 70%). Rf (hexane/EtOAc 1/1) 0.53; dH (400 MHz CDC13) 7.92–7.80 (m, 2H), 7.74–7.70 (m, 3H), 7.58–7.55 (m, 1H), 7.35–7.34 (m, 2H), 7.31–7.28 (m, 2H), 7.23–7.22 (m, 3H), 7.17–7.15 (m, 1H), 7.11–7.05 (m, 2H), 7.00–6.97 (m, 2H), 5.49 (d, J 10.5 Hz, 1H), 5.30 (d, J 9.0 Hz, 1H), 4.69 (d, J 12.5 Hz, 1H), 4.45–4.42 (m, 5H), 4.33–4.20 (m, 3H), 3.97 (s, 2H), 3.75–3.62 (m, 6H), 3.55–3.50 (m, 1H), 3.25 (m, 2H); 2.62 (m, 1H); dC (125 MHz, CDCl3) 168.4, 167.7, 138.3, 138.2, 138.5, 134.2, 134.2, 132.9, 132.3, 132.1, 132.0, 131.8, 128.9, 128.8, 128.6, 128.5, 128.5, 128.4, 128.1, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 123.9, 123.7, 123.5, 99.3, 83.4, 81,9, 79.0, 78.1, 74.8, 73.9, 73.8, 73.5, 73.0, 71.5, 70.1, 68.6, 55.6, 55.2; HRMS (ESI): M+Na+, found 985.2900; C55H5N2O12SNa+ requires 985.2977.
4.7. Phenyl 4,6-O-benzylidene-3-O-benzyl-2-deoxy-2-phthalimido-b-D-glucopyranosyl-(1→4)-6-O-benzyl-2-deoxy-2-phthalimido-1-thio-glucopyranoside S-oxide (29)
A solution of sulfide 28 (1.69 g, 1.76 mmol) in CH2Cl2 (59 mL) was cooled to −78 °C and a solution of mCPBA (77% pure, as commercially available, 434 mg) in CH2Cl2 (39 mL) was added over 1 h. The cloudy reaction mixture was then brought to rt over 45 min and quenched with Na2SO3 (10% aqueous solution). The phases were separated and the organic phase was washed with NaHCO3 (sat.) and brine and dried over Na2SO4. Removal of the solvent in vacuo and column chromatographic purification of the residue (SiO2, hexane/EtOAc 1/1) gave the title compound as colorless solid and as a 1/1 mixture of diastereomers (1.33 g, 1.36 mmol, 77%). Rf (hexane/EtOAc 6/4) 0.30; due to formation of sulfoxide diastereomers NMR spectroscopic is not given.
4.8. Tetrasaccharide 31
Glycosyl acceptor 30 (790 mg, 0.82 mmol), 4-allyl-1,2-dimethoxybenzene (0.88 mL, 5.1 mmol), and 2,6-di-tert-butyl-4-methylpyridine (0.63 g, 3.1 mmol) were dissolved in benzene and concentrated in vacuo to remove traces of water. The residue was dissolved in CH2Cl2 (20.5 mL), MS 4Å (150 mg) was added and the suspension was stirred at rt for 45 min before it was cooled to −78 °C. After addition of Tf2O (0.17 mL, 1.0 mmol) a solution of glycosyl donor 29 (1.0 g, 1.0 mmol) in CH2Cl2 (25.5 mL) was added over 2 h by syringe pump. After these 2 h, the solution was stirred at −60 °C for 1 h, followed by 1 h at −40 °C. The mixture was washed with NaHCO3 (sat.) and the organic phase was dried over Na2SO4. Removal of the solvent in vacuo, followed by column chromatography on SiO2 (hexane/EtOAc 6/4) yielded disaccharide 31 as colorless solid (818 mg, 0.451 mmol, 55%). Rf (hexane/EtOAc 6/4) 0.20; dH (500 MHz D6-DMSO) 7.98–6.80 (m, 48H), 6.75–6.60 (m, 3H), 5.59 (s, 1H), 5.43 (d, J 11.0 Hz, 1H), 5.30 (d, J 9.0 Hz, 1H), 5.12 (d, J 8.5 Hz, 1H), 5.06 (d J 7.5 Hz, 1H), 4.78 (d, J 12.5 Hz, 1H), 4.49–4.26 (m, 10H), 4.15–3.98 (m, 9H), 3.84–3.42 (m, 7H), 3.45–3.33 (m, 4H), 3.14–3.12 (m, 2H), 3.11–3.07 (m, 1H), 2.75–2.70 (m, 1H); dC (125 MHz, D6-DMSO) 168.8, 168.3, 168.0, 167.7, 138.5, 138.3, 138.1, 138.0, 137.7, 137.2, 134.5, 134.3, 134.2, 133.9, 132.2, 133.9, 132.7, 132.4, 132.1, 132.0, 131.8, 129.4, 128.9, 128.6, 128.5, 128.4, 128.3, 128.3, 128.0, 127.9, 127.8, 127.7, 127.6, 127.6, 127.5, 127.4, 127.4, 127.1, 126.3, 124.3, 123.9, 123.7, 123.5, 101.6, 99.9, 99.2, 96.9, 83.4, 82.8, 81.9, 81.3, 78.0, 76.5, 75.7, 74.7, 74.5, 74.4, 74.3, 74.2, 74.2, 73.1, 72.8, 72.8, 71.3, 69.6, 68.5, 68.2, 66.9, 66.2, 56.8, 56.0, 55.9, 55.2; HRMS (ESI): M+H+, found 1814.5944; C104H93N4O24S+ requires 1814.5929.
4.9. Tetraacetate 32
Tetraphthalate 31 (750 mg, 0.413 mmol) was dissolved in a mixture of THF (8.4 mL), MeCN (10.7 mL), EtOH (8.4 mL), and ethylenediamine (1.05 mL) and was stirred under an atmosphere of argon for 48 h at 60 °C. Volatile components were evaporated in vacuo and the residue was dissolved in MeOH (16.8 mL) and H2O (2.7 mL), cooled to 0 °C, and 10 mL of Ac2O were added in portions over 6 h. After evaporation of the volatiles in vacuo the residue was suspended in MeOH (50 mL). The suspension was centrifuged and the pellet dried over night in vacuo. The supernatant was concentrated in vacuo and treated as above (20 mL MeOH) to obtain the tile compound as amorphous solid (422 mg, 289 μmol, 70%). dH (500 MHz CDC13) 8.05–8.03 (m, 1H) 8.00–7.97 (m, 1H), 7.93–7.89 (m, 2H), 7.41–7.37 (m, 6H), 7.30–7.17 (m, 26 H), 5.69 (s, 1H), 4.87 (d, J 12.0 Hz, 1H), 4.80 (d, J 10.5 Hz), 4.70–4.66 (m, 3H), 4.58–4.44 (m, 10 H), 4.28 (d, J 12.0 Hz, 1H), 4.24–4.22 (m, 1H), 3.79–3.65 (m, 10H), 3.59–3.31 (m, 11H), 3.21–3.18 (m, 1H), 3.05–3.03 (m, 1H), 1.82 (s, 3H), 1.79 (s, 3H), 1.78 (s, 3H), 1.77 (s, 3H); dC (125 MHz, CDCl3) 169.9, 169.9, 169.8, 169.7, 139.9, 139.4, 139.4, 139.2, 139.0, 138.3, 135.7, 129.9, 129.5, 128.9, 128.9, 128.8, 128.8, 128.7, 128.6, 128.1, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7, 127.7, 127.1, 126.7, 101.9, 100.8, 100.6, 86.2, 81.3, 80.9, 79.6, 79.2, 78.2, 75.8, 75.0, 74.7, 74.2, 73.9, 73.8, 72.9, 72.9, 72.8, 72.6, 69.8, 69.4, 69.2, 68.3, 66.4, 56.7, 55.5, 54.4, 39.1, 23.8, 23.7, 23.6, 23.6; HRMS (ESI): M+H+, found 1461.5986; C80H93N4O20S+ requires 1461.6098.
4.10. GlcNAc-MurNAc-GlcNAc-MurNAc derivate 33
Compound 32 (150 mg, 103 μmol) was dissolved in DMF (5.15 mL) and cooled to 0 °C. NaH (60% in mineral oil, 102 mg, ca. 25 equiv.) was added and the suspension was vigorously stirred for 10 min until gas evolution had ceased. S-2-bromo propionic acid (93 μL, 1.03 mmol) was added dropwise and the suspension was allowed to reach room temperature over 3 h before acetic acid (0.58 mL, 10 mmol) was carefully added. Volatile components were removed in vacuum over night and the residue was dissolved in benzene (1.5 mL) and MeOH (0.5 mL). This solution was cooled to 0 °C and TMS-diazomethane (2 M in hexane, 1.5 mL) was slowly added. LC/MS analysis at this point ensured that all free acid had been methylated and the reaction was quenched by addition of AcOH (0.2 mL). The mixture was concentrated in vacuum and partitioned between EtOAc and NaHCO3. The organic phase was dried over Na2SO4, concentrated in vacuum, and the residue was purified by column chromatography (SiO2, CH2Cl2/MeOH 96/4) to obtain the title compound as colorless solid (126 mg, 77 μmol, 75%). Rf (CH2Cl2/MeOH 96/4) 0.33; dH (500 MHz CDC13) 7.60–7.59 (d, 1H), 7.52–7.14 (m, 34 H), 5.58 (s, 1H), 4.88 (d, J 12.5 Hz, 1H), 4.79 (d, J 11.5 Hz, 2H), 4.73–4.57 (m, 4H), 4.57–4.34 (m, 10 H), 4.26–4.23 (m, 2H), 4.18 (br, 1H), 4.05–3.99 (m, 2H), 3.89 (m, 1H), 3.78–3.41 (m, 18 H), 3.77 (s, 3H), 3.70 (s, 3H), 3.35–3.30 (m, 1H), 3.30–3.25 (m, 1H), 3.25–3.20 (m, 1H), 2.04 (s, 3H), 1.97 (s, 3H), 1.96 (s, 3H), 1.73 (s, 3H), 1.32 (d J 7.5 Hz, 3H), 1.18 (d J 7.0 Hz, 3H); dC (125 MHz, CDCl3) 176.6, 176.4, 172.7, 172.0, 170.7, 169.9, 162.8, 139.0, 138.6, 138.6, 138.5, 138.0, 137.4, 135.9, 130.7, 129.4, 129.4, 129.0, 129.0, 128.8, 128.7, 128.6, 128.3, 128.2, 128.1, 128.0, 127.8, 127.8, 127.6, 126.9, 126.2, 101.7, 101.5, 100.7, 100.3, 88.8, 82.8, 82.8, 80.4, 79.6, 79.5, 78.1, 77.0, 76.7, 75.0, 74.6, 74.2, 73.9, 73.7, 73.4, 72.0, 71.9, 70.1, 69.3, 69.0, 67.3, 66.0, 55.8, 55.2, 53.8, 52.5, 52.4, 52.2, 36.7, 31.7, 23.9, 23.6, 23.6, 23.2, 19.0, 18.8; HRMS (ESI): M+H+, found 1633.6852; C88H105N4O24S+ requires 1633.6834.
4.11. Determination of the IC50 of moenomycin A
Solutions of PBP1a (E. coli, 20 nM) in 50 mM HEPES pH 7.5, 10 mM CaCl2, 1000 units/mL penicillin G, and 20% DMSO (v/v) were incubated for 20 min with solutions of moenomycin A of different concentrations. In the pre-incubation experiments the PBP1a solutions were first incubated with Gal-Lipid IV (1.36 μM) for 20 min. Reactions were initiated by addition of 14C-Lipid II (4 μM, 3H/1H 1/3) and were quenched after 20 min (MmA only) and 45 min (+ LPIV) with an equal volume of 10% Triton X-100 solution on ice. The mixtures obtained were separated by paper strip chromatography (isobutyric acid/1 M NH4OH 5/3). The paper strips were cut to obtain the top 4/5 (containing monomer) and the lower 1/5 (containing PG). These parts were immersed in EcoLite(+) scintillation cocktail (MP Biomedical) and analyzed by a LS 6500 scintillation counter (Beckman Coulter). The normalized activity (A) was calculated relative to a control reaction (no MmA, no LPIV) and plotted as a function of log(c(MmA)). The IC50 value was extracted with OriginPro 7.0.
4.12. SDS-page assay
Solutions of the enzymes (0.24 μM or 0.48 μM) in 50 mM HEPES pH 7.5, 10 mM CaCl2, 1000 units/mL penicillin G were treated with solutions of MmA in different concentrations for 20 min. The reaction was initiated by addition of 14C-Lipid II (8 μM, 14C/13C 3/1) and quenched after 20 min by heat (90 °C, 5 min). The volatiles were removed by vacuum centrifugation and the residue was taken up in buffer (15% SDS, Lämmli 6X) and analysed by SDS-PAGE.25
Supplementary Material
Scheme 3.
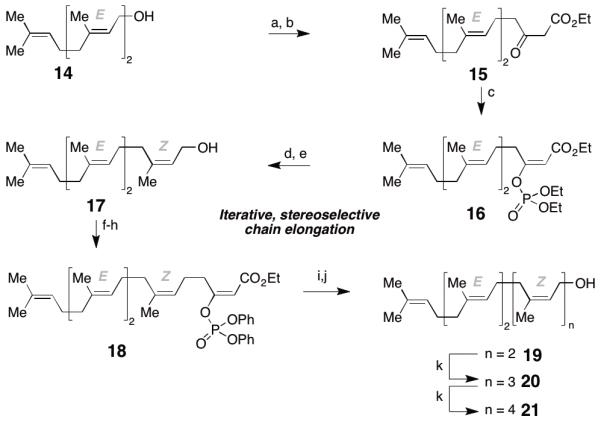
Reagents and conditions: a) PBr3 (0.37 equiv.), Et2O, −30 °C to RT, 96%; b) ethyl acetoacetate (2 equiv.), NaH (2.2 equiv.), BuLi (2.1 equiv.), THF, 0 °C, 87%; c) ClPO(OEt)2 (1.1 equiv.), NEt3 (1.1 equiv.), DMAP (0.1 equiv.), DMPU, 0 °C, 69%; d) CuI (3 equiv.), MeLi (3 equiv.), MeMgCl (5 equiv.) THF; e) DIBAL-H (2.2 equiv.), PhMe, 66% over 2 steps; f) PBr3 (0.37 equiv.), Et2O, −30 °C to RT; g) ethyl acetoacetate (2 equiv.), NaH (2.2 equiv.), BuLi (2.1 equiv.), THF, 0 °C, 70% (2 steps); h) ClPO(OPh)2 (2 equiv.), NEt3 (2 equiv.), DMAP (0.2 equiv.), DMPU, 0 °C, 86%; i) MeMgCl (3 equiv.), Fe(acac)3 (0.05 equiv.), THF/NMP = 20/1; j) DIBAL-H (2.2 equiv.), PhMe, 75% (2 steps); k) see f-j, 45-60% yield over 5 steps.
Scheme 6.
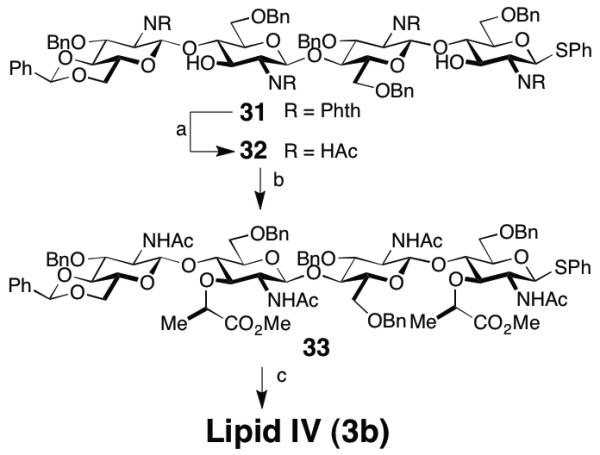
Reagents and conditions: a) ethylene diamine, EtOH/MeCN/THF, 60 °C, 48 h; then: Ac2O, MeOH/H2O, 70%; b) NaH, DMF, S-2-bromopropionic acid, then: TMSCHN2, MeOH, PhH; 75%; c) see ref. 11o
Acknowledgments
Dr. Tania Lupoli and Dr. Matthew Lebar are acknowledged for their help with the biochemical experiments. This research was supported by the National Institutes of Health (GM076710 & GM066174) and by the German Academic Exchange Service (DAAD; postdoctoral fellowship to C. M. G.).
Footnotes
Supporting Information
NMR spectra of new compounds are available in the supporting information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Holtje JV. Microbiol. Mol. Biol. Rev. 1998;62:181. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollmer W, Bertsche U. Biochim. Biophys. Acta. 2008;1778:1714. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 3.a) Arias CA, Murray BE. N. Engl. J. Med. 2009;360:439. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]; b) Cohen ML. Nature. 2000;406:762. doi: 10.1038/35021206. [DOI] [PubMed] [Google Scholar]
- 4.Kahne D, Leimkuhler C, Lu W, Walsh C. Chem. Rev. 2005;105:425. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 5.Walker S, Chen L, Hu Y, Rew Y, Shin D, Boger DL. Chem. Rev. 2005;105:449. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 6.Fisher JF, Meroueh SO, Mobashery S. Chem. Rev. 2005;105:395. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 7.Welzel P. Chem. Rev. 2005;105:4610. doi: 10.1021/cr040634e. [DOI] [PubMed] [Google Scholar]
- 8.a) Halliday J, McKeveney D, Muldoon C, Rajaratnam P, Meutermans W. Biochem. Pharmacol. 2006;71:957. doi: 10.1016/j.bcp.2005.10.030. [DOI] [PubMed] [Google Scholar]; b) Ostash B, Walker S. Curr. Opin. Chem. Biol. 2005;9:459. doi: 10.1016/j.cbpa.2005.08.014. [DOI] [PubMed] [Google Scholar]; c) Wright GD. Science. 2007;315:1373. doi: 10.1126/science.1140374. [DOI] [PubMed] [Google Scholar]; d) Goldman RC, Gange D. Curr. Med. Chem. 2000;7:801. doi: 10.2174/0929867003374651. [DOI] [PubMed] [Google Scholar]
- 9.a) Butaye P, Bevriese LA, Haesebrouck F. Antimicrob. Agents Chemother. 2001;45:1374. doi: 10.1128/AAC.45.5.1374-1378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hentschel S, Kusch D, Sinell H-J. Zentralblatt für Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene, 1. Abteilung, Originale, Reihe B: Hygiene, Betriebshygiene, präventive Medizin. 1979;168:546. [PubMed] [Google Scholar]
- 10.Goldman RC, Gange D. Curr. Med Chem. 2000;7:801. doi: 10.2174/0929867003374651. [DOI] [PubMed] [Google Scholar]
- 11.a) Men H, Park P, Ge M, Walker S. J. Am. Chem. Soc. 1998;120:2484. [Google Scholar]; b) Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl H-G, de Kruijff B. Science. 1999;286:2361. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]; c) Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S. J. Am. Chem. Soc. 2001;123:3155. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]; d) Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S. J. Am. Chem. Soc. 2001;123:3155. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]; e) Schwartz B, Markwalder JA, Wang Y. J. Am. Chem. Soc. 2001;123:11638. doi: 10.1021/ja0166848. [DOI] [PubMed] [Google Scholar]; f) Lazar K, Walker S. Curr. Opin. Chem. Biol. 2002;6:786. doi: 10.1016/s1367-5931(02)00355-1. [DOI] [PubMed] [Google Scholar]; g) VanNieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Winger BE, Hornback WJ, Saha SL, Aikins JA, Blaszczak LC. J. Am. Chem. Soc. 2002;124:3656. doi: 10.1021/ja017386d. [DOI] [PubMed] [Google Scholar]; h) Breukink E, van Heusden HE, Vollmerhaus PJ, Swiezewska E, Brunner L, Walker S, Heck AJ, de Kruijff B. J. Biol. Chem. 2003;278:19898. doi: 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]; i) Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Proc. Natl. Acad. Sci. USA. 2003;100:5658. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Hu Y, Helm JS, Chen L, Ye XY, Walker S. J. Am. Chem. Soc. 2003;125:8736. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]; k) Breukink E, van Heusden HE, Vollmerhaus PJ, Swiezewska E, Brunner L, Walker S, Heck AJR, de Kruijff B. J. Biol. Chem. 2003;278:19898. doi: 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]; l) Chen L, Yuan Y, Helm JS, Hu Y, Rew Y, Shin D, Boger DL, Walker S. J. Am. Chem. Soc. 2004;126:7462. doi: 10.1021/ja047879t. [DOI] [PubMed] [Google Scholar]; m) Breukink E, de Kruijff B. Nat. Rev. Drug Disc. 2006;5:321. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]; n) Perlstein DL, Zhang Y, Wang TS, Kahne DE, Walker S. J. Am. Chem. Soc. 2007;129:12674. doi: 10.1021/ja075965y. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Zhang Y, Fechter EJ, Wang T-SA, Barrett D, Walker S, Kahne DE. J. Am. Chem. Soc. 2007;129:3080. doi: 10.1021/ja069060g. [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Vollmer W, Blanot D, de Pedro MA. FEMS Microbiol. Rev. 2008;32:149. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]; q) Lupoli TJ, Taniguchi T, Wang T-S, Perlstein DL, Walker S, Kahne DE. J. Am. Chem. Soc. 2009;131:18230. doi: 10.1021/ja908916z. [DOI] [PMC free article] [PubMed] [Google Scholar]; r) Perlstein DL, Wang AT-S, Doud EH, Kahne D, Walker S. J. Am. Chem. Soc. 2010;132:48. doi: 10.1021/ja909325m. [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Lupoli TJ, Tsukamoto H, Doud EH, Wang T-SA, Walker S, Kahne D. J. Am. Chem. Soc. 2011;133:10748. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]; t) Wang T-SA, Lupoli TJ, Sumida Y, Tsukamoto H, Wu Y, Rebets Y, Kahne D, Walker S. J. Am. Chem. Soc. 2011;133:8528. doi: 10.1021/ja2028712. [DOI] [PMC free article] [PubMed] [Google Scholar]; u) Shih H-W, Chen K-T, Cheng T-JR, Wong C-H, Cheng W-C. Organic Letters. 2011 doi: 10.1021/ol201806d. doi: 10.1021/ol201806d. [DOI] [PubMed] [Google Scholar]
- 12.a) Casey CP, Marten DF. Synth. Commun. 1973;3:321. [Google Scholar]; b) Casey CP, Marten DF, Boggs RA. Tetrahedron Lett. 1973:2071. [Google Scholar]; c) Casey CP;, Marten DF. Tetrahedron Lett. 1974:925. [Google Scholar]
- 13.a) Sum F-W, Weiler L. Can. J. Chem. 1979;57:1431. [Google Scholar]; b) Alderdice M, Spino C, Weiler L. Tetrahedron Lett. 1984;25:1643. [Google Scholar]
- 14.Hayashi N, Nakada M. Tetrahedron Lett. 2009;50:232. [Google Scholar]
- 15.Tsukamoto H, Kahne D. Bioorg. Med. Chem. Lett. 2011;21:5050. doi: 10.1016/j.bmcl.2011.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain RK, Matta KL. Carbohydr. Res. 1992;226:91. doi: 10.1016/0008-6215(92)84057-y. [DOI] [PubMed] [Google Scholar]
- 17.Averso MC, Barattucci A, Bonaccorsi P. Tetrahedron. 2008;64:7659. [Google Scholar]
- 18.a) Kahne D, Walker S, Cheng Y, Van Engen D. J. Am. Chem. Soc. 1989;111:6881. [Google Scholar]; b) Gildersleeve J, Pascal RA, Jr., Kahne D. J. Am. Chem. Soc. 1998;120:5961. [Google Scholar]; c) Gildersleeve J, Smith A, Sakurai K, Raghavan S, Kahne D. J. Am. Chem. Soc. 1999;121:6176. [Google Scholar]
- 19.Debenham J, Rodebaugh R, Fraser-Reid B. Liebigs Ann. 1997:791. [Google Scholar]
- 20.For a recent study, see: Ohlin M, Johnsson R, Ellervik U. Carbohydr. Res. 2011;346:1358. doi: 10.1016/j.carres.2011.03.032.
- 21.See supporting information for details.
- 22.Galactose-Lipid IV (Gal-Lipid IV) was used since Lipid IV itself was found to be polymerized by GTs. See ref. 11 q,r,t
- 23.Polymerization reactions were run for 45 and 20 min with and without lipid IV, respectively, which leads to maximum conversion in both cases in the absence of moenomycin see ref. 11t
- 24.a) Lovering AL, de Castro LH, Lim D, Strynadka NCJ. Science. 2007;315:1402. doi: 10.1126/science.1136611. [DOI] [PubMed] [Google Scholar]; b) Yuan YQ, Barrett D, Zhang Y, Kahne D, Sliz P, Walker S. P. Natl. Acad. Sci. USA. 2007;104:5348. doi: 10.1073/pnas.0701160104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yuan YQ, Fuse S, Ostash B, Sliz P, Kahne D, Walker S. ACS Chem. Biol. 2008;3:429. doi: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lovering AL, Gretes M, Strynadka NC. Curr. Opin. Struct. Biol. 2008;18:534. doi: 10.1016/j.sbi.2008.07.002. [DOI] [PubMed] [Google Scholar]; e) Heaslet H, Shaw B, Mistry A, Miller AA. J. Struct. Biol. 2009;167:129. doi: 10.1016/j.jsb.2009.04.010. [DOI] [PubMed] [Google Scholar]; f) Sung MT, Lai YT, Huang CY, Chou LY, Shih HW, Cheng WC, Wong CH, Ma C. P. Natl. Acad. Sci. USA. 2009;106:8824. doi: 10.1073/pnas.0904030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett D, Wang TS, Yuan Y, Zhang Y, Kahne D, Walker S. J. Biol. Chem. 2007;282:31964. doi: 10.1074/jbc.M705440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adachi M, Zhang Y, Leimkuhler C, Sun B, LaTour JV, Kahne DE. J. Am. Chem. Soc. 2006;128:14012. doi: 10.1021/ja065905c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Brown RCD, Bataille CJ, Hughes RM, Kenney A, Luker TJ. J. Org. Chem. 2002;67:8079. doi: 10.1021/jo026295b. [DOI] [PubMed] [Google Scholar]; b) Yu JS, Kleckley TS, Wiemer DF. Org. Lett. 2005;7:4803. doi: 10.1021/ol0513239. [DOI] [PubMed] [Google Scholar]; c) Danilov LL, Druzhinia TN, Kalinchuk NA, Maltsev SD, Shibaev VN. Chem. Phys. Lipids. 1989;51:191. doi: 10.1016/0009-3084(89)90006-6. [DOI] [PubMed] [Google Scholar]
- 28.Chiara JL, Garciá Á, Cristóbal-Lumbroso G. J. Org. Chem. 2005;70:4143. doi: 10.1021/jo050185y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.