

Background: Insulin-like growth factor (IGF) II interacts with IR-A and is a more powerful mitogen than insulin.
Results: IGF-II and insulin differ in regulating insulin receptor (IR)-A trafficking and stability.
Conclusion: Compared with insulin, IGF-II induces lower IR-A and downstream effectors activation but protects IR-A and IRS-1 from down-regulation, thereby evoking a sustained mitogenic stimulus.
Significance: These results further elucidate the mechanisms controlling IR-A biological responses.
Keywords: Endocytosis, Growth Factors, Insulin, Insulin-like Growth Factor (IGF), Signal Transduction, IGF-II, Endocytosis, Insulin, Insulin Receptor Isoform A, Signaling
Abstract
The insulin receptor isoform A (IR-A) binds both insulin and insulin-like growth factor (IGF)-II, although the affinity for IGF-II is 3–10-fold lower than insulin depending on a cell and tissue context. Notably, in mouse embryonic fibroblasts lacking the IGF-IR and expressing solely the IR-A (R−/IR-A), IGF-II is a more potent mitogen than insulin. As receptor endocytosis and degradation provide spatial and temporal regulation of signaling events, we hypothesized that insulin and IGF-II could affect IR-A biological responses by differentially regulating IR-A trafficking. Using R−/IR-A cells, we discovered that insulin evoked significant IR-A internalization, a process modestly affected by IGF-II. However, the differential internalization was not due to IR-A ubiquitination. Notably, prolonged stimulation of R−/IR-A cells with insulin, but not with IGF-II, targeted the receptor to a degradative pathway. Similarly, the docking protein insulin receptor substrate 1 (IRS-1) was down-regulated after prolonged insulin but not IGF-II exposure. Similar results were also obtained in experiments using [NMeTyrB26]-insulin, an insulin analog with IR-A binding affinity similar to IGF-II. Finally, we discovered that IR-A was internalized through clathrin-dependent and -independent pathways, which differentially regulated the activation of downstream effectors. Collectively, our results suggest that a lower affinity of IGF-II for the IR-A promotes lower IR-A phosphorylation and activation of early downstream effectors vis à vis insulin but may protect IR-A and IRS-1 from down-regulation thereby evoking sustained and robust mitogenic stimuli.
Introduction
The insulin receptor (IR)4 is expressed in two isoforms: the IR isoform A (IR-A) and isoform B (IR-B). The IR-A is generated by the skipping of exon 11 of the IR gene, which generates a protein that differs from IR-B in only a short stretch of 12 amino acid residues at the carboxyl terminus of the receptor α-subunit (1–4). The two IR isoforms are usually co-expressed and their relative abundance is regulated by several factors, including developmental stage and tissue-specific factors (5, 6). However, IR-A is predominantly expressed in fetal tissues and cancer cells, whereas the IR-B is preferentially expressed in differentiated insulin-responsive tissues (7, 8). Although the IR-B binds insulin with high affinity, the IR-A not only has high affinity for insulin but it also binds IGF-II, although the affinity is 3–10-fold lower than that of insulin depending on a cell and tissue context (7, 9). IGF-II binds instead to IGF type I receptor (IGF-IR) and to IR-A with similar affinities and shares with the homolog ligand IGF-I mitogenic and anti-apoptotic effects (7, 10).
Several studies have now established that IGF-II is capable to elicit biological effects via activation of the IR-A. For instance, in mouse fibroblasts lacking the IGF-IR and expressing solely the IR-A (R−/IR-A cells), IGF-II is a more potent mitogen than insulin (7, 11). In SKUT-1 human leiomyosarcoma cells, which lack functional IGF-IR and express predominantly IR-A, IGF-II is more potent than insulin in inducing cell motility and chemotaxis (12). Moreover, the global profile of gene expression elicited by IGF-II and insulin in R−/IR-A is different (13), with IGF-II being more potent than insulin in regulating specific gene clusters (13). Recent proteomic approaches demonstrated that insulin and IGF-II differ also in their ability to recruit effector proteins to the IR-A (14). However, the molecular mechanisms that may differentially regulate the intensity of the signal promoted by IGF-II and insulin through the IR-A have not been characterized.
Internalization of ligand-activated receptor-tyrosine-kinases (RTKs) occurs through specialized microdomains on the cell surface known as clathrin-coated pits or caveolae. Recent studies suggest an important connection between receptor ubiquitination, endocytosis, and ligand-mediated receptor signaling (15, 16). As shown for the EGFR, the disruption of RTK internalization and degradation results in their inability to attenuate signaling, leading to constitutive activation, prolonged signaling, and transformation (16).
We have previously demonstrated that the Grb10/Nedd4 complex promotes IGF-IR ubiquitination, which enhances ligand-dependent internalization and targets the receptor for degradation (17, 18) thereby regulating IGF-IR-dependent cell proliferation (19).
Because endocytosis provides spatial and temporal regulation of signaling events, we tested whether insulin and IGF-II could affect biological responses by differentially regulating IR-A trafficking. Taking advantage of the unique model of R−/IR-A cells, which lack the IGF-IR (20) and express solely the IR-A (21), here we show that insulin and IGF-II considerably differ in their ability to regulate IR-A and the downstream effectors trafficking and stability. These results suggest that the lower affinity of IGF-II for the IR-A promotes lower IR-A phosphorylation and reduced activation of early downstream effectors compared with insulin but protects IR-A and IRS-1 from negative feedback mechanisms, thereby evoking a sustained and powerful mitogenic stimulus.
EXPERIMENTAL PROCEDURES
Cell Lines
R−/IR-A cells are mouse embryo fibroblasts derived from IGF-IR knock-out mice (20) and expressing solely the human IR-A isoform (21). Cells are maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2 μg/ml of puromycin. These cells were previously well characterized for their mitogenic response to insulin and IGF-II (7, 11). MDA-MB-157 breast cancer cells were from ATTC. R+/IR-A cells (R+ A10) are mouse embryo fibroblasts derived from R-cells by cotransfecting the pECE expression vector containing the human IGF-IR cDNA and the pSV2 plasmid encoding the hygromycin resistance gene. Cells were then cotransfected with the pNTK2 expression vector containing the human IR-A cDNA and the pPDV6+ plasmid encoding the puromycin resistance gene (12). NIH3T3/IR-A cells were generated by retroviral infection of NIH3T3 mouse embryo fibroblasts with the pBABE-Hygro expression vector expressing the IR-A cDNA.
Internalization and Ubiquitination Assays
IR-A internalization was assessed after stimulation with 200 ng/ml of either insulin or IGF-II by enzyme-linked immunosorbent assay (ELISA), as previously described in detail (17, 18), using a monoclonal antibody against the IR α-subunit (Novus Biologicals, diluted 1:1000) and goat anti-mouse alkaline phosphatase-conjugated antibody (diluted 1:1000; Sigma). Antibody binding was visualized by adding 0.25 ml of alkaline phosphatase substrate (Bio-Rad). The reaction was stopped by transferring 0.1 ml of the substrate to a 96-well microtiter plate containing 0.1 ml of 0.4 m NaOH. Plates were read at 405 nm in a microplate reader (Dynex Technologies) using Microplate Manager Software.
For ubiquitination, R−/IR-A cells were plated in triplicate at a density of 3 × 105 cells/well in 6-well plates and transiently transfected with 4 μg/well of the 8 HA-tagged ubiquitin construct (17, 18) using Lipofectamine 2000 (Invitrogen). After 24 h cells were shifted to serum-free medium (SFM) for 24 h and then stimulated for 30 min with 50 ng/ml of either insulin or IGF-II, supplemented with 20 μm of the proteasome inhibitor MG132 (Calbiochem) and 100 μm leupeptin/pepstatin as lysosomal inhibitors (Roche Applied Science) to accumulate the ubiquitinated species. Cell lysates were pooled and 1 mg of proteins were immunoprecipitated in HNTG buffer (20 mm HEPES (pH 7.5), 150 mm NaCl, 0.1% Triton X-100, 10% glycerol, 0.2 mm sodium orthovanadate, protease inhibitor mixture supplemented with MG132 and leupeptin/pepstatin (at the above mentioned concentrations) with anti-IR monoclonal antibodies. Filters were immunoblotted with anti-ubiquitin and anti-HA monoclonal antibodies (Covance) to detect ubiquitinated proteins. Membranes were then reprobed with anti-phosphotyrosine monoclonal antibodies (BD Transduction Laboratories) to evaluate IR-A activation. The IR was detected using polyclonal antibodies from Santa Cruz Biotechnology.
Insulin Analogs and Cell Growth Experiments
The B26 peptide N-methylated (NMe) [NMeTyrB26]-insulin analog was previously described (22). The binding affinity for the IR-A was performed according to Gauguin at al. (23) using IM-9 lymphocytes (ATCC), which are rich in IR-A expression. The IM-9 cell line was cultured in RPMI 1640 medium containing 10% fetal bovine serum. For the assay, 2.0 × 106/ml cells were incubated with increasing concentrations of insulin/analog and human [125I]monoiodotyrosyl-A14-insulin (PerkinElmer Life Sciences, 2200 Ci/mmol, 20,000 cpm, about 0.01 nm) for 2.5 h at 15 °C in HEPES binding buffer (100 mm HEPES, 100 mm NaCl, 5 mm KCl, 1.3 mm MgSO4, 1 mm EDTA, 10 mm glucose, 15 mm NaOAc, 1% BSA (w/v), pH 7.6) (500 μl). After incubation, 2 × 200 μl were centrifuged at 13,000 × g for 10 min. Radioactive pellets were counted using a Wizard 1470 Automatic γ Counter (PerkinElmer Life Sciences). Binding data were analyzed by Excel software using a one-site fitting program developed in the laboratory of Dr. Pierre De Meyts (A. V. Groth and R. M. Shymko, Hagedorn Research Institute, Denmark, a kind gift of Pierre De Meyts) and GraphPad Prism 5, and the dissociation constant was determined. The dissociation constant of human 125I-insulin was set up to 0.3 nm. Receptor binding assays were performed under conditions excluding the depletion of free ligand. The software used for the analysis of binding data developed in the laboratory of Dr. Pierre De Meyts takes the potential ligand depletion into account using equations developed by Swillens (24).
Cell proliferation of R−/IR-A cells in the presence of 1, 5, 10, and 30 nm insulin, IGF-II, or [NMeTyrB26]-insulin was determined as previously described from our laboratories (7, 11, 19). Cells were counted at 48 h in Boyden chambers and values are expressed as % increase over SFM.
Analysis of IR, IRS-1, IRS-2, and Shc Levels
Cells were serum-starved for 24 h, and then stimulated with 50 ng/ml of either insulin or IGF-II or 5 nm insulin, IGF-II, or [NMeTyrB26]-insulin for 8 and 24 h. Cells were lysed in RIPA buffer and the level of IR, IRS-1, IRS-2, and Shc was determined by immunoblotting with polyclonal antibodies against the β subunit of either the IR or IGF-IR (Santa Cruz Biotechnology), against IRS-1 or IRS-2 (UBI Millipore) or Shc (Santa Cruz Biotechnology). Normalization of proteins was done by probing the same filter with anti-β-actin polyclonal antibodies (Sigma). Inhibitors of the proteasome (MG132) or the lysosomal pathway (leupeptin/pepstatin) were added at the time of stimulation with growth factors at the concentrations mentioned above.
Targeting of Clathrin and Caveolin-1 by siRNA
Gene silencing of mouse clathrin and caveolin-1 was obtained by RNA interference using small interfering RNA (siRNA). R−/IR-A cells were transfected with vehicle (diethyl pyrocarbonate-treated water), control siRNA (scrambled), or siRNA directed against clathrin heavy chain or caveolin-1 (200 pmol) using the TransIT-siTKO reagents (Mirus Corporation). siRNA oligos specific for mouse clathrin heavy chain, caveolin-1, and controls were siGenome Smart pools, from Thermo Scientific Dharmacon. Twenty-four h after transfection, R−/IR-A cells were starved in SFM for 48 h, then stimulated with either IGF-II or insulin and evaluated for IR internalization by ELISA. The expression of clathrin and caveolin-1 proteins was detected by immunoblot using anti-clathrin (BD Biosciences) or anti-caveolin-1 polyclonal antibodies (Santa Cruz Biotechnology).
Confocal Microscopy
R−/IR-A cells were plated onto coverslips, serum-starved for 24 h, and then stimulated with 200 ng/ml of insulin or IGF-II at various time points at 37 °C. Coverslips were processed for immunofluorescence and confocal analysis, as previously described (18, 25). Antibodies used were: anti-IR polyclonal antibodies (Santa Cruz Biotechnology), anti-EEA1 monoclonal antibodies, and anti-LAMP1 monoclonal antibodies (BD Pharmingen). Secondary antibodies were Alexa Fluor 488 (green) and Alexa Fluor 594 (red) (Molecular Probes). Coverslips were analyzed and photographed on a Leica TCS-SP2 confocal microscope (Leica Microsystems, Wetzlar, Germany) with a ×63 Apo PLA oil immersion objective (NA 1.4) and 60-μm aperture using the LEICA Scan TCS-SP2 software (Leica Microsystems).
Endocytosis Inhibitors
The broad-range endocytosis inhibitor methyl-β-cyclodextrin was used at 10 mm. The clathrin-dependent pathway inhibitor chlorpromazine was used at 15 μm, whereas the clathrin-independent endocytosis inhibitor filipin was used at 1 μg/ml. Serum-starved cells were preincubated for 60 min with methyl-β-cyclodextrin or filipin, for 20 min with chlorpromazine, and then tested for IR-A internalization and signaling.
Detection of Activated Signaling Pathways
Serum-starved R−/IR-A cells were stimulated with insulin, IGF-II, or [NMeTyrB26]-insulin (50 ng/ml) at various time points in the absence or presence of endocytosis inhibitors. The activation of Akt, ERK1/2, and S6 ribosomal protein was analyzed by Western immunoblot using the PathScan Multiplex Western Mixture I (Cell Signaling Technology), which provides a mixture of phospho-specific antibodies for different activated proteins and for ElF4E protein to monitor the loading of the samples (26, 27). The kit also includes phosphoantibodies to p90 RSK, which was undetectable in our experiments. Phosphorylation of the IR-A was detected using anti-phospho-IGF-IRβ (Tyr1135/Tyr1136)/IRβ (Tyr1150/Tyr1151) antibodies (Cell Signaling Technology), whereas IRS-1 phosphorylation was assessed using either anti-phospho-IRS-1 (Tyr612) or phospho-IRS-1 (Ser307) from Upstate Biotechnology.
Densitometric and Statistical Analysis
Densitometric analysis was performed using a Molecular Dynamics densitometer. Data were analyzed using the ImageQuant program at the Kimmel Cancer Center Nucleic Acid Facility. Values are expressed as arbitrary units. For statistical analysis, all experiments were carried out in triplicate and repeated at least three times. Results are expressed as mean ± S.D. All statistical analyses were carried out with SigmaStat for Windows version 3.10 (Systat Software, Inc., Port Richmond, CA). Results were compared using the two-sided Student's t test or two-way ANOVA with Bonferroni's multiple comparison test. Differences were considered statistically significant at p < 0.05.
RESULTS
IGF-II and Insulin Differ in Their Ability to Regulate Cell Surface Levels of IR-A
We initially assessed ligand-induced IR-A internalization from the cell surface by ELISA, as described in previous work from our laboratories (17, 18). Insulin stimulation of R−/IR-A cells induced a clear reduction in the level of cell surface IR-A proteins with a maximum effect after 60 min. In contrast, IGF-II promoted only a modest but statistically significant internalization of the IR-A (Fig. 1A). To account for the differences in molecular weight between insulin and IGF-II we repeated internalization assays using equimolar concentrations (30 nm) of the two ligands. We found a similar rate of receptor internalization (supplemental Fig. S1), thus corroborating the data presented above.
FIGURE 1.
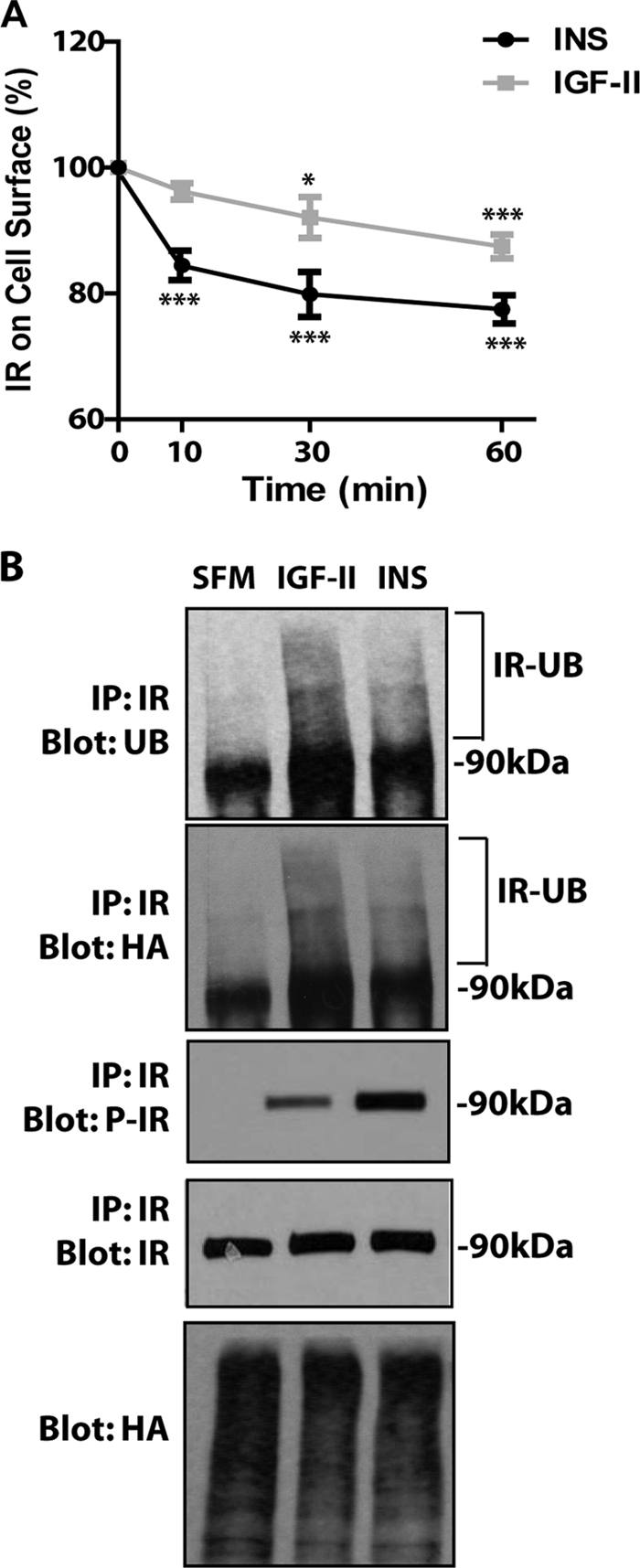
Insulin and IGF-II differentially affect IR-A internalization despite promoting similar levels of IR-A ubiquitination. A, the level of IR-A internalization in R−/IR-A cells was determined by ELISA at different time points after insulin (INS) and IGF-II stimulation, as described under “Experimental Procedures.” Ligands were used at 200 ng/ml to ensure saturating concentrations. Data are the average ± S.D. of three independent experiments. Statistical significance was determined using Student's t test for repeated measures, *, p < 0.05; ***, p < 0.01 (INS or IGF-II versus SFM) and using two-way ANOVA with Bonferroni's multiple comparison test, p < 0.01 (INS versus IGF-II). B, ligand-dependent ubiquitination of the IR-A in R−/IRA cells was assessed as described under “Experimental Procedures” and in previous work from our laboratories (17, 18). The experiment shown is representative of three independent experiments.
We have recently shown that ubiquitination of the IGF-IR regulates ligand-dependent receptor internalization (17, 18). Thus, we performed ubiquitination assays in R−/IR-A cells (17, 18) to test whether the different rates of IR-A internalization induced by IGF-II and insulin was due to a different ability of the two ligands to promote IR-A ubiquitination. After transiently transfecting a HA-ubiquitin construct into R−/IR-A cells, we immunoprecipitated the IR-A and assessed receptor ubiquitination by immunoblot with either an anti-ubiquitin or an anti-HA antibody. Some IR-A ubiquitination was detectable in unstimulated cells but was enhanced after ligand stimulation (Fig. 1B). Significantly, IGF-II stimulation of R−/IR-A cells induced ubiquitination of the IR-A (17, 18) that was even slightly greater than the ubiquitination induced by insulin, as assessed by both anti-ubiquitin antibodies (which detect endogenous and exogenously transfected ubiquitin) and anti-HA antibodies (which detect exogenously transfected ubiquitin). This difference was not due to either a difference in receptor activation, which, as we have previously reported (7, 11) was phosphorylated at higher levels by insulin (Fig. 1B, P-IR), or to differences in transfection efficiency of the HA-ubiquitin plasmid, which was comparable in all conditions (Fig. 1B, Blot: HA).
These results demonstrate that IGF-II and insulin clearly differ in their ability to induce IR-A internalization despite inducing similar levels of receptor ubiquitination. Thus, additional mechanisms (including IR-A phosphorylation) are likely more critical in the regulation of IR-A trafficking.
Affinity of Ligand for IR-A Is Important in Regulating IR-A Phosphorylation, Signaling, Proliferation, and Internalization
Next, we tested in R−/IR-A cells several biological read-outs comparing insulin, IGF-II, and the insulin analog [NMeTyrB26]-insulin, which has been previously described (22). This analog has an IR binding affinity 4-fold lower than insulin and comparable with the affinity of IGF-II (22). Because the affinity of [NMeTyrB26]-insulin for the IR was previously determined in rat adipocytes, which prevalently express the IR-B (22, 28), we first assessed the binding affinity of this insulin analog to the IR-A by performing binding assays in IM-9 cells, which have high IR-A expression (23).
The affinity of [NMeTyrB26]-insulin for the IR-A was about 16% (supplemental Fig. S2A), which is slightly lower than the 21% we previously determined in rat adipocytes (22). The affinity of IGF-II was instead about 7% of human insulin (supplemental Fig. S2A), which is less that 20% expected but in perfect agreement with binding studies recently performed by Gauguin et al. (23) in the same IM-9 lymphocytes. These experiments therefore clearly confirm that IGF-II and the insulin analog [NMeTyrB26]-insulin have similar affinity for the IR-A.
We then tested IR-A phosphorylation in R−/IR-A cells after insulin, IGF-II, and [NMeTyrB26]-insulin stimulation. Insulin at 30 nm (∼200 ng/ml) induced strong IR-A phosphorylation, which was instead reduced after IGF-II stimulation (supplemental Fig. S2B). Importantly, the [NMeTyrB26]-insulin analog induced IR-A phosphorylation at levels similar to IGF-II (supplemental Fig. S2B). To exclude the possibility that the differences in IR-A phosphorylation and therefore internalization between insulin and IGF-II were affected by the concentration of ligands in the supraphysiological range, we also tested IR-A phosphorylation at 1 and 5 nm concentration of ligands, which are in the physiological range for insulin. The phosphorylation of IR-A induced by insulin, IGF-II, and [NMeTyrB26]-insulin in R−/IR-A cells showed a similar pattern at 1 and 5 nm (supplemental Fig. S2B), confirming the validity of our results at 200 ng/ml.
To characterize further the biological properties of [NMeTyrB26]-insulin in comparison with insulin and IGF-II we first determined by immunoblot the activation of Akt and ERK1/2, the two major downstream effectors of IR-A signaling, which are critical for IR-A-dependent proliferation (7, 11).
In agreement with previously published data (7, 11), insulin promoted prolonged Akt activation, which was less pronounced (p < 0.05) after stimulation by both IGF-II and [NMeTyrB26]-insulin at all concentrations tested (supplemental Fig. S2C). On the other end, ERK1/2 activation by all growth factors peaked at 10 min of ligand stimulation (supplemental Fig. S2C) but remained more sustained after insulin stimulation compared with IGF-II and [NMeTyrB26]-insulin at 1 and 5 nm concentrations but was similar (NS) with all three ligands at 30 nm (supplemental Fig. S2C).
As IGF-II is a more potent mitogen than insulin despite its lower affinity for the IR-A and the reduced ability to promote IR-A phosphorylation (7, 11), it is reasonable to assume that the [NMeTyrB26]-insulin analog, which has an affinity in the range of IGF-II, could be as well a more potent mitogen than insulin. Indeed, at all equimolar concentrations tested, [NMeTyrB26]-insulin induced cell growth at significantly higher levels than insulin with values very similar to IGF-II, as determined by growth curves assays in R−/IR-A cells (Fig. 2A). Moreover, the [NMeTyrB26]-insulin analog also induced low levels of IR-A internalization (at all ligand concentrations) in comparison to insulin, with a rate very similar to IGF-II (Fig. 2, B–D).
FIGURE 2.

Ligand affinity for the IR-A modulates cell growth and internalization. A, growth curve experiments were performed as previously described from our laboratories (7, 11, 19). Cells were counted in Boyden chambers after 48 h and values are expressed as % increase over SFM. Data are the averages ± S.D. of three independent experiments. Statistical significance was determined using two-way ANOVA: p < 0.02, insulin versus IGF-II stimulation curve; p < 0.05, insulin versus [NMeTyrB26]-insulin stimulation curve. B–D, the level of IR-A internalization in R−/IR-A cells was determined by ELISA at different time points after insulin (INS), IGF-II, or [NMeTyrB26]-insulin stimulation, as described under “Experimental Procedures.” Data are the averages ± S.D. of three independent experiments. Statistical significance was determined using Student's t test for repeated measures, *, p < 0.05; **, p < 0.01 (INS, IGF-II or [NMeTyrB26]-insulin versus SFM) and using two-way ANOVA with Bonferroni's multiple comparison test, *, p < 0.05; **, p < 0.01 (INS versus IGF-II or [NMeTyrB26]-insulin).
Collectively, these results support the hypothesis that different affinities of the various hormone/ligands could play an important role in modulating receptor phosphorylation, internalization from the cell membrane, and signaling, thereby modulating IR-A downstream biological responses. In addition, these results confirm that the concentration used for the internalization assays (and in subsequent experiments in the manuscript) perfectly recapitulates the results obtained at physiological concentrations.
Insulin but Not IGF-II Induces IR-A and IRS-1 Down-regulation
Next, we determined in R−/IR-A cells whether prolonged stimulation with IGF-II or insulin differentially affected IR-A stability. Total IR-A levels were decreased by 8 h of insulin stimulation and further decreased by 24 h (Fig. 3A). In contrast, IGF-II exerted only a modest effect on IR-A levels, which were reduced by only ∼15% after 24 h. Concurrently, insulin markedly reduced the levels of the docking protein IRS-1 (Fig. 3A), one of the major downstream effectors of IR signaling that leads to cell proliferation and transformation (29, 30). In contrast, IGF-II stimulation had no effect on IRS-1 levels (Fig. 3A). Interestingly, prolonged insulin or IGF-II stimulation of R−/IR-A cells had no effect in regulating the levels of either Shc or IRS-2 (Fig. 3A), two other components of the IR-A downstream signaling pathway (31–33).
FIGURE 3.

Insulin and IGF-II differ in their ability to regulate IR-A and IRS-1 stability. A, R−/IR-A cells were serum starved for 24 h and then stimulated with 50 ng/ml of insulin (INS) or IGF-II for the indicated time points. Proteins levels were determined by immunoblot analysis with specific polyclonal antibodies, as described under “Experimental Procedures.” B, serum-starved R−/IR-A cells were stimulated at the different time points with insulin and IGF-II (50 ng/ml). Serine phosphorylation of IRS-1 was determined by immunoblot using phospho-specific antibodies for Ser307. Total IRS-1 was assessed using anti-IRS-1 polyclonal antibodies. Densitometric analysis is expressed as arbitrary units. B, serine phosphorylation of IRS-1 was assessed by immunoblot using phospho-specific antibodies for serine 307. Blots are representative of three independent experiments. C and D, to assess the stability of the IR-A in the presence of specific inhibitors for either the proteasomal or lysosomal pathway, serum-starved R−/IR-A cells were stimulated for 24 h with 50 ng/ml of insulin (INS) (C) or IGF-II (D) alone or supplemented with 20 μm MG132 (MG) or 100 μm leupeptin/pepstatin (Leu/Pep). IR levels were assessed by immunoblot. The total amount of protein loaded on the gel was monitored using anti-β-actin polyclonal antibodies (A–E). Quantification was performed by densitometry using NIH ImageJ software. The data are presented as mean ± S.D. Statistical significance was determined using two-way ANOVA with Bonferroni's multiple-comparison test. ***, p < 0.001; **, p < 0.01; *, p < 0.05.
Because insulin and IGF-I-dependent IRS-1 degradation is regulated by PI3K-dependent serine phosphorylation of IRS-1 (34, 35), we utilized phospho-specific antibody for IRS-1 Ser307. Insulin stimulation of R−/IR-A cells induced a significant phosphorylation of Ser307 at 10 min and this was sustained for up to 1 h (Fig. 3B). On the contrary, IGF-II promoted phosphorylation of IRS-1 Ser307 with significantly slower kinetics insofar as it reached levels comparable with insulin only after 60 min of IGF-II stimulation (Fig. 3B). These results suggest that the inability of IGF-II to promote IRS-1 degradation correlate to a reduced capacity of IGF-II to promote IR-A-dependent serine phosphorylation of IRS-1.
It has been shown (36) that in HeLa/IR cells insulin-induced IR degradation was inhibited by proteosomal inhibitors. However, it was not established whether IR stability was sensitive to inhibitors of the lysosomal pathway, which is the major route of degradation of RTKs (15, 37). To investigate the pathway(s) responsible for ligand-dependent degradation of the IR-A in R−/IR-A cells, we assessed IR-A levels by immunoblot after 24 h stimulation of insulin or IGF-II alone or in the presence of either the proteosomal inhibitor MG132 (20 μm) or the lysosomal inhibitor pepstatin/leupeptin mixture (100 μm). Treatment with leupeptin/pepstatin was as effective as MG132 in stabilizing the IR-A protein upon prolonged insulin stimulation (Fig. 3C), whereas the difference after IGF-II stimulation was not statistically significant due to the very limited effect of IGF-II on IR-A degradation (Fig. 3D). These results indicate that, in addition to the proteosomal pathway, the lysosomal pathway also plays an important role in down-regulation of the IR-A in R−/IR-A cells.
To confirm the results of the experiments of IR-A and IRS-1 stability at equimolar and physiological concentrations of insulin, we determined IR-A and IRS-1 levels after prolonged stimulation of R−/IR-A cells with 5 nm insulin and IGF-II. We also exposed the cells to the same concentration of [NMeTyrB26]-insulin as an additional control. Although prolonged insulin stimulation induced IR-A degradation, both IGF-II and [NMeTyrB26]-insulin did not affect IR-A levels in R−/IR-A cells (supplemental Fig. S3). Concurrently, 5 nm insulin reduced IRS-1 levels, which were not affected by an equimolar concentration of IGF-II (supplemental Fig. S3). [NMeTyrB26]-Insulin was more effective than IGF-II in promoting IRS-1 degradation although at lower levels than insulin (supplemental Fig. S3). These results strongly support the data obtained at 200 ng/ml (30 nm), and further strengthen the hypothesis that the different abilities of insulin and IGF-II to regulate IR-A and IRS-1 stability contributes to the difference in mitogenic responses between insulin and IGF-II.
We should point out that in the experiments presented in Figs. 1–3 we used mouse embryo fibroblasts null for the IGF-IR and overexpressing solely IR-A. It is important to point out that the results of these experiments may not be equally clear in other cellular models that would coexpress the IGF-IR, which, by binding the IGF-II, could obscure IGF-II action on the IR-A. However, it can be argued that R−/IR-A cells do not exactly represent a physiological or pathological model where the IR-A is expressed in conjunction with the IGF-IR. To address this issue, we performed IGF-II and insulin-mediated internalization assays in NIH3T3/IR-A cells, which express physiological levels of IR-A and endogenous IGF-IR (supplemental Fig. S4A) and in R+/IR-A cells, which are R−/IR-A cells in which the IGF-IR has been reintroduced (supplemental Fig. S4B). In both cell lines insulin promoted IR-A internalization, whereas IGF-II was ineffective (supplemental Fig. S4, A and B). As demonstrated in R−/IR-A cells, also in R+/IR-A, prolonged stimulation with insulin, but not with IGF-II, affected IR-A and IRS-1 stability.
Parallel experiments were also performed in a pathological cell model using MDA-MB-157 breast cancer cells, which coexpress the IGF-IR together with high levels of endogenous IR-A (supplemental Fig. S4C). Here, insulin, but not IGF-II, induced IR-A internalization (supplemental Fig. S4C). However, similarly to previously published data (38, 39), we did not detect degradation of either IR-A or IRS-1 after prolonged insulin stimulation (data not shown) suggesting that aggressive breast cancer cells may escape IR-A degradation and the negative feedback loop regulating IRS-1 stability. These data confirm the results obtained in R−/IR-A cells and indicate that IGF-II and insulin differ in their ability to regulate IR-A internalization even in additional physiological or pathological cell models expressing IR-A in conjunction with IGF-IR.
IR-A Is Internalized through Clathrin-dependent and -independent Pathways
Because the pathways that mediate IR-A internalization are not yet characterized, we assessed ligand-evoked internalization of the IR-A in R−/IR-A cells. Concurrently, we employed small interference RNA (siRNA) strategies to target endogenous expression of clathrin or caveolin-1, two critical components of the clathrin-dependent and -independent pathways, respectively (40–42). Our approach yielded a substantial suppression of endogenous clathrin protein expression (Fig. 4A) and a concurrent significant inhibition of insulin and IGF-II-mediated IR-A internalization (Fig. 4B). Suppression of endogenous caveolin-1 expression (Fig. 4A) was as effective as clathrin depletion but it resulted only in a modest although statistically significant reduction in the level of insulin-dependent internalization of the IR-A (Fig. 4C), whereas the difference in IGF-II-induced internalization was not statistically significant (Fig. 4C). These results suggest that insulin and IGF-II stimulation lead to clathrin-dependent and -independent IR-A internalization, although clathrin-dependent endocytosis may play a prevalent role in the process.
FIGURE 4.

Depletion of endogenous clathrin and caveolin-1 inhibits ligand-dependent internalization of the IR-A. A, gene knockdown for clathrin and caveolin-1 in R−/IR-A cells was achieved by siRNA. Level of clathrin and caveolin-1 in vehicle (Veh), control oligo (Control), and siRNA-treated (siClath or siCav) cells were assessed by immunoblot using anti-clathrin- and anti-caveolin-1-specific polyclonal antibodies. Total protein load was assessed using anti-β-actin polyclonal antibodies. Blots are representative of three independent experiments. Insulin and IGF-II-mediated internalization of the IR-A in R-IR-A cells depleted of endogenous clathrin (B) or caveolin-1 (C) was assessed by ELISA 72 h post-transfection. The data are presented as mean ± S.D. of three independent experiments. Statistical significance was determined using two-way ANOVA with Bonferroni's multiple comparison test. **, p < 0.01; *, p < 0.05 (siRNAs versus oligo-control-treated cells).
IR-A Is Preferentially Degraded via Clathrin-dependent Endocytosis
To determine the contribution of clathrin-dependent and -independent pathways to the regulation of IR-A stability, we utilized clathrin and caveolin-1-specific siRNA, and monitored IR-A levels by immunoblotting analysis. We found that IR-A degradation induced by prolonged insulin stimulation of R−/IR-A cells was significantly inhibited by depletion of clathrin (Fig. 5A). On the contrary, depletion of endogenous caveolin-1 was ineffective in stabilizing IR-A protein levels, whose difference was statistically significant only after 8 h of insulin stimulation (Fig. 5C). The same results were obtained after IGF-II stimulation, although the differences in the level of IR-A protein between clathrin-depleted (Fig. 5B), caveolin-depleted (Fig. 5D), and control-transfected R−/IR-A cells were not statistically significant due to a low level of IR-A degradation induced by IGF-II in these cells. Collectively, these results indicate that insulin-evoked degradation of the IR-A is preferentially regulated by the clathrin-dependent endocytosis pathway.
FIGURE 5.

Clathrin-dependent endocytosis is required for insulin-induced IR-A degradation. Gene knockdown for clathrin and caveolin-1 in R−/IR-A cells was achieved by siRNAs. IR-A levels were determined in clathrin-depleted (A and B) or caveolin-1-depleted (C and D) R−/IR-A cells by immunoblot after stimulation with insulin or IGF-II for 8 and 24 h. IR-A, clathrin, and caveolin-1 expression was assessed using anti-IR (A–D), anti-clathrin (A), and anti-caveolin-1 (B) polyclonal antibodies. Protein load was assessed using anti-β-actin polyclonal antibodies. Results are expressed as average ± S.D. of three independent experiments. Statistical significance was determined using two-way ANOVA with Bonferroni's multiple comparison test. **, p < 0.01.
Insulin and IGF-II Differentially Regulate Sorting of IR-A
To investigate further insulin- and IGF-II-induced sorting of the IR-A, we employed confocal microscopy analysis to test whether the IR-A colocalized with EEA1, a marker of early endosomes, or with LAMP-1, a marker of lysosomes (40). In unchallenged R−/IR-A cells, there was no colocalization of IR-A with EEA1 (Fig. 6A). After IGF-II or insulin stimulation, the IR-A colocalized in EEA1-positive endosomes (Fig. 6A). In contrast, whereas insulin stimulation of R−/IR-A cells induced a significant colocalization of IR-A with LAMP-1, which increased between 2 and 6 h of insulin stimulation (Fig. 6B), we did not detect significant colocalization between the IR-A and LAMP-1 after IGF-II stimulation (Fig. 6B).
FIGURE 6.

Insulin and IGF-II induce different sorting of the IR-A. R−/IR cells were plated onto coverslips and serum starved for 24 h. Cells were then stimulated with 200 ng/ml of either insulin or IGF-II for the indicated time points. Co-localization of the IR-A with either EEA1 (A) or LAMP-1 (B) was assessed by confocal microscopy, as described under “Experimental Procedures.” Insets represent enlarged views (×3) of the boxed regions. Insets include arrows that point to single isolated dots where colocalization between the IR-A and either EEA1 or LAMP-1 is evident. Images were collected on a Leica TCS-SP2 confocal microscope (Leica Microsystems) with a ×63 Apo PLA oil immersion objective (NA 1.4) and 60-μm aperture using LEICA Scan TCS-SP2 software (Leica Microsystems). Images were merged using Photoshop 6. Pictures are representative of at least 10 independent fields from three independent experiments. Fields were selected for the presence of cells with the following criteria: well defined limits, clear identification of nucleus, and absence of intersection with neighboring cells. An average of 300 cells was examined for each condition. Data are representative of ∼90% of the total number of cells examined.
Collectively, these results indicate that insulin and IGF-II significantly differ in their ability to regulate IR-A sorting. Upon insulin stimulation the IR-A is significantly internalized from the cell surface and sorted from early endosomes into the lysosomal/degradative compartment, whereas IGF-II activation of the IR-A promotes less efficient internalization of the receptor, which is sorted initially into early endosomes but does not reach the late endosome/lysosomal compartment, thereby being likely recycled to the cell surface.
Pharmacological Inhibition of IR-A Endocytosis Affects Downstream Signaling
Next, we determined insulin and IGF-II-evoked activation of the Akt and MAPK pathways in R−/IR-A cells, as a read-out of IR-A-induced signaling (7, 11). Concurrently, we utilized well characterized pharmacological inhibitors of the endocytic pathway. Initially, we tested a general inhibitor of endocytosis methyl-β-cyclodextrin, which extracts cholesterol from membranes and affects both clathrin-dependent and -independent pathways (41–43). As control, we tested by ELISA the effect of methyl-β-cyclodextrin on IR-A internalization. Methyl-β-cyclodextrin inhibited both insulin and IGF-II-induced IR-A internalization (Fig. 7A), although the effect after IGF-II stimulation was less evident due to low level of receptor internalization.
FIGURE 7.

Pharmacological inhibition of clathrin-dependent and -independent endocytosis pathways blocks IR-A internalization and regulates IR-A signaling. A, the level of IR-A internalization in R−/IR cells was determined by ELISA. Cells were serum starved for 24 h, pre-treated with 10 mm of methyl-β-cyclodextrin for 1 h, and then stimulated with 200 ng/ml of insulin (INS), IGF-II, or [NMeTyrB26]-insulin alone or supplemented with methyl-β-cyclodextrin. Data are the average ± S.D. of three independent experiments carried out in quadruplicate. Statistical significance was determined using two-way ANOVA, ***, p < 0.0001 (INS versus INS + methyl-β-cyclodextrin and [NMeTyrB26]-insulin versus [NMeTyrB26]-insulin + methyl-β-cyclodextrin); **, p < 0.005 (IGF-II versus IGF-II + methyl-β-cyclodextrin). B, cells were serum starved for 24 h, pre-treated in SFM alone, or supplemented with 10 mm methyl-β-cyclodextrin for 1 h and then stimulated with 50 ng/ml of insulin (INS), IGF-II, or [NMeTyrB26]-insulin alone or supplemented with methyl-β-cyclodextrin for the indicated time points. Effect of methyl-β-cyclodextrin on insulin and IGF-II-dependent IR-A, IRS-1, Akt, ERK1/2, and p70 S6K phosphorylation was assessed by immunoblot using phospho-specific antibodies as described under “Experimental Procedures.” Blots are representative of three independent experiments.
To investigate the impact of endocytosis on IR-A signaling we determined the activation of IRS-1, Akt, ERK1/2, and p70 S6K after insulin or IGF-II stimulation by immunoblot analysis using phosphospecific antibodies. We also assessed IR-A phosphorylation as control to rule out that the inhibitor may affect internalization by interfering with ligand-mediated activation of the IR-A.
Activation of the IR-A by either insulin or IGF-II was not affected by methyl-β-cyclodextrin, even though the inhibitor had some effects on the kinetics of receptor activation (Fig. 7B). Methyl-β-cyclodextrin exposure increased early IR-A phosphorylation (up to 10 min) and slightly decreased the sustained activation of the receptor (30 and 60 min). A similar effect was detectable with IRS-1 tyrosine phosphorylation, which was only affected after prolonged insulin or IGF-II stimulation (Fig. 8B). Significantly, methyl-β-cyclodextrin had instead a major effect on serine phosphorylation of IRS-1 and downstream signaling molecules, abolishing Akt and ERKs activation but only slightly reducing p70 S6K phosphorylation mediated by both insulin and IGF-II.
FIGURE 8.

Pharmacological inhibition of either clathrin-dependent or -independent endocytosis blocks IR-A internalization and specifically affects IR-A signaling. A, R−/IR-A cells were serum starved for 24 h, pre-treated for 1 h with either 1 μg/ml of filipin or 15 μm chlorpromazine, and then stimulated with 200 ng/ml of insulin (INS) or IGF-II alone or in the presence of the specific inhibitors. IR-A internalization was determined by ELISA. Data are the average ± S.D. of three independent experiments carried out in quadruplicates. Statistical significance was determined using Student's t test for repeated measures, *, p < 0.05 (INS versus INS + filipin; INS versus INS + chlorpromazine); *, p < 0.05 (IGF-II versus IGF-II + filipin; IGF-II versus IGF-II + chlorpromazine). B and C, cells were serum-starved for 24 h, pretreated for 1 h in SFM alone, or supplemented with either 1 μg/ml of filipin or 15 μm chlorpromazine and then stimulated with 200 ng/ml of insulin (INS) or IGF-II alone or in the presence of the specific inhibitors. B, effect of chlorpromazine, and C, filipin on insulin and IGF-II-dependent IR-A, IRS-1, Akt, ERK1/2, and p70 S6K phosphorylation was assessed by immunoblot using phosphospecific antibodies. Blots are representative of three independent experiments.
The effect of methyl-β-cyclodextrin on endocytosis and signaling was also assessed in R−/IR-A cells stimulated with equimolar concentrations of the insulin analog [NMeTyrB26]-insulin. Although methyl-β-cyclodextrin was less effective in inhibiting the prolonged IR-A phosphorylation induced by the analog compared with insulin and IGF-II (Fig. 7, A and B), the inhibitory effect on internalization of IRS-1, Akt, ERK1/2, and p70 S6K phosphorylation perfectly recapitulated the one obtained in the presence of insulin and IGF-II (Fig. 7, A and B).
To gain further insight into the role that IR-A internalization plays in regulating IR-A signaling, we assessed whether clathrin-dependent and -independent endocytosis may have different effects in regulating insulin- and IGF-II-evoked activation of IR-A downstream signaling. To this end, we selectively targeted clathrin-dependent and -independent pathways by using inhibitors that specifically inhibit either endocytic pathway. We used the cationic amphiphilic drug chlorpromazine, which specifically inhibits clathrin-dependent endocytosis by affecting the membrane localization of AP2, another essential component of clathrin-coated vesicles (44, 45). To target clathrin-independent endocytosis, we used filipin, a polyene antibiotic that acts as a cholesterol chelator without extracting sterols from membranes (41, 42). ELISAs performed in R−/IR-A cells confirmed that both chlorpromazine and filipin inhibited IR-A internalization from the cell surface after insulin and IGF-II stimulation (Fig. 8A). We then tested by immunoblot analysis the effect of the inhibitors on IR-A activation and downstream signaling. Notably, both chlorpromazine (Fig. 8B) and filipin (Fig. 8C) had no significant effects on IR-A or IRS-1 tyrosine phosphorylation levels at early time points, but they diminished the sustained tyrosine phosphorylation of IRS-1 induced by insulin and IGF-II (after 30 and 60 min). Chlorpromazine increased basal and ligand-induced serine phosphorylation of IRS-1 at early time points but reduced the prolonged phosphorylation particularly after 60 min of ligand stimulation (Fig. 8B). Significantly, chlorpromazine strongly inhibited Akt activation induced by both insulin and IGF-II (Fig. 8B), whereas it promoted a higher basal and a more prolonged activation of ERK proteins. In contrast, filipin severely affected serine phosphorylation of IRS-1 and ERKs activation by both insulin and IGF-II (Fig. 8C), whereas the reduction of Akt activation was more pronounced in IGF-II-stimulated cells than after insulin stimulation, which was affected mostly after 30 and 60 min (Fig. 8C). Both inhibitors slightly increased basal and early activation of p70 S6K induced by both insulin and IGF-II. It is noteworthy that these experiments (and the experiments presented in Fig. 7) were designed to assess the differences in downstream signaling between ligands in the presence or absence of endocytosis inhibitors and that insulin, IGF-II, and [NMeTyrB26]-insulin-stimulated (Fig. 7) samples were run on separate gels. One should be cautious, therefore, in comparing insulin and IGF-II signaling from different blots. This differential signaling was, however, addressed in detail in previous work from our laboratories (7, 11) and as described above.
Altogether, these results clearly indicate that endocytosis does not play a major role in regulating IR-A or IRS-1 early tyrosine phosphorylation, which occurs at the plasma membrane, but it plays a role in sustaining a prolonged receptor and IRS-1 tyrosine phosphorylation. In contrast, serine phosphorylation of IRS-1 seems to be severely regulated by clathrin-independent endocytosis. Moreover, our data suggest that upon insulin and IGF-II stimulation, clathrin-dependent endocytosis is critical for IR-A-dependent activation of Akt, whereas clathrin-independent endocytosis may prevalently regulate IR-A-dependent activation of ERKs and to a lesser extent Akt activation. Interestingly, p70 S6K activation was slightly affected and may be even increased by IR-A internalization inhibition. The relative independence of p70 S6K activation by IR-A internalization is in close agreement with our previous findings demonstrating that, although IGF-II is clearly less potent than insulin in promoting IR-A and Akt phosphorylation, it is approximately as effective as insulin in activating p70 S6K (46).
DISCUSSION
In the present study we tested the hypothesis that insulin and IGF-II could affect IR-A biological responses by differentially regulating IR-A trafficking and stability. For these experiments we used a unique model of mouse embryo fibroblasts lacking the IGF-IR and overexpressing solely the IR-A (11). Our main results can be summarized as follows. (i) Insulin stimulation of R−/IR-A cells promotes IR-A internalization, which is instead only modestly induced by IGF-II stimulation. (ii) The difference in internalization is not due to IR-A ubiquitination, which is comparable in IGF-II and insulin-stimulated R−/IR-A cells. (iii) The insulin analog [NMeTyrB26]-insulin, which has lower affinity than insulin for IR-A, promotes IR-A phosphorylation, internalization, and proliferation at levels comparable with IGF-II. (iv) Prolonged stimulation of R−/IR-A cells with insulin, but not with IGF-II or [NMeTyrB26]-insulin, targets the IR-A for degradation through both proteosomal and lysosomal pathways. (v) The different rates of receptor internalization mediated by insulin and IGF-II are conserved in three additional non-transformed and transformed cell lines where the IR-A is coexpressed at different levels with the IGF-IR. (vi) IRS-1 is down-regulated as well after prolonged insulin exposure but not after IGF-II exposure. (vii) Upon insulin or IGF-II stimulation, the IR-A is internalized through clathrin-dependent and -independent pathways, but only the clathrin-dependent internalization is required for IR-A degradation. (viii) Clathrin-dependent and -independent endocytosis differentially regulates the activation of IR-A downstream effectors.
Previously published work (47) has established that the IR, similarly to other RTKs, is internalized from the cell surface upon ligand stimulation. The majority of work has been concentrated on endocytosis of the IR through clathrin-coated vesicle-mediated internalization (47, 48), but reports have also suggested that additional pathways may be important for IR internalization (49). More recent data have in fact pointed out a role of caveolae in mediating rapid insulin-dependent internalization of the IR (45, 50). The majority of these experiments were performed in adipocytes, which preferentially express the IR-B isoform (7, 8) and exclusively upon insulin stimulation. Our study provides the first characterization of IR-A internalization in the presence of both insulin and IGF-II and provides strong evidence that contributes to elucidate the differences in downstream biological effects mediated by insulin and IGF-II through the IR-A.
Ligand-mediated ubiquitination of RTKs is a critical step in promoting receptor endocytosis (51), and recent data from our laboratories has demonstrated that IGF-I-mediated ubiquitination of the IGF-IR regulates receptor internalization and degradation (17, 18). Compared with insulin, IGF-II stimulation of the IR-A induces only a modest internalization of the receptor, which cannot be explained by a difference in the level of IR-A ubiquitination, as in fact IGF-II induced a slightly higher level of IR-A ubiquitination compared with insulin. These results therefore suggest that IR-A ubiquitination is not essential in promoting receptor internalization and that additional mechanisms may be more critical in the regulation of early events of IR-A endocytosis.
Recent biochemical evidence from several laboratories including ours suggests that RTKs may not be exclusively polyubiquitinated (addition of a polyubiquitin chain to a single lysine residue) but also monoubiquitinated at multiple sites (multiubiquitinated) with different effects on receptor endocytosis, down-regulation, or signaling (18, 52, 53). In the present work, the ubiquitination experiments only compared the total levels of IR-A ubiquitination induced by either insulin or IGF-II but did not address qualitatively the type of ubiquitin modifications on IR-A. We can therefore speculate that insulin and IGF-II may promote different type of ubiquitin modifications on the IR-A (polyubiquitination versus multiubiquitination), which differentially regulate internalization and sorting of the IR-A. Moreover, insulin and IGF-II may induce different kinetics of IR-A ubiquitination/deubiquitination, an additional biological mechanism that may potentially affect receptor internalization and sorting.
Previous work from our laboratories (7, 11) and the present article (cf. Fig. 1 and supplemental Fig. S2) demonstrate that in R−/IR-A cells insulin is more potent than IGF-II in inducing phosphorylation of the IR-A. Collectively, these results suggest that IR-A phosphorylation levels may play a more critical role than ubiquitin in regulating receptor internalization and sorting of the IR-A en route to degradation. This hypothesis is also supported by recent data demonstrating that insulin promotes higher levels of IR-A phosphorylation and internalization compared with the IR-B (54). In addition, the insulin analog [NMeTyrB26]-insulin, which induces IR-A phosphorylation at levels comparable with those evoked by IGF-II, promotes IR-A internalization at very low levels compared with insulin with a rate similar to IGF-II-induced internalization.
The differences in IR-A phosphorylation, internalization, and signaling induced by insulin and IGF-II correlate with the differences in affinity for the IR-A. The experiments performed with the insulin analog [NMeTyrB26]-insulin with an affinity for the IR-A similar to IGF-II support the hypothesis that the affinity of the different ligands for the IR-A may play an important role in determining receptor fate, signaling, and downstream biological responses. However, it is important to point out that additional mechanisms may contribute to this process, including differences of occupancy time and stability of the ligand-receptor complex in the acidifying endosomal compartments, which may affect the recycling process (55). Previous studies from Hansen et al. (56) have reported increased mitogenicity of insulin analogs with slow ligand dissociation rate. For several reasons these studies are not readily comparable with our data. These authors measured kdiss (dissociation rate constant), which does not exactly represent the ligand binding affinity, which is best expressed by Kd (as measured in this study). Moreover, more recent data have demonstrated that one of the highly mitogenic analogs used in the Hansen paper ([B-Asp10]-insulin/X10) binds not only the IR but also the IGF-IR and induces cell proliferation and transformation by activating both IGF-IR and IR (57, 58). Because Hansen et al. (56) tested mitogenesis in CHO-K1 cells and demonstrated that these cells actually express higher levels of IGF-IR (50,000 binding sites) compared with IR (3,000 binding sites), it is likely that the high mitogenic properties of some of these analogs could be prevalently or partially mediated through the IGF-IR, instead of the IR, clearly overestimating the mitogenic potential of these analogs through the IR.
However, because the insulin analog used here has not been rigorously characterized in previous and our current experiments, further studies are required in support of our model. Experiments with different insulin analogs with a wider range of affinities for the IR-A are currently underway to more accurately characterize the contribution that the relative affinity of the various ligands/analogs may play in regulating IR-A phosphorylation, endocytosis, and mitogenic responses.
The confocal analysis demonstrated that IGF-II-activated IR-A follows the same fate of insulin-activated IR-A up to the early endosomal compartment, where the IR-A colocalizes with EEA1 following exposure to either insulin or IGF-II. However, after prolonged insulin stimulation, a significant fraction of the IR-A is then sorted into the lysosomal compartment and targeted for degradation, whereas the majority of IGF-II-activated IR-A does not co-localize with the lysosomal marker LAMP-1. These results suggest that upon IGF-II stimulation, the IR-A may be sorted from early endosomes into a recycling compartment where it is routed back to the plasma membrane. The nature of this recycling compartment and whether the internalized IR-A can recycle back to the cell membrane from early endosomes in a RAB4-dependent fashion (rapid recycling route), or through RAB11-positive endosomes (slow recycling route) (59) remain to be elucidated.
The differential effects of IGF-II and insulin on IR-A internalization and sorting are reminiscent of the EGFR, where different EGFR ligands, such as amphiregulin, TGF-α, HB-EGF, or epiregulin, have diverse effects on the regulation of receptor endocytosis, sorting, and degradation (60–62). Sustained IGF-II stimulation of R−/IR-A cells fails to appreciably modulate IR-A levels and fails to induce degradation of IRS-1, a critical regulator of IR-A-induced mitogenesis (29, 30). Insulin and IGF-I-dependent IRS-1 degradation are regulated by PI3K-dependent phosphorylation of IRS-1 on serine residues, which is a negative feedback regulatory mechanism to control the activity of the IR and IRS-1 after prolonged ligand stimulation (34, 35). Our data clearly show that both insulin and IGF-II evoke tyrosine phosphorylation of IRS-1 but they differ in the ability to promote serine phosphorylation of IRS-1, which may regulate IRS-1 degradation. Thus, these results support the hypothesis that the inability of IGF-II to promote IRS-1 degradation may depend on the reduced capacity of IGF-II to promote IR-A-dependent serine phosphorylation of IRS-1. We cannot exclude that additional mechanisms may also contribute to determine IRS-1 escape from the negative feedback loop that targets it for degradation.
The pathways that regulate endocytosis of the EGFR are well characterized, as are many proteins that play a critical role in the regulation of receptor internalization and sorting (15, 16, 37, 51). However, proteins that regulate IR-A internalization and sorting have not been so far identified. We have previously established that the adapter protein Grb10 binds the ubiquitin ligase Nedd4 (63) and promotes IGF-IR ubiquitination and internalization through clathrin-dependent and -independent pathways (17, 18). Grb10 binds the IR in an insulin-dependent fashion (64–67) and Grb10 depletion by shRNA in HeLa cells inhibits insulin-dependent IR ubiquitination and degradation (36). However, whether insulin and IGF-II may differentially affect Grb10 binding to the IR-A isoform and whether Grb10 may regulate IR-A internalization and sorting remains to be established. Because IGF-II induces lower levels of IR-A phosphorylation compared with insulin, we can hypothesize that the stronger mitogenic activity induced by IGF-II over insulin may not only be attributed to a reduced ability to promote receptor internalization and degradation (cf. Figs. 1–3) but also to a decreased ability of negative regulators of IR signaling, such as Grb10, to bind the IR-A after IGF-II stimulation. Clathrin-dependent and -independent routes of EGFR internalization have significantly different effects on receptor fate and signaling abilities (41). Clathrin-mediated endocytosis plays no major role on EGFR degradation but rather induces receptor recycling and sustained signaling, whereas the clathrin-independent pathway promotes EGFR degradation (41). Our results suggest instead that insulin-dependent IR-A degradation requires clathrin-dependent endocytosis, whereas both clathrin-dependent and -independent pathways may contribute to the regulation of IR-A downstream signaling induced by both insulin and IGF-II.
Endocytosis does not play a major role in regulating IR-A or IRS-1 early tyrosine phosphorylation, which occurs at the plasma membrane, but it may play a role in regulating the prolonged tyrosine phosphorylation, which may require IR-A and IRS-1 internalization into early endosomes. In contrast, serine phosphorylation of IRS-1 requires IR-A endocytosis through a clathrin-independent pathway, as it is severely reduced after exposure to filipin. Clathrin-dependent endocytosis is critical instead for IR-A-dependent activation of Akt, whereas clathrin-independent endocytosis may prevalently regulate IR-A-dependent activation of ERKs and provides a minor contribution to Akt activation.
In summary, our results suggest that the lower affinity of IGF-II for the IR-A may on one hand promote lower IR-A phosphorylation and activation of early downstream effectors compared with insulin but may also protect IR-A and IRS-1 from negative feedback down-regulation mechanisms, thereby evoking a more potent mitogenic stimulus. In addition, the reduced IR-A activation promoted by IGF-II may protect the receptor from the action of negative regulators, such as Grb10, which bind the IR in a ligand-dependent manner.
Supplementary Material
Acknowledgment
We thank Dr. Renato Baserga for the kind gift of R−/IR-A cells and helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 DK068419 (to A. M.), the Benjamin Perkins Bladder Cancer Fund and the Martin Greitzer Fund (to A. M.), and grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC Grant 10625), AIRC project Calabria 2011 and Fondazione Cassa di Risparmio di Calabria e Lucania, Programmi di Ricerca di Rilevante Interesse Nazionale-Ministero dell' Istruzione, dell' Università e della Ricerca (PRIN-MIUR) Grant 2008BKRFBH_005 (to A. B.), and Research Project of the Academy of Sciences of the Czech Republic Grant Z40550506 (to J. J.).

This article contains supplemental Figs. S1–S4.
- IR
- insulin receptor
- IGF-II
- insulin-like growth factor 2
- IGF-IR
- IGF receptor I
- RTK
- receptor-tyrosine kinase
- IRS-1
- insulin receptor substrate 1
- SFM
- serum-free medium
- ANOVA
- analysis of variance
- S6K
- S6 kinase.
REFERENCES
- 1. Seino S., Bell G. I. (1989) Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 159, 312–316 [DOI] [PubMed] [Google Scholar]
- 2. Mosthaf L., Grako K., Dull T. J., Coussens L., Ullrich A., McClain D. A. (1990) Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 9, 2409–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Papa V., Gliozzo B., Clark G. M., McGuire W. L., Moore D., Fujita-Yamaguchi Y., Vigneri R., Goldfine I. D., Pezzino V. (1993) Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 53, 3736–3740 [PubMed] [Google Scholar]
- 4. Yamaguchi Y., Flier J. S., Benecke H., Ransil B. J., Moller D. E. (1993) Ligand-binding properties of the two isoforms of the human insulin receptor. Endocrinology 132, 1132–1138 [DOI] [PubMed] [Google Scholar]
- 5. Kosaki A., Webster N. J. (1993) Effect of dexamethasone on the alternative splicing of the insulin receptor mRNA and insulin action in HepG2 hepatoma cells. J. Biol. Chem. 268, 21990–21996 [PubMed] [Google Scholar]
- 6. Webster N. J., Kong Y., Sharma P., Haas M., Sukumar S., Seely B. L. (1997) Differential effects of Wilms tumor WT1 splice variants on the insulin receptor promoter. Biochem. Mol. Med. 62, 139–150 [DOI] [PubMed] [Google Scholar]
- 7. Frasca F., Pandini G., Scalia P., Sciacca L., Mineo R., Costantino A., Goldfine I. D., Belfiore A., Vigneri R. (1999) Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 19, 3278–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belfiore A. (2007) The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 13, 671–686 [DOI] [PubMed] [Google Scholar]
- 9. Belfiore A., Malaguarnera R. (2011) Insulin receptor and cancer. Endocr. Relat. Cancer 18, R125–147 [DOI] [PubMed] [Google Scholar]
- 10. Krywicki R. F., Yee D. (1992) The insulin-like growth factor family of ligands, receptors, and binding proteins. Breast Cancer Res. Treat. 22, 7–19 [DOI] [PubMed] [Google Scholar]
- 11. Morrione A., Valentinis B., Xu S. Q., Yumet G., Louvi A., Efstratiadis A., Baserga R. (1997) Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 94, 3777–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pandini G., Frasca F., Mineo R., Sciacca L., Vigneri R., Belfiore A. (2002) Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 277, 39684–39695 [DOI] [PubMed] [Google Scholar]
- 13. Frasca F., Pandini G., Vigneri R., Goldfine I. D. (2003) Insulin and hybrid insulin/IGF receptors are major regulators of breast cancer cells. Breast Dis. 17, 73–89 [DOI] [PubMed] [Google Scholar]
- 14. Morcavallo A., Gaspari M., Pandini G., Palummo A., Cuda G., Larsen M. R., Vigneri R., Belfiore A. (2011) Research resource. New and diverse substrates for the insulin receptor isoform A revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol. Endocrinol. 25, 1456–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Fiore P. P., De Camilli P. (2001) Endocytosis and signaling. An inseparable partnership. Cell 106, 1–4 [DOI] [PubMed] [Google Scholar]
- 16. Seto E. S., Bellen H. J., Lloyd T. E. (2002) When cell biology meets development. Endocytic regulation of signaling pathways. Genes Dev. 16, 1314–1336 [DOI] [PubMed] [Google Scholar]
- 17. Vecchione A., Marchese A., Henry P., Rotin D., Morrione A. (2003) The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol. Cell Biol. 23, 3363–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monami G., Emiliozzi V., Morrione A. (2008) Grb10/Nedd4-mediated multiubiquitination of the insulin-like growth factor receptor regulates receptor internalization. J. Cell Physiol. 216, 426–437 [DOI] [PubMed] [Google Scholar]
- 19. Morrione A., Valentinis B., Resnicoff M., Xu S., Baserga R. (1997) The role of mGrb10α in insulin-like growth factor I-mediated growth. J. Biol. Chem. 272, 26382–26387 [DOI] [PubMed] [Google Scholar]
- 20. Sell C., Dumenil G., Deveaud C., Miura M., Coppola D., DeAngelis T., Rubin R., Efstratiadis A., Baserga R. (1994) Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 14, 3604–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miura M., Surmacz E., Burgaud J. L., Baserga R. (1995) Different effects on mitogenesis and transformation of a mutation at tyrosine 1251 of the insulin-like growth factor I receptor. J. Biol. Chem. 270, 22639–22644 [DOI] [PubMed] [Google Scholar]
- 22. Jirácek J., Záková L., Antolíková E., Watson C. J., Turkenburg J. P., Dodson G. G., Brzozowski A. M. (2010) Implications for the active form of human insulin based on the structural convergence of highly active hormone analogues. Proc. Natl. Acad. Sci. U.S.A. 107, 1966–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gauguin L., Klaproth B., Sajid W., Andersen A. S., McNeil K. A., Forbes B. E., De Meyts P. (2008) Structural basis for the lower affinity of the insulin-like growth factors for the insulin receptor. J. Biol. Chem. 283, 2604–2613 [DOI] [PubMed] [Google Scholar]
- 24. Swillens S. (1995) Interpretation of binding curves obtained with high receptor concentrations. Practical aid for computer analysis. Mol. Pharmacol. 47, 1197–1203 [PubMed] [Google Scholar]
- 25. Monami G., Gonzalez E. M., Hellman M., Gomella L. G., Baffa R., Iozzo R. V., Morrione A. (2006) Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex. Cancer Res. 66, 7103–7110 [DOI] [PubMed] [Google Scholar]
- 26. Lovat F., Bitto A., Xu S. Q., Fassan M., Goldoni S., Metalli D., Wubah V., McCue P., Serrero G., Gomella L. G., Baffa R., Iozzo R. V., Morrione A. (2009) Proepithelin is an autocrine growth factor for bladder cancer. Carcinogenesis 30, 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Metalli D., Lovat F., Tripodi F., Genua M., Xu S. Q., Spinelli M., Alberghina L., Vanoni M., Baffa R., Gomella L. G., Iozzo R. V., Morrione A. (2010) The insulin-like growth factor receptor I promotes motility and invasion of bladder cancer cells through Akt- and mitogen-activated protein kinase-dependent activation of paxillin. Am. J. Pathol. 176, 2997–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Záková L., Kazdová L., Hanclová I., Protivínská E., Sanda M., Budesínský M., Jirácek J. (2008) Insulin analogs with modifications at position B26. Divergence of binding affinity and biological activity. Biochemistry 47, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 29. Sun X. J., Crimmins D. L., Myers M. G., Jr., Miralpeix M., White M. F. (1993) Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell. Biol. 13, 7418–7428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rose D. W., Saltiel A. R., Majumdar M., Decker S. J., Olefsky J. M. (1994) Insulin receptor substrate 1 is required for insulin-mediated mitogenic signal transduction. Proc. Natl. Acad. Sci. U.S.A. 91, 797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sasaoka T., Rose D. W., Jhun B. H., Saltiel A. R., Draznin B., Olefsky J. M. (1994) Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J. Biol. Chem. 269, 13689–13694 [PubMed] [Google Scholar]
- 32. White M. F. (1998) The IRS-signaling system. A network of docking proteins that mediate insulin action. Mol. Cell. Biochem. 182, 3–11 [PubMed] [Google Scholar]
- 33. White M. F. (2002) IRS proteins and the common path to diabetes. Am. J. Physio.l. Endocrinol. Metab. 283, E413–422 [DOI] [PubMed] [Google Scholar]
- 34. Haruta T., Uno T., Kawahara J., Takano A., Egawa K., Sharma P. M., Olefsky J. M., Kobayashi M. (2000) A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14, 783–794 [DOI] [PubMed] [Google Scholar]
- 35. Lee A. V., Gooch J. L., Oesterreich S., Guler R. L., Yee D. (2000) Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26 S proteasome and blocked by phosphatidylinositol 3′-kinase inhibition. Mol. Cell. Biol. 20, 1489–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramos F. J., Langlais P. R., Hu D., Dong L. Q., Liu F. (2006) Grb10 mediates insulin-stimulated degradation of the insulin receptor. A mechanism of negative regulation. Am. J. Physiol. Endocrinol. Metab. 290, E1262–1266 [DOI] [PubMed] [Google Scholar]
- 37. Polo S., Pece S., Di Fiore P. P. (2004) Endocytosis and cancer. Curr. Opin. Cell Biol. 16, 156–161 [DOI] [PubMed] [Google Scholar]
- 38. Hwang D. L., Papoian T., Barseghian G., Josefsberg Z., Lev-Ran A. (1985) Absence of down-regulation of insulin receptors in human breast cancer cells (MCF-7) cultured in serum-free medium. Comparison with epidermal growth factor. J. Recept. Res. 5, 27–43 [DOI] [PubMed] [Google Scholar]
- 39. Mountjoy K. G., Finlay G. J., Holdaway I. M. (1987) Abnormal insulin-receptor down-regulation and dissociation of down-regulation from insulin biological action in cultured human tumor cells. Cancer Res. 47, 6500–6504 [PubMed] [Google Scholar]
- 40. Le Roy C., Wrana J. L. (2005) Clathrin- and non-clathrin-mediated endocytic regulation of cell signaling. Nat. Rev. Mol. Cell Biol. 6, 112–126 [DOI] [PubMed] [Google Scholar]
- 41. Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., Di Fiore P. P. (2008) Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15, 209–219 [DOI] [PubMed] [Google Scholar]
- 42. Sigismund S., Woelk T., Puri C., Maspero E., Tacchetti C., Transidico P., Di Fiore P. P., Polo S. (2005) Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. U.S.A. 102, 2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen C. L., Hou W. H., Liu I. H., Hsiao G., Huang S. S., Huang J. S. (2009) Inhibitors of clathrin-dependent endocytosis enhance TGFβ signaling and responses. J. Cell Sci. 122, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao D., Ehrlich M., Henis Y. I., Leof E. B. (2002) Transforming growth factor-β receptors interact with AP2 by direct binding to β2 subunit. Mol. Biol. Cell 13, 4001–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fagerholm S., Ortegren U., Karlsson M., Ruishalme I., Strålfors P. (2009) Rapid insulin-dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS One 4, e5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sacco A., Morcavallo A., Pandini G., Vigneri R., Belfiore A. (2009) Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 150, 3594–3602 [DOI] [PubMed] [Google Scholar]
- 47. Fan J. Y., Carpentier J. L., Gorden P., Van Obberghen E., Blackett N. M., Grunfeld C., Orci L. (1982) Receptor-mediated endocytosis of insulin. Role of microvilli, coated pits, and coated vesicles. Proc. Natl. Acad. Sci. U.S.A. 79, 7788–7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paccaud J. P., Siddle K., Carpentier J. L. (1992) Internalization of the human insulin receptor. The insulin-independent pathway. J. Biol. Chem. 267, 13101–13106 [PubMed] [Google Scholar]
- 49. McClain D. A., Olefsky J. M. (1988) Evidence for two independent pathways of insulin-receptor internalization in hepatocytes and hepatoma cells. Diabetes 37, 806–815 [DOI] [PubMed] [Google Scholar]
- 50. Gustavsson J., Parpal S., Karlsson M., Ramsing C., Thorn H., Borg M., Lindroth M., Peterson K. H., Magnusson K. E., Strâlfors P. (1999) Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 13, 1961–1971 [PubMed] [Google Scholar]
- 51. Acconcia F., Sigismund S., Polo S. (2009) Ubiquitin in trafficking. The network at work. Exp. Cell Res. 315, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 52. Haglund K., Sigismund S., Polo S., Szymkiewicz I., Di Fiore P. P., Dikic I. (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 5, 461–466 [DOI] [PubMed] [Google Scholar]
- 53. Mosesson Y., Shtiegman K., Katz M., Zwang Y., Vereb G., Szollosi J., Yarden Y. (2003) Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J. Biol. Chem. 278, 21323–21326 [DOI] [PubMed] [Google Scholar]
- 54. Giudice J., Leskow F. C., Arndt-Jovin D. J., Jovin T. M., Jares-Erijman E. A. (2011) Differential endocytosis and signaling dynamics of insulin receptor variants IR-A and IR-B. J. Cell Sci. 124, 801–811 [DOI] [PubMed] [Google Scholar]
- 55. Zapf A., Hsu D., Olefsky J. M. (1994) Comparison of the intracellular itineraries of insulin-like growth factor-I and insulin and their receptors in Rat-1 fibroblasts. Endocrinology 134, 2445–2452 [DOI] [PubMed] [Google Scholar]
- 56. Hansen B. F., Danielsen G. M., Drejer K., Sørensen A. R., Wiberg F. C., Klein H. H., Lundemose A. G. (1996) Sustained signalling from the insulin receptor after stimulation with insulin analogs exhibiting increased mitogenic potency. Biochem. J. 315, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Milazzo G., Sciacca L., Papa V., Goldfine I. D., Vigneri R. (1997) ASPB10 insulin induction of increased mitogenic responses and phenotypic changes in human breast epithelial cells. Evidence for enhanced interactions with the insulin-like growth factor-I receptor. Mol. Carcinog. 18, 19–25 [DOI] [PubMed] [Google Scholar]
- 58. Hansen B. F., Kurtzhals P., Jensen A. B., Dejgaard A., Russell-Jones D. (2011) Insulin X10 revisited. A super-mitogenic insulin analog. Diabetologia 54, 2226–2231 [DOI] [PubMed] [Google Scholar]
- 59. Grant B. D., Donaldson J. G. (2009) Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baldys A., Göoz M., Morinelli T. A., Lee M. H., Raymond J. R., Jr., Luttrell L. M., Raymond J. R., Sr. (2009) Essential role of c-Cbl in amphiregulin-induced recycling and signaling of the endogenous epidermal growth factor receptor. Biochemistry. 48, 1462–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stern K. A., Place T. L., Lill N. L. (2008) EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem. J. 410, 585–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roepstorff K., Grandal M. V., Henriksen L., Knudsen S. L., Lerdrup M., Grøvdal L., Willumsen B. M., van Deurs B. (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morrione A., Plant P., Valentinis B., Staub O., Kumar S., Rotin D., Baserga R. (1999) mGrb10 interacts with Nedd4. J. Biol. Chem. 274, 24094–24099 [DOI] [PubMed] [Google Scholar]
- 64. Laviola L., Giorgino F., Chow J. C., Baquero J. A., Hansen H., Ooi J., Zhu J., Riedel H., Smith R. J. (1997) The adapter protein Grb10 associates preferentially with the insulin receptor as compared with the IGF-I receptor in mouse fibroblasts. J. Clin. Invest. 99, 830–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu F., Roth R. A. (1995) Grb-IR, a SH2-domain-containing protein that binds to the insulin receptor and inhibits its function. Proc. Natl. Acad. Sci. U.S.A. 92, 10287–10291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dong L. Q., Du H., Porter S. G., Kolakowski L. F., Jr., Lee A. V., Mandarino L. J., Fan J., Yee D., Liu F., Mandarino J. (1997) Cloning, chromosome localization, expression, and characterization of an Src homology 2 and pleckstrin homology domain-containing insulin receptor binding protein hGrb10γ. J. Biol. Chem. 272, 29104–29112 [DOI] [PubMed] [Google Scholar]
- 67. Hansen H., Svensson U., Zhu J., Laviola L., Giorgino F., Wolf G., Smith R. J., Riedel H. (1996) Interaction between the Grb10 SH2 domain and the insulin receptor carboxyl terminus. J. Biol. Chem. 271, 8882–8886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.