

Background: Cross-linking of TCR to CTLA-4 attenuates TCR signaling and inhibits T activation.
Results: A novel bispecific fusion protein comprising CD80 mutant (CD80w88a) and LAG-3 was designed to promote TCR and CTLA-4 cross-linking via MHCII.
Conclusion: TCR and CTLA-4 cross-linking resulted in T cell inhibition and Foxp3+ regulatory T cell differentiation.
Significance: This novel bispecific protein may represent a new class of therapeutics for immune modulation.
Keywords: Biotechnology, Cellular Immune Response, Protein Design, T Cell Biology, T-cell Receptor, Bispecific Biologics, CTLA-4, Cross-linking, Regulatory T Cells, TGF-beta
Abstract
Cross-linking of ligand-engaged cytotoxic T lymphocyte antigen-4 (CTLA-4) to the T cell receptor (TCR) during the early phase of T cell activation attenuates TCR signaling, leading to T cell inhibition. To promote this event, a bispecific fusion protein comprising a mutant mouse CD80 (CD80w88a) and lymphocyte activation antigen-3 was engineered to concurrently engage CTLA-4 and cross-link it to the TCR. Cross-linking is expected to be attained via ligation of CTLA-4 first to MHCII and then indirectly to the TCR, generating a CTLA-4-MHCII-TCR trimolecular complex that forms between T cells and antigen-presenting cells during T cell activation. Treating T cells with this bispecific fusion protein inhibited T cell activation. In addition, it induced the production of IL-10 and TGF-β and attenuated AKT and mTOR signaling. Intriguingly, treatment with the bispecific fusion protein also directed early T cell differentiation into Foxp3-positive regulatory T cells (Tregs). This process was dependent on the endogenous production of TGF-β. Thus, bispecific fusion proteins that engage CTLA-4 and co-ligate it to the TCR during the early phase of T cell activation can negatively regulate the T cell response. Bispecific biologics with such dual functions may therefore represent a novel class of therapeutics for immune modulation. These findings presented here also reveal a potential new role for CTLA-4 in Treg differentiation.
Introduction
Cytotoxic T lymphocyte associated antigen-4 (CTLA-4),3 which is a well established negative regulator of the T cell response and is also known as CD152, is critical for the maintenance of T cell homeostasis and self-tolerance (1–6). The mechanisms by which CTLA-4 exerts its immune inhibitory function are multifaceted and can occur directly via conventional effector T cells or indirectly via regulatory T cells (7–10).
CTLA-4 is homologous to the co-stimulatory molecule CD28 and shares the same ligands, CD80 (B7.1) and CD86 (B7.2) (3, 11), which are expressed on the surface of antigen-presenting cells (APCs). However, differential binding of CD80/CD86 on APCs to CD28 and CTLA-4 on effector T cells leads to opposing outcomes, with CD28 triggering T cell activation and CTLA-4 causing T cell inhibition. One mechanism by which CTLA-4 may induce T cell inhibition involves recruitment of the Src homology-2 domain-containing protein tyrosine phosphatase-1 (12) and protein phosphatase 2A (13, 14) to the vicinity of the T cell receptor (TCR) in the immune synapse (15, 16). This recruitment may result in dephosphorylation of the signaling molecules within the TCR complex (17) and consequent termination of T cell activation.
The timing of CD80/CD86 engagement of CD28 or CTLA-4 during T cell activation dictates the outcome of the effector T cell response. Because CD28 is constitutively expressed on T cells and the expression of CTLA-4 is only induced following T cell activation, peaking 2–3 days later (18), extensive T cell activation would have occurred prior to CTLA-4 engagement. Hence, the main role of CTLA-4 is to act as a safeguard against an excessive T cell response rather than to inhibit T cell activation. However, early engagement of CTLA-4 by its ligand and its subsequent cross-linking to the TCR can prematurely dampen TCR signaling, causing T cell inhibition and hyporesponsiveness, or anergy. This concept has been validated experimentally using a variety of methods, including the following: (i) cross-linking T cell-activating antibodies (anti-CD3/anti-CD28) using an agonistic anti-CTLA-4 antibody by co-immobilization on a bead or via a secondary antibody (19–21); (ii) molecularly engineering a surface-linked agonistic scFv against CTLA-4 on an APC (17, 22, 23); and (iii) chemically cross-linking antibodies that recognize specific antigens on an APC to an agonistic anti-CTLA-4 antibody (24–26).
For effective inhibition of T cell activation, the configuration of TCR and CTLA-4 ligands (MHCII and CD80/CD86 or agonist antibodies to CTLA-4) is also important. These ligands must be provided in the cis configuration and expressed on the same APC that activates the T cell (17). Hence, effective CTLA-4-mediated inhibition of T cells requires both proper configuration of the effector molecules and correct timing of their interactions. These requirements are met when the peptide-MHCII complex (pMHCII) and CD80/CD86 on APCs bind to the TCR and CTLA-4 on T cells during the immune synapse-formation phase of T cell activation (17, 27).
To develop a protein mimic that can inhibit T cell activation, a bispecific fusion protein comprising moieties that selectively bind and activate CTLA-4 and co-ligate it to the TCR was generated. The fusion construct was designed to cross-link MHCII to CTLA-4; both are then drawn to the TCR, generating the CTLA-4-MHCII-TCR trimolecular complex within immune synapses. Preferential binding of the bispecific fusion protein to CTLA-4 over CD28 was attained using mutant CD80 (CD80w88a, referred to hereafter as CD80wa), which contains alanine instead of tryptophan at amino acid 88 (numbered in mouse CD80), as the ligand. CD80wa binds CTLA-4 but exhibits minimal affinity for CD28 (28). Lymphocyte activation gene-3 (LAG-3), a natural ligand of MHCII, was selected as the other binding component of the bispecific fusion protein (29, 30). We show that a fusion protein with such bifunctionality effectively inhibits T cell activation and stimulates anti-inflammatory cytokines IL-10 and TGF-β production. More importantly, this bispecific fusion protein also directed T cell differentiation into highly suppressive Foxp3+ Tregs. This did not occur when the well established co-stimulatory inhibitor CTLA-4Ig was used instead (31, 32). Therefore, early engagement of CTLA-4 and cross-linking of CTLA-4 to the TCR during T cell activation could actively influence T cell differentiation. Such bispecific fusion proteins might thus represent a novel class of biologics that could be used to control excessive T cell responses in autoimmune diseases.
EXPERIMENTAL PROCEDURES
Animals
Wild-type C57BL/6, BALB/c, and Foxp3-enhanced green fluorescent protein (Foxp3-EGFP) knock-in mice (CD90.2+) were purchased from The Jackson Laboratory. Animal experiments were conducted in accordance with the guidelines issued by the U. S. Department of Health and Human Services (NIH publication no. 86-23) and were approved by Genzyme's Institutional Animal Care and Use Committee.
Antibodies and Reagents
Functional grade or fluorescently labeled anti-mouse CD3 (clone 145-2C11), CD4, CD28 (clone 37.51), CTLA-4 (UC9-4F10), CTLA-4 (9H10), CD80 (B7-1), CD62L, CD44, CD25, and Foxp3 antibodies were purchased from eBioscience or BD Biosciences. Anti-TGF-β (clone 1D11) and control mouse IgG1 isotype (clone 13C4) were from Genzyme Corporation. Goat anti-mouse CD80 and LAG-3 polyclonal antibodies, CTLA-4-Fc, CD28-Fc, and CD80-Ig were purchased from R&D Systems. Mouse LAG-3Ig was obtained from Enzo Lifesciences, Inc. Horse anti-goat IgG coupled to horseradish peroxidase was purchased from Abcam, and goat anti-rabbit IgG and anti-mouse IgG coupled to horseradish peroxidase were purchased from Pierce. Protein G-Sepharose was obtained from BioVision. CFSE, ultralow Ig FBS, EX-Cell 325 serum-free media and other cell culture media were from Invitrogen. Non-cytolytic form of mouse CTLA-4Ig and all common chemicals were obtained from Sigma unless otherwise noted.
Bispecific Fusion Protein Construction, Protein Expression, and Purification
Plasmids encoding a bispecific fusion protein comprising the extracellular domain of CD80w88a, LAG-3 extracellular domains 1 and 2, and the Fc of mouse IgG2a (CD80wa-LAG-3-Fc, referred to hereafter as BsB throughout this work), and encoding a fusion protein comprising CD80wa and the Fc of mouse IgG2a without LAG-3 (CD80wa-Fc, referred to hereafter as BsBΔ) were synthesized by Genscript and cloned into the in-house protein expression vector pGZ6. Stable Chinese hamster ovary cell lines expressing the fusion proteins were generated following selection with methotrexate at 100 nm. Stable cell pools were expanded under selection in EX-Cell 325 media containing 1% ultralow IgG FBS (Invitrogen) or lacking serum. Fusion proteins in pooled culture media were purified using a protein G column followed by size exclusion chromatography on Superose 6 column to remove aggregates.
Isolation of Naïve T Cells
Naive T cells from the spleens and lymph nodes of either wild-type C57BL/6 or Foxp3-EGFP knock-in mice were purified by magnetic separation followed by flow cytometry. Cells were first negatively selected by magnetic cell separation (Miltenyi Biotech). Naïve T cells from female C57BL/6 mice were sorted as CD4+CD25−CD62LhiCD44low cells, whereas naïve T cells from Foxp3-EGFP knock-in mice were sorted as CD4+CD25−CD62LhiCD44lowGFP− cells. All cell populations were >98% pure.
T Cell Inhibition and Treg Induction Assays
In a typical assay, 105 naïve T cells were mixed in round-bottomed 96-well plates with 105 irradiated APCs from allogenic mice pre-activated for 48 h with 1 μg/ml LPS. The test constructs BsB, BsBΔ, mouse IgG2a, LAG-3Ig or mouse CTLA-4Ig were added to the cultured cells at different concentrations (up to 100 μg/ml) as indicated in the figures. A typical concentration of BsB used in these studies was 50 μg/ml, at which the effect appeared to be saturated. For T cell inhibition assays, cells were cultured for 2 days, followed by media collection for IL-2 measurement using the respective ELISA kits according to the manufacturer's instructions. To assay Treg induction, the cells were cultured for 5 days before being analyzed by flow cytometry. Media were collected for analysis using IL-10 and TGF-β ELISA kits per the manufacturer's instructions. To assess the role of CTLA-4 and TGF-β in Treg induction, blocking antibodies to CTLA-4 (anti-CTLA-4, clone 9H10), TGF-β antibody (1D11), or an IgG isotype control was added to the culture at 10 μg/ml, respectively.
To assess T cell proliferation, naïve T cells from female C57BL/6 mice were sorted and labeled with 5 μm CFSE for 5 min at 37 °C. The cells were then washed to remove unbound CFSE and used in T cell inhibition and Treg induction assays as described above. Cells were cultured for 5 days to allow them to divide before being analyzed by flow cytometry for CFSE dilution. For detection of Foxp3 in T cells from C57BL/6 mice, cells were stained for surface markers before being permeabilized with Fix/Perm buffer (eBioscience) and stained with PE-Cy7 conjugated anti-Foxp3 antibody (clone FJK-16s, eBioscience).
Treg Restimulation Assay
Antibody co-immobilization was used in the assays for Treg induction and restimulation. Briefly, anti-CD3 (1 μg/ml), anti-CD28 (5 μg/ml), and BsB or mouse IgG (all at 10 μg/ml) were co-immobilized onto 96-well plates at 4 °C overnight. The next morning, the plates were washed with PBS and blocked with culture media for 1 h before seeding with GFP− naïve T cells. The GFP+ Tregs in the BsB co-coated wells after 5 days of culture were then purified by flow cytometry, reseeded into wells co-coated with antibodies and BsB or IgG, as described above, and recultured for 5 days before flow cytometry analysis.
T Cell Suppression Assay
Tregs were induced by BsB as described above. To induce Tregs by TGF-β and IL-2, the mixed lymphocyte reactions were performed in the presence of TGF-β at 5 ng/ml and IL-2 at 25 ng/ml. GFP+ Tregs (CD90.2+) were purified by flow cytometry and mixed with CFSE-labeled fresh naïve responder T cells isolated from CD90.1+ congenic C57BL/6 mice, as well as fresh allogeneic BALB/c APCs, at the indicated ratios in a Transwell® (Corning) or a normal culture well. After culturing the cells for 5 days, proliferation of responder T cells, indicated by CFSE dilution, was assessed by flow cytometry. To assess the effects of cytokines on Treg suppression, control, anti-TGF-β, or anti-IL-10 antibodies at 10 μg/ml were included in the suppression assay.
In Situ AKT and mTOR Phosphorylation Assay
To avoid the influence of APCs on measurements of AKT and mTOR phosphorylation, purified naïve T cells were activated by co-immobilization of anti-CD3, anti-CD28, and BsB or mouse IgG, as described for the restimulation assay. However, this protocol varied from the previously detailed protocol in that the T cells were harvested after 18 h of stimulation. Intracellular phosphorylated AKT (pSer743) and phosphorylated mTOR (Cell Signaling Technology) were then probed using fluorescently labeled antibodies and analyzed by flow cytometry.
RESULTS
Design of Bispecific Fusion Protein That Engages CTLA-4 and Cross-links It to TCR via MHCII
To generate a bispecific fusion protein that selectively and agonistically engages CTLA-4 and simultaneously ligates it to the TCR, mutant CD80 (CD80wa) that binds CTLA-4 but has minimal affinity for CD28 (28) was fused with LAG-3, a natural ligand of MHCII (29, 30). CD80wa was joined to LAG-3 using a linker composed of nine glycines, which in turn was attached to the Fc portion of mouse IgG2a to purportedly increase its circulating half-life (Fig. 1A, BsB). In response to a ligand of this configuration, CTLA-4 engagement and ligation to the TCR were expected to occur indirectly, via formation of the trimolecular complex (CTLA-4-MHCII-TCR) in the immune synapses during early T cell activation (Fig. 1B). Conceptually, outside of the context of the immune synapse, binding of the bispecific fusion protein to either CTLA-4 or MHCII alone or to both CTLA-4 and MHCII should not lead to inhibition of T cell activity. The engagement of CTLA-4 by CD80wa was designed to trigger CTLA-4 signaling via the recruitment of phosphatases to the cytoplasmic tail of CTLA-4. Meanwhile, binding of LAG-3 to MHCII was intended to bring CTLA-4 into the proximity of the cognate TCR, which binds the pMHCII complex in the immune synapse (Fig. 1B). The combination of these two binding events was expected to deliver an inhibitory signal to the TCR. A control fusion protein comprising CD80wa and IgG2a Fc was also constructed (Fig. 1A, BsBΔ), which should not be capable of cross-linking CTLA-4 to the TCR (Fig. 1C) as it lacks LAG-3.
FIGURE 1.
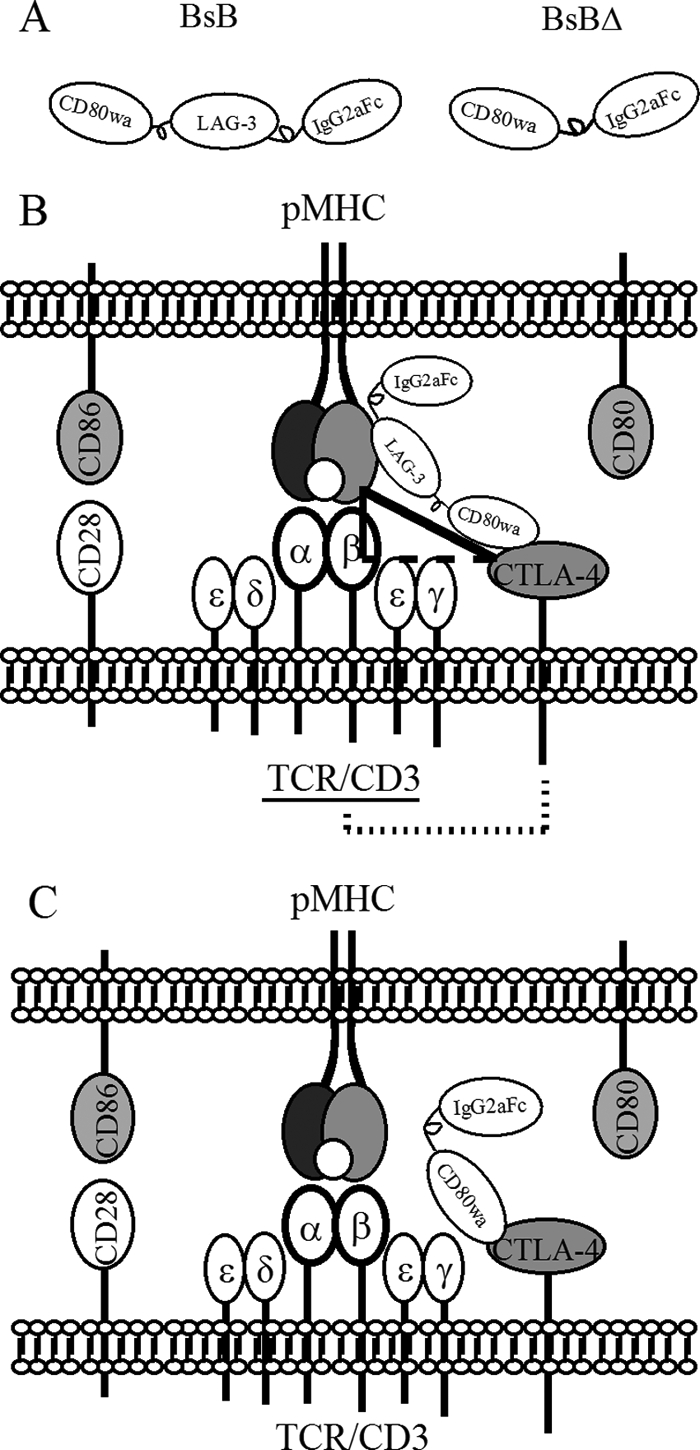
Designs of BsB and BsBΔ. A, schematic drawings of the BsB and BsBΔ fusion proteins. B, schematic drawing of pMHCII, the TCR and co-stimulatory molecules in the immune synapse, as well as the proposed scheme for BsB-mediated cross-linking of CTLA-4 to the TCR via the CTLA-4-MHC II-TCR trimolecular complex. The fusion protein engages CTLA-4 and indirectly ligates the TCR via binding to MHCII in the immune synapse. The two solid sides of the triangle denote cross-linking of MHCII and CTLA-4 as well as MHCII and TCR; the dashed side depicts ligation of CTLA-4 to TCR. The dotted line indicates inhibition of TCR signaling by BSB-engaged CTLA-4. C, schematic drawing showing that the action of BsBΔ is similar to that of BsB except it is unable to ligate the TCR.
The test and control fusion proteins were expressed in Chinese hamster ovary cells and purified with affinity chromatography on a protein G column. Aggregates were removed using size exclusion chromatography (supplemental Fig. S1A). As expected, both fusion proteins appeared as dimers on non-reducing SDS-PAGE gels (BsB, 200 kDa; BsBΔ, 140 kDa) and as monomers (BsB, 100 kDa; BsBΔ, 70 kDa) on reducing SDS-PAGE gels (supplemental Fig. S1B). Their identities were further confirmed by Western blotting, using antibodies against LAG-3 and CD80 (data not shown). The differential binding of BsB to CTLA-4 and CD28 was also confirmed by BIAcore analysis (supplemental Fig. S1C). Whereas CD80Ig was able to bind both CTLA-4 and CD28 as expected, BsB only bound CTLA-4 but not CD28. The binding of BsB to MHCII on APCs was also confirmed by ELISA (data not shown). Thus, BsB has the expected bifunctionality to both CTLA-4 and MHCII.
BsB Inhibits T Cell Activation in Allogenic Mixed Lymphocyte Reaction
The relative ability of BsB and BsBΔ to inhibit T cell activation was assessed in an allogenic mixed lymphocyte reaction by measuring the production of IL-2. Naïve CD4+CD25−CD62LhighCD44low T cells that had been purified from BALB/c mice were mixed with APCs isolated from C57BL/6 mice in the presence or absence of the BsB or BsBΔ. Murine IgG2a and CTLA-4Ig, a co-stimulation inhibitor that binds to CD80/86 and blocks their binding to CD28, were included as negative and positive controls, respectively. Inclusion of BsB but not BsBΔ, in the mixed lymphocyte reaction inhibited IL-2 production albeit not to the same extent as that achieved by CTLA-4Ig (Fig. 2A). This difference was likely the result of BsB-mediated T cell inhibition occurring later than CTLA-4Ig-mediated inhibition. More specifically, for BsB, inhibition only occurred after CTLA-4 was up-regulated following T cell activation. The inability of BsBΔ to reduce IL-2 production strongly suggests that engagement of CTLA-4 alone is insufficient to prevent T cell activation because concurrent cross-linking to the TCR is required. To exclude the possibility that the LAG-3 portion of BsB plays a role in T cell inhibition, we tested LAG-3Ig in this assay and verified that it did not inhibit T cell activation (data not shown).
FIGURE 2.

Inhibition of IL-2 production in allogenic T cell activation and T cell proliferation by BsB in a mixed lymphocyte reaction. A, naïve T cells from C57BL/6 mice and LPS-treated and irradiated BALB/c APCs were mixed with the test constructs for 2 days. Culture media were then harvested and assayed for IL-2. Only BsB and CTLA-4Ig inhibited T cell activation, as indicated by a decreased amount of IL-2 in the media. B, T cell proliferation as assessed by CFSE dilution. Mixed lymphocyte reactions were set up as described in A; however, naive T cells were prelabeled, and the cells cultured for 5 days to allow cell divisions to occur as indicated by CFSE dilution. mCTLA-4Ig inhibited both IL-2 production and cell proliferation, whereas BsB inhibited IL-2 production but increased T cell proliferation. The figure is representative of more than five independent but similar studies.
T cell inhibition was also followed by prelabeling naïve T cells with CFSE prior to their mixing with APCs. Cells were cultured for 5 days to allow for sufficient cycles of cell division to occur so that cell proliferation could be monitored by flow cytometry for CFSE dilution. Unexpectedly, although BsB inhibited IL-2 production (Fig. 2A and supplemental Fig. S2), cell proliferation was not inhibited but increased instead in a concentration-dependent manner (Fig. 2B, BsB panels) when compared with the IgG control. As expected, CTLA-4Ig inhibited T cell proliferation by ∼60% (Fig. 2B, mIgG and mCTLA-4Ig panels). The increase in T cell proliferation and decrease in IL-2 production in BsB-treated samples seemed contradictory initially; however, we later found that this was due to expansion of induced Tregs (Fig. 3A and supplemental Fig. S3A).
FIGURE 3.

Induction of Foxp3+ Tregs and IL-10 and TGF-β production by BsB and involvement of CTLA-4 and TGF-β in Treg induction. A, allogenic mixed lymphocyte reactions were set up as described in the legend to Fig. 2, using naïve CD4+CD62LhiCD25−GFP− cells that had been isolated from Foxp3-EGFP knock-in mice in the presence of the test constructs. Five days post-activation, CD4+ T cells were analyzed for GFP expression by flow cytometry. Tregs were gated as GFP+ and CD25+ cells. Only BsB treatment led to GFP expression, indicating induction of Foxp3+ Tregs (middle left panel). Culture media were collected for cytokine analysis (right panels), which revealed elevated IL-10 and TGF-β levels in the presence of BsB. B, mCTLA-4Ig, mLAG-3Ig, BsBΔ + mLAG-3Ig failed to induced Tregs. C, CTLA-4 engagement is required for Treg induction. Inclusion of a blocking antibody to CTLA-4 reduced Tregs by ∼50%. D, requirement of autocrine TGF-β for Treg induction is indicated by the complete blockade of Treg induction in the presence of a blocking antibody to TGF-β. The data are representative of numerous independent but similar studies. In both C and D, naïve T cells from wildtype C57BL/6 mice were used as these studies were performed to concurrently evaluate Treg proliferation using CFSE labeling, which was not compatible with EGFP.
BsB Directs T Cell Differentiation into Tregs
Early termination of TCR signaling by withdrawal of antigen stimulation, inhibition of mTOR signaling, suboptimal TCR stimulation due to a low affinity antigen, or weak co-stimulation during T cell activation have been shown to induce Foxp3+ expression and skew T cell differentiation toward a Treg phenotype (33–35). As BsB forces early engagement of the TCR by activation-induced CTLA-4 with consequent attenuation of TCR signaling, its ability to generate Foxp3+ Tregs was also investigated. Naïve CD4+CD62LhighGFP− T cells prepared from Foxp3-EGFP knock-in mice (36) were mixed with LPS-treated allogenic APCs in the presence of BsB or BsBΔ. Flow cytometry analysis of the cells after 5 days of culture revealed a large number of CD4+CD25+GFP+ T cells among the BsB-treated cells (Fig. 3A, middle left panel) but not among cells treated with mouse IgG2a (Fig. 3A, top left panel) or the BsBΔ control (Fig. 3A, bottom left panel), suggesting that these CD4+CD25+GFP+ T cells were Foxp3+ Tregs. To confirm this finding, cell culture media were collected and assayed for the signature Treg cytokines, IL-10 and TGF-β (37). Large amounts of IL-10 and TFG-β were detected in the media of BsB-treated cells (Fig. 3A, right panels) but not in media of cells treated with BsBΔ or mIgG2a. Interestingly, CTLA-4Ig did not induce generation of GFP+ Tregs (Fig. 3B) or IL-10 and TGF-β production (supplemental Fig. S3B), suggesting that the mechanism by which CTLA-4Ig curtails the T cell response is different from that of BsB. LAG-3Ig alone or in combination with BsBΔ also failed to induce generation of GFP+ Tregs (Fig. 3B), suggesting that BsB-mediated cross-linking of CTLA-4 with the TCR was required for Treg induction.
To determine whether BsB-induced Tregs are proliferative and contributed to the observed proliferation noted in Fig. 2B, naïve T cells from C57BL/6 mice were purified and prelabeled with CFSE before mixing with allo-APCs for Treg induction. As shown in supplemental Fig. S3A, the Foxp3+ Tregs are highly proliferative as indicated by several rounds of cell division/CFSE dilution. The extent of induction of Foxp3+ Tregs and production of IL-10 and TGF-β were dependent on the concentrations of BsB used (supplemental Fig. S3, A and B).
Induction of Tregs by BsB Requires CTLA-4 and Autocrine TGF-β
To explore whether CTLA-4 engagement is involved in Treg induction, a CTLA-4 blocking antibody (9H10) was added to the Treg induction culture. As shown in Fig. 3C, addition of a CTLA-4 blocking antibody reduced the number of Foxp3+ Tregs by ∼50%. As BsB and the blocking antibody could concurrently bind to CTLA-4, a complete inhibition of Treg induction in this assay is not expected.
The concurrent detection of elevated levels of IL-10 and TGF-β following treatment with BsB raised the possibility that these cytokines, TGF-β in particular, may have played a role in facilitating the generation of Tregs (Fig. 3A). To address this possibility, culture media were collected over a period of 5 days and analyzed for cytokine and Foxp3+ Treg content. Elevated IL-10 and TGF-β levels were detected as early as day 2 post-treatment, and Foxp3+ Tregs were detected after day 3 (data not shown). These results suggest that the endogenous production of TGF-β, presumably stimulated by BsB, likely plays a role in Treg differentiation. Addition of an anti-TGF-β antibody (clone 1D11), but not an isotype control IgG (clone 13C4), to the Treg induction assay completely blocked the appearance of Foxp3+ Tregs (Fig. 3D). This implies that the early engagement of CTLA-4 and its subsequent cross-linking to the TCR by BsB likely stimulates endogenous TGF-β production, which in turn encourages Treg differentiation. Cross-linking of CTLA-4 and the TCR has been previously reported to induce TGF-β production (38), although Treg differentiation was not assessed in this study.
BsB-induced Tregs Are Functionally Suppressive in Cell-cell Contact-dependent Manner
To assess whether the BsB-induced Tregs were functionally suppressive, BsB-induced Tregs and TGF-β-induced Tregs, which served as a positive control, were purified using flow cytometry and mixed with CFSE-labeled congeneic responder T cells at different ratios and allogenic APCs. Cells were co-cultured for 5 days in either Transwells or regular culture wells, after which the proliferation of responder T cells (Tresp) was analyzed using flow cytometry. As summarized in Fig. 4A and supplemental Fig. S4, both BsB- and TGF-β-induced Tregs cultured in regular culture wells almost completely inhibited the proliferation of the responder T cells. The potency of the suppressive activity of the BsB-induced Tregs was comparable with that of TGF-β-induced Tregs. In contrast, Tregs generated by either BsB or TGF-β did not significantly inhibit the proliferation of responder T cells when the T cells were separated from the Tregs in a Transwell. This finding suggests that Treg suppressive activity depends on cell-cell contact and is not mediated by secreted cytokines or other factors. Supporting this notion, we demonstrated that inclusion of an antibody to IL-10 (clone JES5–2A5) in the regular culture well did not affect the suppressive activity of either the BsB- or the TGF-β-induced Tregs (Fig. 4B). The addition of an antibody to TGF-β (1D11) also did not affect the suppressive activity of BsB-induced Tregs, although it partially reduced suppression by TGF-β-induced Tregs (Fig. 4B).
FIGURE 4.

Suppressive function of BsB-induced Tregs. A, BsB- or TGF-β-induced Tregs were purified by flow cytometry and mixed with CFSE-labeled naïve Tresp prepared from C57BL/6 mice at the indicated ratios in Transwells (hatched columns) or regular culture wells (filled columns). LPS-treated allogenic BALB/c APCs were added to stimulate T cell activation. The results (mean + S.E.) indicate the percentage of proliferating Tresp, based on a CFSE dilution without Tregs (Tresp + APC only) set to 100%. B, anti-IL-10 and anti-TGF-β antibodies were added to cells in regular culture wells at a Tresp:Treg ratio of 1:1 to determine the contribution of cytokines to T cell proliferation. The anti-TGF-β antibody partially inhibited the suppressive function of TGF-β-induced Tregs (left panel) but did not affect BsB-induced Tregs (right panel). The figure is representative of more than three independent but similar studies.
Induction of Tregs by BsB May Involve Attenuation of AKT/mTOR Signaling Pathway
Recent reports have indicated that the AKT and mTOR signaling pathways play important roles in determining T cell fate. The presence of constitutively active AKT in T cells diminishes Treg differentiation in a rapamycin-sensitive manner (34), suggesting that the AKT and mTOR signaling pathways intersect to influence Treg fate. Moreover, T cells deficient in mTOR differentiate to Tregs more readily than normal control T cells (33). An obligatory role for the co-inhibitory molecules PD-1/PD-L1 in controlling adaptive Treg development by antagonizing AKT/mTOR has also been reported (39). To determine whether these pathways are also involved in BsB-mediated induction of Tregs, anti-CD3 and anti-CD28 antibodies were co-immobilized with BsB, mIgG, or PD-L1 on 96-well plates, onto which naïve T cells were seeded. Eighteen hours post-activation, the cells were stained with fluorescently labeled antibodies against phosphorylated AKT and mTOR and analyzed by flow cytometry. Phosphorylation of both AKT and mTOR was attenuated by BsB and PD-L1 co-immobilization (Fig. 5), suggesting that signaling events mediated CTLA-4 and PD-L1 inhibitory molecules may converge at some point along the AKT/mTOR signaling pathway during T cell activation to regulate Treg differentiation.
FIGURE 5.

Down-regulation of AKT and mTOR phosphorylation by BsB. Naïve T cells were cultured in round-bottomed 96-well plates co-coated with anti-CD3, anti-CD28, and BsB, mouse IgG (mIgG), or mouse PD-L1 (mPD-L1) for 18 h. Cells deemed not activated were cultured in wells coated with IgG only. The phosphorylation status of AKT and mTOR was then monitored by flow cytometry after staining with fluorescently labeled antibodies to phosphorylated AKT and mTOR. MFI denotes mean fluorescent intensity. This figure represents one of three independent experiments.
Exposure to BsB Sustains Foxp3+ Expression in Induced Tregs
In vitro induced Tregs, unlike fully committed natural Tregs, are reportedly less stable and can lose Foxp3+ expression upon extended culture in the absence of the initial inducer (e.g. TGF-β or retinoic acid) (40). In the current work, BsB-induced Tregs showed similar instability, with some cells losing Foxp3+ expression following repeated culture (Fig. 6 and data not shown). To test whether restimulation by BsB could prolong Foxp3+ expression, Tregs were first induced by coating 96-well plates with both anti-CD3/anti-CD28 antibodies and BsB. Purified Tregs were then subjected to an additional round of culture in the presence or absence of BsB. Restimulation of the purified Tregs with BsB allowed for maintenance of a large population (∼93% of total Tregs) of Foxp3+ Tregs (Fig. 6, bottom right panel), compared with ∼40% Foxp3 expression in response to the IgG control (Fig. 6, upper right panel).
FIGURE 6.

Sustained Foxp3+ expression in Tregs in response to continuous stimulation with BsB. Round-bottomed 96-well plates were co-coated with anti-CD3, anti-CD28 and BsB or mouse IgG. Naïve T cells from Foxp3-EGFP knock-in mice were cultured for 5 days to induce Tregs (left panels), which were then purified from the BsB-treated cells (red square) and restimulated in another round of culture in co-coated wells, as above, for 5 days, before analysis by flow cytometry for GFP+ cells. Reculturing of purified Tregs with the mouse IgG control for 5 days resulted in a loss of Foxp3+ expression in ∼60% of cells (upper right quadrant of upper right panel), whereas <7% of the Tregs recultured with BsB had lost Foxp3+ expression (upper right quadrant of bottom right panel). This figure represents one of three independent experiments.
DISCUSSION
T cell inhibition via cross-linking of CTLA-4 to the TCR represents an attractive and practical alternative to the blockade of co-stimulation, such as by CTLA-4Ig. However, the efficacy of this approach is critically dependent on the efficiency with which the activating TCR ligates the ligand-engaged CTLA-4. Moreover, both the configuration of the participating molecules (i.e. the TCR and CTLA-4 ligands on the APC need to be in the cis configuration) and the timing of the intervention (i.e. the early phase of T cell activation) need to be taken into account (17). To meet these spatial and temporal prerequisites, a bispecific fusion protein (BsB) capable of engaging CTLA-4 expressed on activating T cells and concurrently binding MHCII on APCs was generated. Cross-linking of CTLA-4 to TCR was expected to be indirect and mediated by the CTLA-4-MHCII-TCR trimolecular complex formed within the immune synapse during T cell activation (Fig. 1B). Therefore, BsB-mediated T cell inhibition was predicted to be highly restricted to T cells undergoing activation.
Consistent with previous reports using alternative strategies to cross-link CTLA-4 and the TCR, BsB efficiently inhibited T cell activation as indicated by reduced IL-2 production. The critical requirement for BsB-mediated cross-linking of CTLA-4 to the TCR to achieve this inhibition was demonstrated clearly by the lack of inhibition in response to the control fusion protein (BsBΔ). More specifically, BsBΔ retained the ability to engage CTLA-4 but was unable to effect cross-linking via MHCII because it lacked the LAG-3 moiety (Figs. 2A and 3A), or when the LAG-3 was provided but separated from CD80wa as in the molecule LAG-3Ig (Fig. 3B). These results suggest that the inhibitory effect of BsB on IL-2 production was not the result of direct activation of CTLA-4 via engagement by CD80wa or interference of MHCII presentation. The T cell-inhibiting activity of BsB on IL-2 production was similar to that of CTLA-4Ig, which blocks CD28-mediated co-stimulation during T cell activation (31, 32). However, BsB exhibited additional advantageous properties that CTLA-4Ig lacks. Specifically, BsB stimulated IL-10 and TGF-β production and, perhaps more intriguingly, directed T cells toward differentiation into Tregs that are capable of continuously expanding in vitro, thus providing additional effectors for immune suppression. CTLA-4Ig did not effect Treg induction, consistent with a role for the CD28 co-stimulatory pathway in Treg homeostasis. Indeed, it has been reported that CTLA-4Ig can decrease the number of Tregs when employed in animal models of autoimmune disease (41).
The observed decrease in IL-2 levels in BsB-treated cultures and the simultaneous increase in the proliferation of BsB-treated T cells (Fig. 2) may appear contradictory at first. We speculate that because the Treg population induced by BsB expresses a high level of CD25 (the high affinity α-chain of the IL-2 receptor complex), amounts of the produced IL-2 may have been utilized in a paracrine or autocrine manner.
BsB appeared to dampen T cell activation and promote Treg differentiation through down-regulation of the AKT/mTOR signaling pathway. This is particularly interesting in light of the role of another pair of co-inhibitory molecules, PD-1/PD-L1, in Treg differentiation. Sharpe and colleagues (39) have reported that PD-1/PD-L1 also enhances Treg differentiation through attenuation of AKT/mTOR signaling. This finding suggests that signaling by these two co-inhibitory pathways, mediated by CTLA-4 and PD-1, may converge at some common node(s) to safeguard against an excessive immune response. This may be accomplished by switching off T cell activation and stimulating the anti-inflammatory sentinels, the Tregs. The contribution of CTLA-4 appears to be more prominent because CTLA-4 engagement stimulates TGF-β production and induces Treg differentiation in the absence of exogenous TGF-β, whereas PD-1 engagement only minimally induces Treg differentiation in the absence of exogenous TGF-β. Sharpe and colleagues (39) have suggested that PD-1/PD-L1 may act to augment the effects of TGF-β on Treg induction. It is conceivable that the endogenous production of TGF-β stimulated by CTLA-4 via BsB engagement may act on PD-1 to synergistically promote and enhance Treg differentiation.
A recent report indicated that the degree of TCR activation by antigens is correlated with the extent of mTOR signaling and Treg differentiation (42). In this study, a low dose of antigen resulted in weak TCR signaling through the AKT/mTOR pathway and consequent production of more Tregs. Interestingly, in the presence of rapamycin, a specific inhibitor of mTOR, maximal induction of Foxp3 expression was achieved within 10–48 h post-T cell activation (35). These kinetics are akin to those CTLA-4 expression on activating T cells (18), supporting the contention that BsB (and by extension, PD-1/PD-L1) modulates T cell differentiation by dampening AKT/mTOR signaling.
Cross-linking CTLA-4 to the TCR during T cell activation has previously been reported to lead to TGF-β production (38). In our studies, both IL-10 and TGF-β were produced in response to BsB but only TGF-β was determined to be necessary for Treg induction. In support of this, the addition of an anti-TGF-β antibody to the culture system inhibited Treg differentiation. The mechanism by which BsB induces TGF-β production remains unclear but requires BsB-mediated cross-linking of CTLA-4 to the TCR, as the control fusion protein, BsBΔ, lacked the ability to elicit TGF-β production and Treg differentiation.
The finding that CTLA-4 is constitutively expressed on Tregs has led to assessments of its role in Treg function, development, and homeostasis in several experimental settings. A critical role for CTLA-4 in the suppressive function of Tregs has been clearly established (43–45). However, its role in Treg development and homeostasis is less well defined. CTLA-4 does not appear to be obligatory for Treg generation because these cells are present in CTLA-4 KO mice (46) and are even found at increased numbers (47), suggesting it may be providing a negative feedback signal to maintain Treg homeostasis.
Our data provides the first demonstration that CTLA-4 plays an active role in Treg differentiation or at least in the induction of adaptive Tregs. This only manifests when CTLA-4 is engaged and cross-linked to the TCR during the early phase of T cell activation. Our observations suggest a potential new therapeutic paradigm for inducing Tregs, and thus immune tolerization, for the treatment of autoimmune diseases. Indeed, preliminary studies conducted in our laboratory have shown that BsB can delay the development of type I diabetes in non-obese diabetic mice (data not shown). Also, antigen specific Tregs can be induced by BsB. When naïve T cells purified from OT-II ovalbumin peptide 323–339-specific TCR transgenic mice were used in the Treg induction assay with ovalbumin peptide 323–339 loaded congenic APCs, antigen-specific Tregs were generated successfully (data not shown). These findings are consistent with reports by other investigators who targeted this same pathway in order to treat autoimmune diseases, such as type I diabetes and thyroiditis. For example, Bluestone and colleagues (22) engineered a B cell surface linked CTLA-4 agonistic scFv that protected non-obese diabetic mice from developing type I diabetes. Chemically conjugating an agonistic CTLA-4 antibody to an antibody recognizing surface molecules on target or dendritic cells also protected animals from developing thyroiditis (24, 48). Interestingly, antigen-specific CD4+CD25+CTLA-4+ and CD4+CD25−TGF-β1+ adaptive Tregs have also been detected in animals treated with dendritic cells coated with a bispecific antibody to CD11c and CTLA-4 (24). However, it is our opinion that the strategy of developing a bispecific fusion protein, proposed here, represents a more practical approach for drug development due to its scalability and manufacturability.
Supplementary Material
Acknowledgments
We thank John Williams and Melanie Ruzek for helpful discussions. We are very grateful to Elizabeth Masterjohn, William Brondyk, Jean McLarty, Tim Boire, Mark Levine, Dewan Haque, and Andy Zhou for kind help with protein expression and purification and Julie Bird and Huawei Qiu for BIAcore binding analysis. We also thank Ronald Scheule for critical reading of the manuscript.
All of the authors are employees of Genzyme Corporation.

This article contains supplemental Figs. S1–S4.
- CTLA-4
- cytotoxic lymphocyte-associated antigen-4
- mTOR
- mammalian target of rapamycin
- Treg
- regulatory T cell
- TCR
- T cell receptor
- EGFP
- enhanced GFP
- APC
- antigen presenting cell
- MHCII
- major histocompatibility complex class II
- LAG-3
- lymphocyte activation antigen 3
- PD-1
- programmed cell death 1
- PD-L1
- PD-1 ligand
- Foxp3
- forkhead box P3
- Tresp
- responder T cells
- CSFE
- carboxyfluorescein diacetate succimidyl ester.
REFERENCES
- 1. Eagar T. N., Karandikar N. J., Bluestone J. A., Miller S. D. (2002) The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur. J. Immunol. 32, 972–981 [DOI] [PubMed] [Google Scholar]
- 2. Karandikar N. J., Vanderlugt C. L., Walunas T. L., Miller S. D., Bluestone J. A. (1996) CTLA-4: A negative regulator of autoimmune disease. J. Exp. Med. 184, 783–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krummel M. F., Allison J. P. (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tivol E. A., Borriello F., Schweitzer A. N., Lynch W. P., Bluestone J. A., Sharpe A. H. (1995) Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 [DOI] [PubMed] [Google Scholar]
- 5. Walunas T. L., Bluestone J. A. (1998) CTLA-4 regulates tolerance induction and T cell differentiation in vivo. J. Immunol. 160, 3855–3860 [PubMed] [Google Scholar]
- 6. Walunas T. L., Lenschow D. J., Bakker C. Y., Linsley P. S., Freeman G. J., Green J. M., Thompson C. B., Bluestone J. A. (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413 [DOI] [PubMed] [Google Scholar]
- 7. Fife B. T., Bluestone J. A. (2008) Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182 [DOI] [PubMed] [Google Scholar]
- 8. Ise W., Kohyama M., Nutsch K. M., Lee H. M., Suri A., Unanue E. R., Murphy T. L., Murphy K. M. (2010) CTLA-4 suppresses the pathogenicity of self-antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat. Immunol. 11, 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jain N., Nguyen H., Chambers C., Kang J. (2010) Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 107, 1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paterson A. M., Sharpe A. H. (2010) Taming tissue-specific T cells: CTLA-4 reins in self-reactive T cells. Nat. Immunol. 11, 109–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Linsley P. S., Greene J. L., Brady W., Bajorath J., Ledbetter J. A., Peach R. (1994) Human B7–1 (CD80) and B7–2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1, 793–801 [DOI] [PubMed] [Google Scholar]
- 12. Guntermann C., Alexander D. R. (2002) CTLA-4 suppresses proximal TCR signaling in resting human CD4+ T cells by inhibiting ZAP-70 Tyr319 phosphorylation: A potential role for tyrosine phosphatases. J. Immunol. 168, 4420–4429 [DOI] [PubMed] [Google Scholar]
- 13. Baroja M. L., Vijayakrishnan L., Bettelli E., Darlington P. J., Chau T. A., Ling V., Collins M., Carreno B. M., Madrenas J., Kuchroo V. K. (2002) Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 168, 5070–5078 [DOI] [PubMed] [Google Scholar]
- 14. Chuang E., Fisher T. S., Morgan R. W., Robbins M. D., Duerr J. M., Vander Heiden M. G., Gardner J. P., Hambor J. E., Neveu M. J., Thompson C. B. (2000) The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 13, 313–322 [DOI] [PubMed] [Google Scholar]
- 15. Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L. (1999) The immunological synapse: A molecular machine controlling T cell activation. Science 285, 221–227 [DOI] [PubMed] [Google Scholar]
- 16. Pentcheva-Hoang T., Egen J. G., Wojnoonski K., Allison J. P. (2004) B7–1 and B7–2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413 [DOI] [PubMed] [Google Scholar]
- 17. Griffin M. D., Hong D. K., Holman P. O., Lee K. M., Whitters M. J., O'Herrin S. M., Fallarino F., Collins M., Segal D. M., Gajewski T. F., Kranz D. M., Bluestone J. A. (2000) Blockade of T cell activation using a surface-linked single-chain antibody to CTLA-4 (CD152). J. Immunol. 164, 4433–4442 [DOI] [PubMed] [Google Scholar]
- 18. Jago C. B., Yates J., Câmara N. O., Lechler R. I., Lombardi G. (2004) Differential expression of CTLA-4 among T cell subsets. Clin. Exp. Immunol. 136, 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blair P. J., Riley J. L., Levine B. L., Lee K. P., Craighead N., Francomano T., Perfetto S. J., Gray G. S., Carreno B. M., June C. H. (1998) CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-xL induction. J. Immunol. 160, 12–15 [PubMed] [Google Scholar]
- 20. Krummel M. F., Allison J. P. (1996) CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 183, 2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walunas T. L., Bakker C. Y., Bluestone J. A. (1996) CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 183, 2541–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fife B. T., Griffin M. D., Abbas A. K., Locksley R. M., Bluestone J. A. (2006) Inhibition of T cell activation and autoimmune diabetes using a B cell surface-linked CTLA-4 agonist. J. Clin. Invest. 116, 2252–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffin M. D., Holman P. O., Tang Q., Ashourian N., Korthäuer U., Kranz D. M., Bluestone J. A. (2001) Development and applications of surface-linked single chain antibodies against T-cell antigens. J. Immunol. Methods 248, 77–90 [DOI] [PubMed] [Google Scholar]
- 24. Li R., Perez N., Karumuthil-Melethil S., Prabhakar B. S., Holterman M. J., Vasu C. (2007) Enhanced engagement of CTLA-4 induces antigen-specific CD4+CD25+Foxp3+ and CD4+CD25- TGF-β 1+ adaptive regulatory T cells. J. Immunol. 179, 5191–5203 [DOI] [PubMed] [Google Scholar]
- 25. Rao S., Vasu C., Martinez O., Kaithamana S., Prabhakar B. S., Holterman M. J. (2001) Targeted delivery of anti-CTLA-4 antibody down-regulates T cell function in vitro and in vivo. Clin. Immunol. 101, 136–145 [DOI] [PubMed] [Google Scholar]
- 26. Vasu C., Prabhakar B. S., Holterman M. J. (2004) Targeted CTLA-4 engagement induces CD4+CD25+CTLA-4high T regulatory cells with target (allo)antigen specificity. J. Immunol. 173, 2866–2876 [DOI] [PubMed] [Google Scholar]
- 27. Darlington P. J., Baroja M. L., Chau T. A., Siu E., Ling V., Carreno B. M., Madrenas J. (2002) Surface cytotoxic T lymphocyte-associated antigen 4 partitions within lipid rafts and relocates to the immunological synapse under conditions of inhibition of T cell activation. J. Exp. Med. 195, 1337–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y., Guo Y., Huang A., Zheng P., Liu Y. (1997) CTLA-4-B7 interaction is sufficient to costimulate T cell clonal expansion. J. Exp. Med. 185, 1327–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baixeras E., Huard B., Miossec C., Jitsukawa S., Martin M., Hercend T., Auffray C., Triebel F., Piatier-Tonneau D. (1992) Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 176, 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Triebel F., Jitsukawa S., Baixeras E., Roman-Roman S., Genevee C., Viegas-Pequignot E., Hercend T. (1990) LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bluestone J. A., St Clair E. W., Turka L. A. (2006) CTLA4Ig: Bridging the basic immunology with clinical application. Immunity 24, 233–238 [DOI] [PubMed] [Google Scholar]
- 32. Linsley P. S., Nadler S. G. (2009) The clinical utility of inhibiting CD28-mediated costimulation. Immunol. Rev. 229, 307–321 [DOI] [PubMed] [Google Scholar]
- 33. Delgoffe G. M., Kole T. P., Zheng Y., Zarek P. E., Matthews K. L., Xiao B., Worley P. F., Kozma S. C., Powell J. D. (2009) The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haxhinasto S., Mathis D., Benoist C. (2008) The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 205, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sauer S., Bruno L., Hertweck A., Finlay D., Leleu M., Spivakov M., Knight Z. A., Cobb B. S., Cantrell D., O'Connor E., Shokat K. M., Fisher A. G., Merkenschlager M. (2008) T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. U.S.A. 105, 7797–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haribhai D., Lin W., Relland L. M., Truong N., Williams C. B., Chatila T. A. (2007) Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol. 178, 2961–2972 [DOI] [PubMed] [Google Scholar]
- 37. Cools N., Van Tendeloo V. F., Smits E. L., Lenjou M., Nijs G., Van Bockstaele D. R., Berneman Z. N., Ponsaerts P. (2008) Immunosuppression induced by immature dendritic cells is mediated by TGF-β/IL-10 double-positive CD4+ regulatory T cells. J. Cell Mol. Med. 12, 690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen W., Jin W., Wahl S. M. (1998) Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor β (TGF-β) production by murine CD4+ T cells. J. Exp. Med. 188, 1849–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Francisco L. M., Salinas V. H., Brown K. E., Vanguri V. K., Freeman G. J., Kuchroo V. K., Sharpe A. H. (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Selvaraj R. K., Geiger T. L. (2007) A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-β. J. Immunol. 178, 7667–7677 [DOI] [PubMed] [Google Scholar]
- 41. Tang Q., Henriksen K. J., Boden E. K., Tooley A. J., Ye J., Subudhi S. K., Zheng X. X., Strom T. B., Bluestone J. A. (2003) Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J. Immunol. 171, 3348–3352 [DOI] [PubMed] [Google Scholar]
- 42. Turner M. S., Kane L. P., Morel P. A. (2009) Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J. Immunol. 183, 4895–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bour-Jordan H., Bluestone J. A. (2009) Regulating the regulators: Costimulatory signals control the homeostasis and function of regulatory T cells. Immunol. Rev. 229, 41–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Friedline R. H., Brown D. S., Nguyen H., Kornfeld H., Lee J., Zhang Y., Appleby M., Der S. D., Kang J., Chambers C. A. (2009) CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J. Exp. Med. 206, 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 [DOI] [PubMed] [Google Scholar]
- 46. Tang Q., Boden E. K., Henriksen K. J., Bour-Jordan H., Bi M., Bluestone J. A. (2004) Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34, 2996–3005 [DOI] [PubMed] [Google Scholar]
- 47. Schmidt E. M., Wang C. J., Ryan G. A., Clough L. E., Qureshi O. S., Goodall M., Abbas A. K., Sharpe A. H., Sansom D. M., Walker L. S. (2009) Ctla-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J. Immunol. 182, 274–282 [DOI] [PubMed] [Google Scholar]
- 48. Vasu C., Wang A., Gorla S. R., Kaithamana S., Prabhakar B. S., Holterman M. J. (2003) CD80 and CD86 C domains play an important role in receptor binding and co-stimulatory properties. Int. Immunol. 15, 167–175 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.