

Background: Deletion of N. gonorrhoeae virulence factor ng1686 results in increased sensitivity to H2O2 and PMN-mediated killing.
Results: NG1686 has endopeptidase and carboxypeptidase activities.
Conclusion: NG1686 is a M23B family zinc metallopeptidase with bifunctional activity.
Significance: This is the first demonstration of a metallopeptidase affecting both resistance to H2O2 and PMN-mediated killing in any bacterium.
Keywords: Antioxidants, Bacterial Pathogenesis, Cell Wall, Oxidative Stress, Peptidoglycan, Metallopeptidase, Hydrogen Peroxide, Neisseria gonorrhoeae
Abstract
Symptomatic gonococcal infection, caused exclusively by the human-specific pathogen Neisseria gonorrhoeae (the gonococcus), is characterized by the influx of polymorphonuclear leukocytes (PMNs) to the site of infection. Although PMNs possess a potent antimicrobial arsenal comprising both oxidative and non-oxidative killing mechanisms, gonococci survive this interaction, suggesting that the gonococcus has evolved many defenses against PMN killing. We previously identified the NG1686 protein as a gonococcal virulence factor that protects against both non-oxidative PMN-mediated killing and oxidative killing by hydrogen peroxide. In this work, we show that deletion of ng1686 affects gonococcal colony morphology but not cell morphology and that overexpression of ng1686 does not confer enhanced survival to hydrogen peroxide on gonococci. NG1686 contains M23B endopeptidase active sites found in proteins that cleave bacterial cell wall peptidoglycan. Strains of N. gonorrhoeae expressing mutant NG1686 proteins with substitutions in many, but not all, conserved metallopeptidase active sites recapitulated the hydrogen peroxide sensitivity and altered colony morphology of the Δng1686 mutant strain. We showed that purified NG1686 protein degrades peptidoglycan in vitro and that mutations in many conserved active site residues abolished its degradative activity. Finally, we demonstrated that NG1686 possesses both dd-carboxypeptidase and endopeptidase activities. We conclude that the NG1686 protein is a M23B peptidase with dual activities that targets the cell wall to affect colony morphology and resistance to hydrogen peroxide and PMN-mediated killing.
Introduction
The obligate human pathogen Neisseria gonorrhoeae (the gonococcus) is the sole causative agent of the sexually transmitted infection gonorrhea, which affects more than 700,000 individuals yearly in the United States and over 88 million worldwide (1). Gonococci infect healthy individuals, causing urethritis in men and cervicitis in women. If left untreated, infection can result in extensive reproductive tract scarring and potentially lead to sterility, pelvic inflammatory disease, and ectopic pregnancy in women (2, 3). Gonococci produce no exotoxins, and this cellular damage is effected by the release of peptidoglycan (PG)4 fragments and lipooligosaccharide, which elicit an inflammatory immune response in the host (4), resulting in damage to host cells (5, 6).
Symptomatic gonococcal infection is characterized by the influx of polymorphonuclear leukocytes (PMNs) to the site of infection. The resulting purulent exudate, consisting almost exclusively of PMNs with attached or internalized gonococci, is the clinical hallmark of a gonococcal infection. PMNs typically kill microorganisms through the combined action of reactive oxygen species (ROS) (e.g. hydrogen peroxide (H2O2), superoxide, and hypochlorous acid) and antimicrobial proteins (e.g. lysozyme, cathepsins, and cationic antimicrobial peptides) (7). Despite this potent two-pronged attack, many gonococci found in the purulent exudate remain viable and cultivable (2), suggesting that the gonococcus has evolved mechanisms to circumvent PMN-mediated killing. Accordingly, the gonococcal genome encodes many antioxidant gene products, some of which have been experimentally shown to detoxify various ROS directly (e.g. catalase, cytochrome c peroxidase, and a manganese-dependent ROS quenching system) (8–11). Other gonococcal gene products repair specific types of cellular damage caused by ROS (e.g. peptide methionine sulfoxide reductase and DNA recombinational repair enzymes) (12, 13). Finally, two efflux pump systems confer resistance to cationic antimicrobial peptides (14, 15) and aid in infection of the murine genital tract (16). A growing body of literature suggests that PMNs kill gonococci primarily by ROS- independent means (16–20). It has additionally been shown that gonococci actively abrogate the oxidative burst of PMNs when growing bacteria predominate in a culture but not when bacteria are non-growing (21). Thus, although gonococci are likely to encounter ROS during human infection and may use ROS production as a signal for late stages of infection, gonococci are relatively unaffected by ROS.
We had previously performed a microarray analysis to detect N. gonorrhoeae genes altered in expression in response to the oxidative damaging agent H2O2 for the purpose of identifying novel virulence factors (22). Of the 75 up-regulated genes, several had been previously shown to be important for protection against oxidative damage in gonococci; however, over one-quarter of these genes were predicted to encode proteins with unknown function. Insertional inactivation of a subset of the up-regulated genes (recN, ng1686, and ng554) revealed that mutant strain Δng554 exhibited increased sensitivity to high levels of H2O2. In contrast, strains recN and Δng1686 showed increased sensitivity to both H2O2 and PMN-mediated killing, with strain Δng1686 exhibiting extreme sensitivity to H2O2. Although BLAST searches revealed no characterized homologs of the NG1686 protein, NG1686 contains the active sites of the M23B family of zinc metallopeptidases, and members of this family act to cleave cell wall PG, suggesting that PG could be a substrate of NG1686.
PG is a complex macromolecule that is an essential component of most bacterial cell walls. PG protects cells from osmotic lysis and helps to regulate cell size and shape (reviewed in Ref. 23). Gram-negative PG is composed of glycan chains formed by repeating subunits of N-acetylglucosamine and N-acetylmuramic acid, which are linked by β-1,4-glycosidic bonds. The muramic acid moiety has an amide-linked peptide side chain usually composed of the four amino acids l-alanine, d-glutamic acid, meso-diaminopimelic acid (DAP), and d-alanine or the first three of those without the terminal alanine. Interpeptide cross-bridges formed between adjacent side chains cross-link the glycan chains, reinforcing the PG structure. Although the stability of PG is essential for cell viability, PG is also a dynamic structure that undergoes constant remodeling. Accordingly, bacteria contain peptidoglycanases with a variety of substrate specificities that cleave the bacterial cell wall at prescribed locations and times (reviewed in Ref. 23). In addition to roles in cell wall growth, turnover, and cell separation, peptidoglycanases have also been suggested to contribute to bacterial pathogenesis (24, 25) by generating inflammatory PG fragments (5), releasing virulence factors (26), or altering cellular morphology (27, 28).
In this work, we investigated the pleiotropic effects of the Δng1686 mutation on N. gonorrhoeae. We show that NG1686 influences the colony morphology but not the cellular morphology of gonococci and that NG1686 is located within the gonococcal periplasm. Gonococcal strains with mutations in some, but not all, conserved M23B active site residues of NG1686 recapitulate the peroxide-sensitive and altered colony morphology phenotypes of the Δng1686 null mutant. Finally, we demonstrate that NG1686 degrades peptidoglycan in vitro and that it possesses both dd-carboxypeptidase and endopeptidase activities. Importantly, NG1686 differs from the classical M23B family of metallopeptidases in certain active site residues, suggesting that it may represent a new family of M23B metallopeptidases. The unique qualities may be key features in the ability of NG1686 to mediate hydrogen peroxide resistance and survival of killing by PMNs.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of ng1686
Site-directed mutants of ng1686 were created using the QuikChange multisite-directed mutagenesis kit (Stratagene) as described. Primers employed to introduce mutations are listed in supplemental Table S1. A plasmid construct containing the ng1686 gene under control of its endogenous promoter in the vector pCR-Blunt (pBlunt/1686) was used as a substrate to mutate each putative M23B active site residue individually to an alanine, yielding constructs pBlunt/H295A, pBlunt/D299A, pBlunt/H373A, and pBlunt/H375A. Double mutants for each group of putative active sites were also created, yielding constructs pBlunt/H295A/D299A and pBlunt/H373A/H375A. Mutant genes were sequenced to ensure that only the desired mutations were introduced. Mutant ng1686 gene sequences were released from the vector with a BamHI digest followed by fill-in with Klenow, subsequent digestion with PacI, gel purification with the Qiaquick kit, and cloning into PacI/PmeI-digested vector pGCC5 (29). These resulting mutant ng1686 genes, under control of their endogenous promoter, were subsequently recombined into an irrelevant ectopic chromosomal locus of strain Δng1686. All mutant constructs were verified by both Southern blot and PCR amplification with subsequent sequencing of the mutant ng1686 alleles from the gonococcal chromosome.
Protein Isolation from N. gonorrhoeae and Western Blot Analysis
Protein was isolated from gonococci grown on solid medium as described previously (30) or from gonococci grown in liquid as detailed in the supplemental material. Detection by Western blot analysis is detailed in the supplemental material.
Construction of Regulatable ng1686 Construct in N. gonorrhoeae
For construction of an IPTG-regulatable ng1686 construct, DNA beginning 26 bp upstream of the ATG start codon, including the putative RBS of ng1686, was PCR-amplified with primers 1686-6 and 1686-5-Pac (supplemental Table S1) using Pfu polymerase. The resulting PCR product was cloned into pCR-Blunt, yielding the construct pCR-Blunt/1686ORF. The ng1686-associated DNA was released with a PacI/SpeI double digest and cloned into PacI/SpeI-digested plasmid pKH35 (31), yielding construct pKH35/1686ORF. This construct was then recombined into strains FA1090nv and Δ1686nv, yielding strains FA1090nv/Plac1686 and Δ1686nv/Plac1686. Proper strain construction was confirmed by PCR and Southern blot analysis.
H2O2 Sensitivity Assays
H2O2 resistance assays were performed as described previously (13) but with the modification of growth of N. gonorrhoeae detailed in the supplemental material.
Construction of NG1686 and Related Site-directed Mutant Proteins for Overexpression
The protein coding sequences of ng1686 and the six site-directed mutants, not including the signal sequences, were amplified by PCR with primers 1686-For-Nhe and 1686-Rev-Eco (supplemental Table S1), cloned into pCR-Blunt (Invitrogen), and sequenced to verify that no unwanted mutations had been introduced. The genes were released from these plasmids with a NheI-EcoRI double digest and ligated to the NheI-EcoRI sites in pET28a, yielding constructs pET/HIS-1686, pET/HIS-H295A, pET/HIS-D299A, pET/HIS-H295A/D299A, pET/HIS-H373A, pET/HIS-H375A, and pET/HIS-H373A/H375A.
Anti-NG1686 Antibody Production
HIS-1686 (NG1686) protein was purified from Escherichia coli BL21(DE3) cells overexpressing pET/HIS-1686 by Creative Dynamics, Inc. (Port Jefferson Station, NY). Anti-1686 polyclonal antibodies were generated in a rabbit and purified using a protein G column by the same company.
PG Isolation
From E. coli
PG was isolated from E. coli strain TOP10 (Invitrogen) using the technique described by Zahrl et al. (32). 1 liter of cells in stationary phase was harvested, washed with 40 ml of 10 mm Tris-HCl (pH 6), and resuspended in 30 ml of the same buffer. The resuspended cells were added dropwise to 300 ml of boiling 4% SDS, followed by an additional 45 min of boiling. PG sacculi were collected by ultracentrifugation at 200,000 × g for 20 min at 20 °C. The resulting pellet was resuspended in 150 ml of 2 m NaCl and incubated overnight at room temperature. After ultracentrifugation, sacculi were washed with water and resuspended in 20 ml of 0.1 mm MgCl2. The suspension was treated with 50 μg/ml DNase, 50 μg/ml RNase A, and 200 μg/ml α-amylase (Roche Applied Science) for 90 min at 37 °C. Pronase was added to a final concentration of 200 μg/ml and further incubated at 60 °C for 60 min. Enzymes were inactivated by the addition of SDS to 8% final concentration and 15 min of boiling. PG was collected by ultracentrifugation followed by two washes with water. Pellet was resuspended in 5 ml of water, and the yield was quantified by lyophilizing 1 ml of the sample and weighing. Purified PG was stored at −20 °C.
From N. gonorrhoeae for Zymogram Analysis, HPLC-based Assays, and LC/MS-based Assays
PG was isolated from N. gonorrhoeae strain FA1090 or KH530 (pacA mutant) as described earlier (33) but with the following modifications. The final ultracentrifugation step was eliminated, and the PG preparation was suspended in 500 μl of 25 mm sodium phosphate buffer (pH 6). To inactivate any PG-associated enzymes that may degrade PG, the PG preparation was incubated with either trypsin (5–7.5 μg) or Pronase (60–100 μg) at 37 °C for 2–16 h. To inactivate trypsin and Pronase, the PG was boiled in 25 mm sodium phosphate buffer (pH 6) containing 4% SDS for 1–1.5 h. The PG preparation was centrifuged for 30 min at 30,000 × g at 15 °C, and the pellet was washed five times by suspension in 10 ml of phosphate buffer prior to suspension in 500 μl of 50 mm sodium phosphate buffer (pH 7.5).
Zymogram Analysis of PG Degradation
Zymogram analysis was performed adapting protocols described by others (34, 35). E. coli BL21(DE3) cell extracts carrying pET/HIS-1686, the site-directed mutant NG1686 proteins, or purified NG1686 protein were subjected to electrophoresis on a 12% SDS gel containing 0.05% (w/v) E. coli murein sacculi, 0.1% (w/v) N. gonorrhoeae murein sacculi, or 0.2% (w/v) lyophilized Micrococcus lysodeikticus cells (Sigma). Gels were run at 4 °C at 75 V. Following electrophoresis, gels were rinsed with water twice for 30 min each at room temperature with gentle shaking to remove SDS. Gels were transferred to renaturing buffer (0.5% Triton X-100, 25 mm Tris-HCl (pH 7.5)) for 2 × 30 min, and renaturing was continued overnight (∼16 h) at room temperature. Fresh renaturing buffer was added, and gels were shifted to 37 °C for 2.5 h. Gels were stained with 0.1% methylene blue dissolved in 0.01% KOH for 60 min and destained with water to visualize zones of PG clearing. Gels were finally stained with Coomassie Brilliant Blue to allow visualization of protein in the gel.
Solubilization of PG Sacculi
N. gonorrhoeae PG (2 μl, ∼140,000 cpm) labeled with [3H]glucosamine was added to 1.5 μg of purified enzyme in 1.2 ml of a 25 mm Tris-HCl (pH 7.5), 0.5% Triton X-100 buffer in the presence or absence of 1 mm phenanthroline. The reaction was incubated at 37 °C. Samples of 200 μl were taken at various time points and added to 500 μl of 20% TCA and 20 μl of unlabeled PG to serve as carrier. Samples were placed on ice for 30 min and then centrifuged for 30 min at 48,000 × g at 4 °C. Peptidoglycan in 500 μl of each soluble fraction was determined by scintillation counting.
Characterization of NG1686 Reaction Products
For the HPLC-based assays containing NG1686 and H373A/H375A, reactions contained 50 mm sodium phosphate buffer (pH 7.5), 75 μl of purified FA1090 PG, and 1.5 μm enzyme in a total reaction volume of 150 μl. The reactions were incubated overnight at 37 °C. For the assays of HIS-1686 in the presence of zinc or EDTA, reactions contained 50 mm sodium phosphate buffer (pH 7.5), 25 μl of purified FA1090 PG, 1.5 μm NG1686, and either 0.5 μm ZnSO4 or 1 mm EDTA in a total reaction volume of 150 μl; the reactions were incubated for 5 h at 37 °C. To stop the reactions, samples were boiled for 10–15 min. The insoluble material was removed by centrifugation, and the supernatants were applied to Centricon 10,000 MWCO spin columns, which had been prewashed with 50 mm sodium phosphate buffer (pH 7.5). HPLC analysis was carried out using a Prevail (Alltech) C18 HPLC column (5 μm, 25 × 4.6 mm). Reaction products were separated using a 0–15% gradient of 60% acetonitrile, 0.05% TFA over 60 min at a flow rate of 1 ml/min, and the elution of the products was monitored at 210 nm.
LC/MS analysis of soluble reaction products was performed at the University of Wisconsin-Madison Biotechnology Center. Reaction mixtures analyzed by LC/MS were set up as described above except that 75 μl of FA1090 PG was used. The filtered reaction products were analyzed at the University of Wisconsin-Madison Biotechnology Center using a Zorbax SB-C18 column (1.8 μm, 2.1 × 50 mm) run on an Agilent 1200 HPLC with a linear gradient of 99.9% water, 0.1% formic acid to 99.9% acetonitrile, 0.1% formic acid over 60 min at a flow rate of 0.25 ml/min. Peaks were analyzed using an Agilent LC/MSD TOF using electrospray ionization in positive ion mode.
Analysis of NG1686 Reaction Products Digested with Mutanolysin
Reaction mixtures contained 50 mm sodium phosphate buffer (pH 7.5), 75 μl of purified KH530 PG, 1.5 μm NG1686, and 0.5 μm ZnSO4 in a total reaction volume of 200 μl. The reactions were incubated on a rotator overnight at 37 °C. NG1686 was heat-killed by boiling for 5 min, and the insoluble material was removed by centrifugation. The soluble products were pooled and filtered using a Centricon 10,000 MWCO spin column, and the filtrate was divided into two aliquots. To one sample, 10 μl of a stock solution of mutanolysin (1 mg/ml) was added; to the second sample, 10 μl of water was added as a control. The reaction mixtures were incubated on a rotator at 37 °C for 2 h and then filtered using a Centricon 10,000 MWCO spin column. LC/MS analysis was performed following the same protocol described above.
Determination of Peptidoglycan Cleavage Site
Reaction mixtures contained 50 mm sodium phosphate buffer (pH 7.5), 75 μl of purified FA1090 PG, 1.5 μm NG1686, and 0.5 μm ZnSO4 in a total reaction volume of 200 μl. Control reactions containing H373A/H375A or EDTA (1 mm) were also set up in parallel, as was a reaction lacking PG. Following overnight incubation at 37 °C, the samples were boiled for 5 min to stop the reactions, and the soluble products were filtered using a Centricon 10,000 MWCO spin column. The samples were derivatized with 1-fluoro-2,4-dinitrobenzene (FDNB) using a published protocol (36) with slight modifications. To each sample, 50 μl of 10% K2B7O4 (pH 9) and 1 μl of FDNB were added in a total reaction volume of 500 μl. After the reactions were incubated at 65 °C for 45 min in the dark, the dinitrophenyl (DNP) derivatives were hydrolyzed in 4 n HCl at 95–100 °C for 12 h in the dark. The hydrolyzed DNP products were dried under vacuum, resuspended in 30% acetonitrile, and analyzed by HPLC using a Prevail (Alltech) C18 HPLC column (5 μm, 25 × 4.6 mm) in a column heater set at 40 °C. DNP products were separated using a 0–100% gradient of 60% acetonitrile, 0.01% TFA over 60 min at a flow rate of 0.5 ml/min, and the elution of the products was monitored at 365 nm. Peaks of interest were collected, dried under vacuum, and analyzed by ESI-MS at the University of Wisconsin-Madison Biotechnology Center. ESI-MS was carried out in both positive and negative ion modes.
Carboxypeptidase Activity Assay
To assay NG1686 for carboxypeptidase activity, the synthetic substrate N-acetyl-muramoyl-l-alanyl-d-glutamyl-l-diaminopimelyl-d-alanyl-d-alanine (MurNAc-pentapeptide) (supplemental Fig. S4) was purchased from the University of Warwick Peptidoglycan Synthesis Facility (37, 38). Reactions contained 50 mm sodium phosphate buffer (pH 7.5), 1.5 μm ZnSO4, 0.4 mm MurNAc-pentapeptide, and 1.5 μm NG1686 in a total volume of 250 μl. Control reactions lacking MurNAc-pentapeptide or containing HIS-H373A/H375A or EDTA were also set up in parallel. Reactions were incubated on a rotator overnight at 37 °C and then filtered using a Centricon 10,000 MWCO spin column. To each sample, 25 μl of 10% K2B7O4 (pH 9) and 0.5 μl of FDNB were added in a total reaction volume of 250 μl, and the reactions were incubated at 65 °C for 30 min in the dark. DNP products were dried under vacuum, resuspended in 30% acetonitrile, and analyzed by HPLC as described above for the cleavage site experiment. Peaks of interest were collected, dried under vacuum, and analyzed by ESI-MS in negative ion mode at the University of Wisconsin-Madison Biotechnology Center.
RESULTS
NG1686 Is a Periplasmic Protein
NG1686 has a canonical signal sequence (residues 1–31) and a putative signal peptide cleavage site, suggesting that it is secreted across the cytoplasmic membrane. Kyte-Doolittle hydropathy analysis predicted that NG1686 is largely a hydrophilic protein, apart from a short hydrophobic region at the N terminus of the protein corresponding to the signal sequence (data not shown) (39). Additionally, NG1686 lacks a canonical lipobox motif (see the LipoP Server Web site), indicating that the protein is not likely to be membrane-associated. To experimentally determine the location of NG1686, strains FA1090 and FA1090Δng1686 (referred to as Δ1686 or Δng1686), which contains a deletion of the ng1686 gene, and the complement strain Δ1686/1686+ (22) were grown in liquid medium, the supernatant and total cellular protein fractions were isolated, and a Western blot analysis was performed. A ∼46 kDa band of equal intensity was detected from cell extracts of strains FA1090 and Δ1686/1686+ but not from cell extracts of strain Δng1686 (Fig. 1), demonstrating that the complement strain expresses equivalent amounts of NG1686 as the parent strain FA1090. Moreover, no NG1686 protein was observed in the concentrated supernatant fractions from strain FA1090 or Δ1686/1686+ (data not shown). The presence of a signal sequence combined with a lack of detectable protein in the culture supernatant strongly suggests that the NG1686 protein is located in the periplasm.
FIGURE 1.

Western blot analysis of NG1686 protein expression in parent, mutant, and complement strains. SDS-polyacrylamide gels containing 10 μg of total cellular protein per lane of strains FA1090, Δ1686 (Δng1686), and Δ1686/1686+ were run and transferred to PVDF membrane, with subsequent Western blot analysis using anti-1686 antiserum in the ECL Plus detection kit.
Deletion of ng1686 Affects Colony Morphology but Not Cell Morphology or Sensitivity to Cell Wall-targeting Antibiotics
We previously noted that the Δng1686 mutant exhibits a larger and flatter colony morphology relative to strain FA1090 (22), which is complemented in strain Δ1686/1686+ (Fig. 3A) (data not shown). Because NG1686 encodes a protein with sequence similarity to the M23B family of endopeptidases, many of which act to degrade cell walls, we sought to determine whether the overall cell morphology or cell size of the Δng1686 mutant was altered. Transmission electron microscopy of strains FA1090 and Δng1686 was used to visualize the size of individual cells grown on solid medium. Measurement of >100 individual cells of each strain revealed no difference in cell size between cells of the two strains (data not shown). Moreover, no qualitative difference in the overall morphology of individual cells was noted in the transmission electron microscopy analysis, suggesting that NG1686 does not affect septum formation or cell separation (data not shown).
FIGURE 3.

Phenotypes of M23B metallopeptidase active site point mutants of NG1686 expressed in N. gonorrhoeae. A, colony morphology of N. gonorrhoeae strains containing M23B point mutants. Representative stereomicroscope observations of strains using a Nikon SMZ-10A stereomicroscope after 24 h of growth on solid medium were recorded using a Polaroid camera model DMC Ie. B, dose-response curve of H2O2 resistance after 15 min of stains carrying NG1686 M23B point mutants. Cells were treated with varying doses of H2O2 for 15 min and serially diluted into medium containing catalase. Error bars, S.E. of six independent experiments. Strains are statistically the same as strain Δ1686 at 20 and 50 mm H2O2 doses (*, p > 0.05) and statistically the same as strain Δ1686/1686+ at all H2O2 doses (†, p > 0.05) by Student's t test. C, Western blot analysis of NG1686 M23B point mutant protein expression. SDS-polyacrylamide gels containing 10 μg of total cellular protein per lane were run and transferred to PVDF membrane. Lane 1, Δ1686/1686+; lane 2, H295A; lane 3, D299A; lane 4, H295A/D299A; lane 5, H373A; lane 6, H375A; lane 7, H373A/H375A.
Mutation of ng1686 does not affect the general cellular permeability of gonococci because resistance to the oxidative damaging agents paraquat and diamide and the antibiotics naladixic acid, chloramphenicol, and streptomycin is not altered in the Δng1686 mutant strain (22). To specifically test the resistance of strains FA1090, Δng1686, and Δ1686/1686+ to antibiotics that act on the cell wall, we measured the minimum inhibitory concentration of these three strains to the antibiotics ampicillin, ceftazidime, vancomycin, and polymyxin B (data not shown). All three strains showed identical resistance profiles to the antibiotics. These results suggest that NG1686 does not affect general cell wall integrity in a way such as to influence resistance to antibiotics, including antibiotics that target the cell wall.
NG1686 Does Not Act to Detoxify H2O2
We have previously observed that deletion of ng1686 in the gonococcus results in increased sensitivity to H2O2 (22), which could suggest that NG1686 acts to detoxify H2O2. Therefore, we tested whether overexpression of NG1686 was able to decrease the sensitivity of a strain to H2O2. An IPTG-inducible version of ng1686 was created by cloning the ng1686 gene and its associated ribosome binding site into plasmid pKH35 (31) under control of lac promoter regulatory sequences, resulting in NG1686 protein expression that is dependent on the addition of IPTG. This construct was then recombined into an ectopic locus in the chromosome of strain FA1090nv and the corresponding Δng1686 mutant strain, Δ1686nv (the subscript “nv” in the designation indicates that a strain cannot undergo pilin antigenic variation (40)), yielding strains FA1090nv/Plac1686 and Δ1686nv/Plac1686, respectively. A 2–3-fold increase in NG1686 protein levels was observed in strain FA1090nv/Plac1686 (in the presence of 1 mm IPTG) relative to the parent strain FA1090nv, as determined by Western blot analysis (Fig. 2A). Very low levels of NG1686 protein were detected from strain Δ1686nv/Plac1686 in the absence of IPTG (∼70-fold lower than in strain FA1090nv), confirming that expression from the regulated lac promoter is leaky (Fig. 2A). Next, we measured the effect of modulating NG1686 protein levels on the H2O2 sensitivity of the strains. In the absence of IPTG, strain Δ1686nv/Plac1686 showed a 10-fold increase in H2O2 sensitivity relative to strain FA1090nv and showed a 10–100-fold decrease in sensitivity relative to strain Δ1686nv (Fig. 2B). There was no decrease in the H2O2 sensitivity of strain FA1090nv/Plac1686, which expresses 2–3 times more NG1686 protein than strain FA1090nv (Fig. 2). Taken together, these results indicate that low level sensitivity to H2O2 is mediated by extremely small amounts of NG1686 and that overexpression of NG1686 does not result in decreased sensitivity to H2O2. These results suggest that NG1686 does not directly detoxify H2O2 to influence survival.
FIGURE 2.

NG1686 protein expression in and H2O2 resistance of IPTG-regulatable ng1686 strains. A, representative Western blot analysis of NG1686 protein expression in strains containing IPTG-regulatable ng1686 construct pKH35/Plac1686. SDS-polyacrylamide gels containing 10 μg of total protein per lane were run, transferred, and developed as described in the legend to Fig. 1. B, dose-response curve of H2O2 resistance after 15 min of strains containing pKH35/1686. Cells were treated with varying doses of H2O2 for 15 min and serially diluted into medium containing catalase. The relative survival at each dose was calculated as viable cfu divided by total cfu (receiving no H2O2). Error bars, S.E. of 2–4 independent experiments. Strain Δ1686nv/Plac1686 (designated Δ1686nv/1686, in the absence of IPTG (−IPTG) is statistically the same as strain Δ1686nv at 20 and 50 mm H2O2 doses (*, p > 0.05) and statistically different from strain FA1090nv (†, p < 0.04) at the same doses by Student's t test. Indicated strains are statistically the same as strain FA1090nv at all doses (‡, p > 0.05).
Subset of M23B Active Site Residues Is Required for NG1686 Phenotypes in Gonococcus
The NG1686 protein contains conserved active sites of the M23B family of zinc metallopeptidases, as classified by the MEROPS peptidase database. The active site residues of this family occur in the motifs HXXXD and HXH, with the histidine shown in boldface type serving as the catalytic residue (41), and these residues correspond to amino acids 295–299 and 373–375 of NG1686. To test the contribution of these M23B active sites to NG1686 functions, the DNAs encoding each of these residues were mutated individually and in combination to encode alanine residues, and the mutant genes were subsequently recombined into strain Δng1686, yielding the single mutant strains H295A, D299A, H373A, and H375A and the double mutant strains H295A/D299A and H373A/H375A. We then tested whether the mutant genes were able to restore the parental colony morphology and H2O2 sensitivity to strain Δ1686. Strains D299A, H295A/D299A, H375A, and H373A/H375A recapitulated the altered colony morphology of strain Δ1686, whereas the colony morphology of strains H373A and H295A recapitulated that of the complement strain Δ1686/1686+ (Fig. 3A). The H2O2 sensitivity phenotype of the mutant strains mirrored the observed colony morphologies, with strains D299A, H295A/D299A, H375A, and H373A/H375A showing statistically the same H2O2 sensitivity as strain Δng1686 and strains H373A and H295A showing statistically the same sensitivity as the complement strain Δ1686/1686+ (Fig. 3B). These results indicate that the His-295 and His-373 residues of the NG1686 protein are not essential for activity and therefore suggest that NG1686 may have activities different from those of other M23B family proteins.
Western blot analysis of NG1686 protein levels in the Δ1686/1686+ and six point mutant strains revealed similar levels of NG1686 protein in all strains. The largest difference was in strain H373A/H375A, which showed 3-fold less protein than strain Δ1686/1686+ (Fig. 3C). Because strain Δ1686nv/Plac1686, in the absence of IPTG, expressed ∼70-fold less NG1686 protein than strain FA1090nv yet still showed H2O2 sensitivity (Fig. 2), the small differences in protein levels are not responsible for the observed differences in H2O2 sensitivity and colony morphology. Taken together, these data show that some, but not all, of the defined M23B active site residues are required for NG1686 function in gonococci.
NG1686 Degrades E. coli and N. gonorrhoeae PG in Vitro
There were no consistent changes in protein profiles recorded between the parent strain FA1090 and the Δng1686 mutant in a two-dimensional gel analysis (data not shown), and several active site residues required for the ng1686 phenotypes corresponded to the M23B clade of metallopeptidase, indicating that the target of NG1686 could be pepidoglycan.
To determine whether PG is a target of NG1686, we assayed the hydrolytic activity of NG1686 using zymogram gels containing PG. E. coli bacterial cell lysates overproducing NG1686 and each of the site-directed mutant proteins as well as the purified NG1686 protein were run on zymogram gels containing PG purified from either E. coli (Fig. 4, A and D) or N. gonorrhoeae (Fig. 4C). We observed zones of clearing that corresponded to the location of the NG1686 protein (Fig. 4), as determined by subsequent staining of the gel with Coomassie Blue (Fig. 4, B and E), as well as to lysozyme, which was added to lyse the bacterial cells for cell extracts (Fig. 4, A, C, and D). In contrast, zymogram gels containing desiccated Micrococcus lysodeikticus did not show zones of clearing dependent on NG1686 protein but did for lysozyme, indicating that this Gram-positive PG is not cleaved by the NG1686 protein (data not shown). There are several factors that could contribute to the comparatively low activity of NG1686 observed relative to that of lysozyme. First, the site of PG cleavage (e.g. PG backbone or PG cross-links) influences the zone size that is created by peptidoglycanase activity. Because lysozyme degrades the PG backbone, the resulting PG monomers can easily diffuse out of the gel. Second, NG1686 has a molecular mass that is about 3-fold larger than lysozyme, and this may impede its diffusion in the gel; third, the efficiency of protein renaturation within the zymogram gel also influences zone size.
FIGURE 4.

Zymogram analysis of peptidoglycan hydrolase activity. A, equal amounts of E. coli cell extract carrying pET28a/NG1686 or pET28a/H373A/H375A or purified NG1686 protein were separated by SDS-PAGE, renatured in the E. coli PG-containing gel, and stained with methylene blue. Zones of clearing corresponding to either lysozyme or NG1686 protein represent PG degradation and are indicated with arrows. B, gel subsequently stained with Coomassie shows equivalent amounts of purified NG1686 and NG1686 proteins derived from E. coli cell lysates were loaded on the gel. C, equal amounts (5 μl) of E. coli cell extracts carrying NG1686 M23B point mutant constructs were run on an N. gonorrhoeae PG-containing gel and stained with methylene blue. Zones of clearing are indicated with arrows. D, differing amounts of E. coli cell extracts carrying NG1686 M23B point mutant constructs, indicated at the bottom of E, were run on an E. coli PG-containing gel and stained with methylene blue. E, gel from D subsequently stained with Coomassie to show the relative amounts of NG1686 mutant proteins loaded on gel.
To demonstrate the relative abilities of the mutant proteins to cleave E. coli PG, we ran differing amounts of cell lysates on zymogram gels (Fig. 4, D and E). E. coli lysates carrying construct pET/HIS-D299A, pET/HIS-H295A/D299A, or pET/HIS-H373A/H375A showed no zone of clearing at the location of NG1686 protein migration (Fig. 4, D and E); lysates carrying construct pET/HIS-H295A or pET/HIS-H373A showed less robust zones of clearing relative to the pET/HIS-NG1686 lysate; and the pET/HIS-H375A lysate showed an extremely faint zone of clearing at the location of the protein (Fig. 4D). We attribute the additional bands with PG degradative activities to proteins present in E. coli because they directly correlate with the amount of lysate loaded (Fig. 4, D and E) and are not present in the lanes containing purified protein (Fig. 4A). Identical results were observed for zymogram gels containing gonococcal PG (Fig. 4C).
To further demonstrate the ability of NG1686 to degrade PG, we performed PG sacculi solubilization studies using purified NG1686 protein or the double point mutant H373A/H375A protein. NG1686 converted radiolabeled insoluble gonococcal sacculi to soluble PG fragments, whereas the mutant H373A/H375A protein lacked this ability (Fig. 5). The soluble PG fragments released from gonococcal sacculi by NG1686 were further analyzed by LC/MS. This enabled detection of 22 reaction products with masses consistent with PG fragments, nine of which were the most abundant (Fig. 6 and Table 1). Importantly, these products were absent in NG1686 reactions containing the zinc chelating agent EDTA or phenanthroline (supplemental Figs. S2 and S3) and reactions containing the mutant H373A/H375A enzyme (supplemental Fig. S1). We have shown that NG1686 degrades PG by both zymogram gel and PG sacculi solubilization studies. Moreover, we have demonstrated that NG1686 activity is inhibited by phenanthroline, EDTA, and mutation of certain M23B active residues. These in vitro data provide strong evidence that NG1686 is a M23B zinc metallopeptidase whose target is PG.
FIGURE 5.
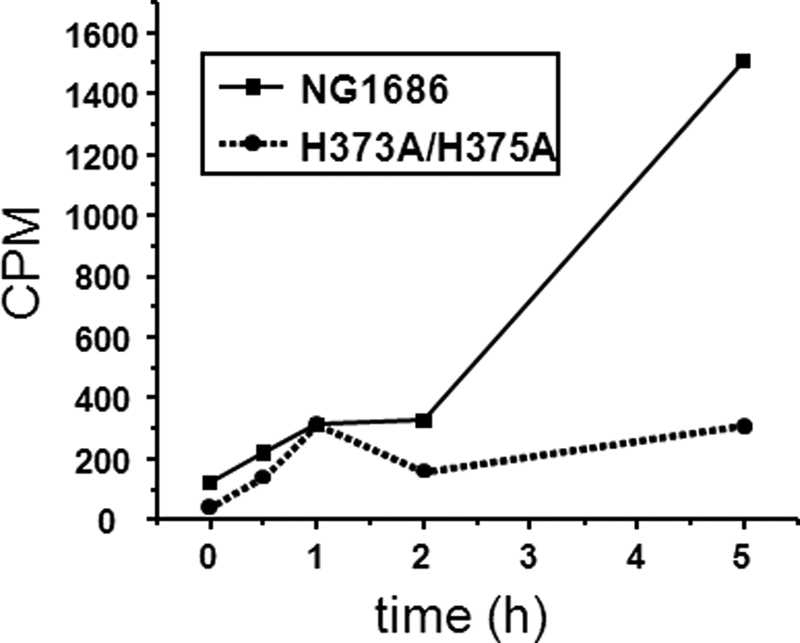
Degradation of radiolabeled PG by purified NG1686 and H373A/H375A proteins. Gonococcal PG, NG1686 protein, and H373A/H375A protein were incubated together. Samples of the reactions were collected at the indicated times, and the insoluble, macromolecular PG was precipitated by the addition of TCA and collected by centrifugation. The soluble fragments were quantified by scintillation counting. Values are the average of two independent experiments.
FIGURE 6.

LC/MS characterization of soluble reaction products produced by digestion of PG by purified NG1686 protein. Base peak chromatogram showing soluble reaction products from the NG1686 enzymatic reaction (mass range, 800–3900 Da). See Table 1 for a description of the numbered peaks. These ions were not detected in control reactions containing NG1686 and EDTA (1 mm) or the H373A/H375A mutant protein. cps, counts per second.
TABLE 1.
Proposed structures of peaks revealed by LC/MS (shown in Fig. 6) produced by digestion of PG by NG1686
| Peaka | Mass | Proposed structureb |
|---|---|---|
| Da | ||
| Monomers | ||
| 1 | 850.34 | Tri (anh) |
| 2 | 921.38 | Tetra (anh) |
| Dimersc | ||
| 3 | 1771.73 | Tri-tetra (anh) or Tetra-tri (anh) |
| 4 | 1842.76 | Tetra-tetra (anh) |
| 5 | 1813.74 | Tri-tetra (anh) OAc or Tetra-tri (anh) OAc |
| 6 | 1884.78 | Tetra-tetra (anh) OAc |
| Trimersd | ||
| 7 | 2806.16 | Tetra-tetra-tetra (anh) OAc |
| 8 | 2848.17 | Tetra-tetra-tetra (anh) di-OAc |
| Tetramerd | ||
| 9 | 3811.55 | Tetra-tetra-tetra-tetra (anh) tri-OAc |
a Peaks correspond to Fig. 6.
b Tri, disaccharide tripeptide; tetra, disaccharide tetrapeptide (disaccharide = N-acetylglucosamine-N-acetylmuramic acid); anh, 1,6-anhydromuramic acid; OAc, O-acetylation on N-acetylmuramic acid.
c Dimers could be glycosidic or peptide dimers.
d Trimers and tetramer could contain all glycosidic linkages or both glycosidic and peptide linkages.
NG1686 Possesses both Endopeptidase and Carboxypeptidase Activities
If NG1686 is an endopeptidase, as predicted from the homology, then soluble PG fragments released from sacculi by digestion with NG1686 should not contain peptide cross-links. Thus, multimeric PG fragments produced by NG1686 (listed in Table 1) should only contain glycosidic linkages. To determine whether the multimeric PG products released by NG1686 contained glycosidic or peptide linkages, we subjected the soluble NG1686 reaction products to digestion with mutanolysin, an N-acetylmuramidase that cleaves the β-1,4-linkages between N-acetylmuramic acid and N-acetylglucosamine residues of PG. Consistent with mutanolysin digestion, the NG1686 soluble products released PG monomers with reducing ends (Table 2). PG monomers with reducing ends were not detected in the control reaction, which was treated with water instead of mutanolysin. In addition, the mutanolysin-treated sample contained significantly more anhydro monomers than the control. No multimeric PG fragments were detected in the doubly digested sample, suggesting that NG1686 cleaved at peptide linkages. These results demonstrate that NG1686 digested sacculi to release PG strands with no peptide cross-links. Mutanolysin could then convert all of these products into the various PG monomers that make up in each strand. These results thus demonstrate that NG1686 acts as an endopeptidase.
TABLE 2.
Reaction products from the sequential digestion of PG by NG1686 and mutanolysin
| Peak | Calculated mass | Observed mass (Da) |
|
|---|---|---|---|
| 1686 + mutanolysin | 1686 + water | ||
| Da | Da | ||
| Monomers | |||
| Tria (anhb) | 850.3 | 850.4 | 850.4 |
| Tetrac (anh) | 921.4 | 921.4 | 921.4 |
| Tetra (redd) | 939.4 | 939.4 | NDe |
| Tetra (OAc) (red) | 981.4 | 981.4 | ND |
| Pentaf (anh) | 992.4 | 992.4 | ND |
| Penta (red) | 1010.4 | 1010.4 | ND |
| Penta (OAc) (red) | 1052.4 | 1052.5 | ND |
| Dimers | |||
| Tetra-tri (anh) or tri-tetra (anh) | 1771.7 | ND | 1771.8 |
| Tetra (OAc)-tri (anh) or tri (OAc)-tetra (anh) | 1813.7 | ND | 1813.7 |
| Tetra-tetra (anh) | 1842.8 | ND | 1842.8 |
| Tetra (OAc)-tetra (anh) | 1884.8 | ND | 1884.8 |
| Penta-tetra (anh) or tetra-penta (anh) | 1913.8 | ND | 1913.8 |
| Penta (OAc)-tetra (anh) or tetra (OAc)-penta (anh) | 1955.8 | ND | 1955.8 |
| Trimer | |||
| Tetra (OAc)-tetra or (OAc)-tetra (anh) | 2848.2 | ND | 2848.2 |
a Tri, disaccharide tripeptide.
b anh, 1,6-anhydromuramic acid.
c Tetra, disaccharide tetrapeptide.
d red, N-acetylmuramic acid with reducing end.
e ND, not detected.
f Penta, disaccharide pentapeptide (disaccharide = N-acetylglucosamine-N-acetylmuramic acid).
To determine the cleavage site of NG1686, we treated the soluble reaction products with the derivatizing agent FDNB and analyzed the hydrolyzed reaction products by HPLC. FDNB reacts with free amino groups and thus can identify new amino groups freed by the cleavage with a peptidase. In the reaction containing NG1686, we detected two peaks that were absent in the control reactions (Fig. 7). Analysis of the elution products by ESI-MS revealed masses consistent with mono-DNP-DAP and DNP-Ala. Mono-DNP-DAP was detected in positive ion mode (Theo. +[H+]: 357.105; Obs. +[H+]: 357.105), and DNP-Ala was detected in negative ion mode (Theo. −[H+]: 254.041; Obs. −[H+]: 254.042). The formation of mono-DNP-DAP indicates that NG1686 cleaves PG at cross-links between DAP-d-Ala or DAP-DAP residues. The formation of DNP-d-Ala was unexpected and raised the possibility that NG1686 might have dd-carboxypeptidase activity in addition to endopeptidase activity.
FIGURE 7.

HPLC analysis of FDNB-derivatized reaction mixtures containing NG1686 (A), H373A/H375A (B), or NG1686 + EDTA (C) and NG1686 reaction lacking PG (D). The arrows indicate elution products that were collected, dried, and analyzed by ESI-MS. Based on ESI-MS and the elution of standards, the first arrow corresponds to the mono-DNP-DAP; the second arrow corresponds to DNP-alanine. mAU, milliabsorbance units.
To probe the potential dd-carboxypeptidase activity of NG1686, we incubated the enzyme with the synthetic substrate MurNAc-pentapeptide (supplemental Fig. S4). The reactions were derivatized with FDNB and analyzed by HPLC followed by ESI-MS. The reaction containing NG1686 and MurNAc-pentapeptide resulted in a peak not observed in any of the control reactions (Fig. 8). This peak had an elution time that matched the elution of DNP-alanine standard, and ESI-MS analysis in negative ion mode showed that this elution product had a mass consistent with DNP-Ala (Theo. −[H+]: 254.041; Obs. −[H+]: 254.044). This experiment demonstrates that NG1686 can degrade small soluble PG fragments and that it cleaves the terminal Ala from the pentapeptide PG fragment. Thus, it is clear that NG1686 has both dd-carboxypeptidase activity and endopeptidase activity (Fig. 9).
FIGURE 8.

HPLC analysis of FDNB-derivatized reaction mixtures containing d-alanine (A), MurNAc-pentapeptide + NG1686 (B), or MurNAc-pentapeptide + H373A/H375A (C) and NG1686 reaction lacking substrate (D). The arrows indicate the elution products corresponding to DNP-alanine. mAU, milliabsorbance units.
FIGURE 9.

Sites of PG cleavage by NG1686. Shown is a schematic representation of sites cleaved by the endopeptidase and dd-carboxypeptidase activities of NG1686.
DISCUSSION
This study reveals the ability of the N. gonorrhoeae virulence factor NG1686 to degrade PG and demonstrates that NG1686 belongs to the M23B family of zinc metallopeptidases. We show that NG1686 is a bifunctional enzyme, possessing both endopeptidase and dd-carboxypeptidase activities (Fig. 9). We therefore conclude that the lack of NG1686 PG degradative activity in the Δng1686 mutant manifests itself in the varied phenotypes seen in this strain, specifically altered colony morphology, extreme sensitivity to H2O2, and sensitivity to PMN-mediated killing. This is the first suggestion that a PG-degrading enzyme has roles in resistance to H2O2 and PMN-mediated killing.
The most well studied members of the M23B family of zinc metallopeptidases are LytM and lysostaphin, both of which cleave the pentaglycine cross-bridges found in staphylococcal PG (41). The NG1686 protein clearly belongs to this family, possessing all of the active site residues of this family (HXXXD, HXH), demonstrating the ability to degrade PG from both E. coli and N. gonorrhoeae in vitro, and being sensitive to metal chelation by EDTA or phenanthroline (supplemental Figs. S2 and S3). However, NG1686 must differ fundamentally from these well characterized M23B class endopeptidases because neither E. coli nor N. gonorrhoeae PG contains pentaglycine cross-bridges and because NG1686 does not require the HXH motif for activity. The crystal structure of LytM, an M23B autolysin from Staphylococcus aureus, identifies the His and Asp residues of the HXXXD motif and the second His of the HXH motif as ligands for the Zn2+ ion; the first His of the HXH motif is the catalytic residue (41). Mutagenesis of these active sites completely abolishes the activity of the LytM protein (41). In contrast, mutation of the analogous residues in the NG1686 protein revealed that only the Asp-299 residue is absolutely critical for activity in vitro, whereas mutation of the His-295, His-373, and His-375 residues resulted in decreased activity (Fig. 4). These results demonstrate that the His-373 residue is not the catalytic residue and suggest that NG1686 is a structurally distinct member of the M23B family of metallopeptidases.
The recent elucidation of the crystal structure of the NMB0315 protein from N. meningitidis (98% sequence identity; 99% sequence similarity to the NG1686 protein from N. gonorrhoeae) allowed us to interpret the site-directed mutant data of NG1686 in the context of the newly solved three-dimensional structure of this meningococcal protein (42). In the crystal structure of NMB0315, the metal ion is coordinated by three spatially adjacent residues (His-295, Asp-299, and His-375) and two water molecules. Residue His-373 interacts with the metal ion through one of the water molecules (42). Consistent with the crystal structure, mutation of residues His-295, Asp-299, His-373, and His-375 in NG1686 resulted in decreased or no observed activity of NG1686 by zymogram (Fig. 4), although the H295A and H373A mutants were able to mediate the colony morphology and H2O2 resistance phenotypes of N. gonorrhoeae as well as the parental NG1686 protein (Fig. 3). Interestingly, although the domain structure of NMB0315 suggests that NMB0315 is in an autoinhibited conformational state (42), our in vitro data argue either for the lack of autoinhibition or autoinhibition that is overcome upon the addition of appropriate substrate (e.g. PG or a peptide) with no need for further processing of the NG1686 to activate the protein.
The HdpA protein from Helicobacter pylori was recently shown to also carry dual carboxypeptidase and endopeptidase activities (28). Whereas site-directed mutagenesis of one of the predicted active site residues (H259A) of HdpA disrupted activity of HdpA, the analogous H295A mutation of NG1686 had little effect on NG1686 phenotypes. Therefore, although NG1686 and HdpA may share dual activities, the residues responsible for activity differ between the proteins. The two other known gonococcal PG endopeptidases, PBP3 and PBP4, also have both endopeptidase and dd-carboxypeptidase activity, although they are not members of the M23B family (43, 44). It is unknown what factors determine which activity of NG1686 is performed or what the mechanism is behind the dual action of NG1686. Therefore, it would be interesting to both determine the crystal structure of NG1686 in the context of its different substrates and separately test the effect of the mutations on the endopeptidase and dd-carboxypeptidase activities of NG1686 in vitro.
The large colony morphology phenotype of the Δng1686 mutant is the easiest to reconcile with the demonstrated peptidoglycanase activities of NG1686. The loss of a protein that acts on PG has previously been shown to affect both the colony morphology (45, 46) and the cellular morphology of gonococci (45, 47). Much recent work has sought to define the cellular roles of these M23B/LytM family endopeptidase enzymes in a variety of other Gram-negative bacteria (27, 48–51). Several M23B family proteins function in the cleavage of septal PG to allow for efficient cell separation (51–54). In H. pylori, four M23B family proteins have been found to affect PG cross-linking, contributing to the helical curvature of this bacterium and its ability to colonize the stomach; however, these proteins do not affect cell separation in H. pylori (27, 28). In contrast, the ng1686 mutant does not show an altered cellular morphology or cell separation defect. Moreover, HPLC analysis of the PG from strains FA1090 and Δng1686 revealed no discernable differences in the type or amount of PG components in mutanolysin-digested PG isolated from strain FA1090 or Δng1686 (supplemental Fig. S5), indicating that NG1686 does not affect PG structure overall.
Our current hypothesis is that NG1686 causes subtle, localized, changes in the PG structure. Localized actions of peptidoglycanases have been proposed to explain the requirement of specific peptidoglycanases for the function of various trans-envelope machines, such as flagella or type III or IV secretion systems (55–57). None of these are present in N. gonorrhoeae strain FA1090, so if NG1686 is acting in the localized degradation of PG, it is unclear what the localized breaks in the PG would facilitate.
There are several hypotheses that explain how the lack of PG degradative ability results in the susceptibility of the Δng1686 strain to both H2O2 and PMN-mediated killing. The most concise explanation has both phenotypes resulting from the NG1686 protein acting on a common substrate. Therefore, the simplest hypothesis is that deletion of ng1686 affects the overall permeability of the cell wall. If this were the case, the Δng1686 strain should be more susceptible to killing by a variety of chemicals. However, we previously only observed increased susceptibility to killing by H2O2 and the inorganic peroxide cumene hydroperoxide (22) and not to other oxidants or antibiotics, suggesting that small defects in PG structure in the Δng1686 mutant result in discernable phenotypes only for peroxide and PMN-mediated killing. An alternate hypothesis is that NG1686 has more than one cellular target and that the phenotypes of altered colony morphology and increased sensitivity to H2O2 and PMN-mediated killing result from the action of NG1686 on different substrates. Because the results of a two-dimensional gel electrophoresis analysis performed on total cell lysates from strains FA1090 and Δng1686 (data not shown) revealed no gross differences in the protein profiles of the two strains resulting from the loss of ng1686, we do not favor this hypothesis. Therefore, our preferred hypothesis is that NG1686 acts exclusively on PG in the gonococcus.
Criss et al. (20) has confirmed that N. gonorrhoeae is neither affected by oxidative killing mechanisms of PMN (17, 18) nor protected by its antioxidant gene products against killing by PMNs (19), further suggesting that PMNs kill gonococci exclusively through non-oxidative factors, such as cationic antimicrobial peptides or degradative enzymes (14, 16). The Δng1686 mutant is more susceptible to non-oxidative killing by PMNs than the parent strain FA1090 (20, 22). Moreover, this increased sensitivity is only manifest in the Δng1686 bacteria located extracellularly, not Δng1686 bacteria that are internalized by PMNs (20), suggesting that exocytosed PMN factors, such as antimicrobial peptides or DNA nets, are responsible for the enhanced susceptibility of the Δng1686 strain. Our current work also shows that the NG1686 protein is localized inside the gonococcal cell, so the enhanced susceptibility of the Δng1686 strain to extracellular killing by PMNs could reflect susceptibility to a PMN factor that is internalized by gonococci. Although two-dimensional gel electrophoresis did not identify any gonococcal proteins that were substrates of NG1686 (data not shown), the NG1686 protein could additionally act on proteins or peptides produced by PMNs. Thus, if the Δng1686 mutant were unable to cleave some PMN antimicrobial factor, this would render the mutant more susceptible to killing by PMNs. However, the simplest explanation for the influence of NG1686 activity on these diverse phenotypes is that the localized cleavage of PG is required for expression of one or more bacterial factors that directly increase resistance to ROS and non-oxidative PMN killing.
Supplementary Material
Acknowledgments
We thank Dr. Amy Harms, Dr. Melissa Boersma, and Dr. Greg Barrett-Wilt (University of Wisconsin Biotechnology Center) for assistance in the mass spectrometry analyses and Dr. Wayne Anderson (Northwestern University Feinberg School of Medicine) for helpful discussions about crystallography.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 AI44239 and R37 AI33493 (to H. S. S.), R01 AI047958 (to J. P. D.), U19 AI01448-16 (to E. A. S.), and T32 AI055397 (to P. L. K.).

This article contains supplemental Table S1, Figs. S1–S5, Experimental Procedures, and References.
- PG
- peptidoglycan
- PMN
- polymorphonuclear leukocyte
- ROS
- reactive oxygen species
- DAP
- meso-diaminopimelic acid
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- MWCO
- molecular weight cut-off
- FDNB
- 1-fluoro-2,4-dinitrobenzene
- DNP
- dinitrophenyl
- MurNAc-pentapeptide
- N-acetyl-muramoyl-l-alanyl-d-glutamyl-l-diaminopimelyl-d-alanyl-d-alanine
- ESI
- electrospray ionization.
REFERENCES
- 1. World Health Organization (2011) Emergence of multi-drug resistant Neisseria gonorrhoeae. Threat of global rise in untreatable sexually transmitted disease. in WHO/RHR/11.4 Department of Reproductive Health and Research, World Health Organization, Geneva [Google Scholar]
- 2. Hook E. W., 3rd, Holmes K. K. (1985) Gonococcal infections. Ann. Int. Med. 102, 229–243 [DOI] [PubMed] [Google Scholar]
- 3. Edwards J. L., Apicella M. A. (2004) The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin. Microbiol. Rev. 17, 965–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dokter W. H., Dijkstra A. J., Koopmans S. B., Stulp B. K., Keck W., Halie M. R., Vellenga E. (1994) G(Anh)MTetra, a natural bacterial cell wall breakdown product, induces interleukin-1 β and interleukin-6 expression in human monocytes. A study of the molecular mechanisms involved in inflammatory cytokine expression. J. Biol. Chem. 269, 4201–4206 [PubMed] [Google Scholar]
- 5. Melly M. A., McGee Z. A., Rosenthal R. S. (1984) Ability of monomeric peptidoglycan fragments from Neisseria gonorrhoeae to damage human fallopian-tube mucosa. J. Infect. Dis. 149, 378–386 [DOI] [PubMed] [Google Scholar]
- 6. Melly M. A., Gregg C. R., McGee Z. A. (1981) Studies of toxicity of Neisseria gonorrhoeae for human fallopian tube mucosa. J. Infect. Dis. 143, 423–431 [DOI] [PubMed] [Google Scholar]
- 7. Burg N. D., Pillinger M. H. (2001) The neutrophil. Function and regulation in innate and humoral immunity. Clin. Immunol. 99, 7–17 [DOI] [PubMed] [Google Scholar]
- 8. Tseng H. J., Srikhanta Y., McEwan A. G., Jennings M. P. (2001) Accumulation of manganese in Neisseria gonorrhoeae correlates with resistance to oxidative killing by superoxide anion and is independent of superoxide dismutase activity. Mol. Microbiol. 40, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 9. Turner S., Reid E., Smith H., Cole J. (2003) A novel cytochrome c peroxidase from Neisseria gonorrhoeae. A lipoprotein from a Gram-negative bacterium. Biochem. J. 373, 865–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soler-García A. A., Jerse A. E. (2004) A Neisseria gonorrhoeae catalase mutant is more sensitive to hydrogen peroxide and paraquat, an inducer of toxic oxygen radicals. Microb. Pathog. 37, 55–63 [DOI] [PubMed] [Google Scholar]
- 11. Seib K. L., Tseng H. J., McEwan A. G., Apicella M. A., Jennings M. P. (2004) Defenses against oxidative stress in Neisseria gonorrhoeae and Neisseria meningitidis. Distinctive systems for different lifestyles. J. Infect. Dis. 190, 136–147 [DOI] [PubMed] [Google Scholar]
- 12. Skaar E. P., Tobiason D. M., Quick J., Judd R. C., Weissbach H., Etienne F., Brot N., Seifert H. S. (2002) The outer membrane localization of the Neisseria gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 99, 10108–10113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stohl E. A., Seifert H. S. (2006) Neisseria gonorrhoeae DNA recombination and repair enzymes protect against oxidative damage caused by hydrogen peroxide. J. Bacteriol. 188, 7645–7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shafer W. M., Qu X., Waring A. J., Lehrer R. I. (1998) Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. U.S.A. 95, 1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee E. H., Shafer W. M. (1999) The farAB-encoded efflux pump mediates resistance of gonococci to long-chained antibacterial fatty acids. Mol. Microbiol. 33, 839–845 [DOI] [PubMed] [Google Scholar]
- 16. Jerse A. E., Sharma N. D., Simms A. N., Crow E. T., Snyder L. A., Shafer W. M. (2003) A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect. Immun. 71, 5576–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rest R. F., Fischer S. H., Ingham Z. Z., Jones J. F. (1982) Interactions of Neisseria gonorrhoeae with human neutrophils. Effects of serum and gonococcal opacity on phagocyte killing and chemiluminescence. Infect. Immun. 36, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frangipane J. V., Rest R. F. (1992) Anaerobic growth of gonococci does not alter their Opa-mediated interactions with human neutrophils. Infect. Immun. 60, 1793–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seib K. L., Simons M. P., Wu H. J., McEwan A. G., Nauseef W. M., Apicella M. A., Jennings M. P. (2005) Investigation of oxidative stress defenses of Neisseria gonorrhoeae by using a human polymorphonuclear leukocyte survival assay. Infect. Immun. 73, 5269–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Criss A. K., Katz B. Z., Seifert H. S. (2009) Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leukocytes. Cell Microbiol. 11, 1074–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Criss A. K., Seifert H. S. (2008) Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol. 10, 2257–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stohl E. A., Criss A. K., Seifert H. S. (2005) The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 58, 520–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Höltje J. V. (1998) Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62, 181–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boneca I. G. (2005) The role of peptidoglycan in pathogenesis. Curr. Opin. Microbiol. 8, 46–53 [DOI] [PubMed] [Google Scholar]
- 25. Cloud-Hansen K. A., Peterson S. B., Stabb E. V., Goldman W. E., McFall-Ngai M. J., Handelsman J. (2006) Breaching the great wall. Peptidoglycan and microbial interactions. Nat. Rev. Microbiol. 4, 710–716 [DOI] [PubMed] [Google Scholar]
- 26. Rosenthal R. S., Nogami W., Cookson B. T., Goldman W. E., Folkening W. J. (1987) Major fragment of soluble peptidoglycan released from growing Bordetella pertussis is tracheal cytotoxin. Infect. Immun. 55, 2117–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sycuro L. K., Pincus Z., Gutierrez K. D., Biboy J., Stern C. A., Vollmer W., Salama N. R. (2010) Peptidoglycan cross-linking relaxation promotes Helicobacter pylori's helical shape and stomach colonization. Cell 141, 822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonis M., Ecobichon C., Guadagnini S., Prévost M. C., Boneca I. G. (2010) A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation, and virulence. Mol. Microbiol. 78, 809–819 [DOI] [PubMed] [Google Scholar]
- 29. Mehr I. J., Seifert H. S. (1998) Differential roles of homologous recombination pathways in Neisseria gonorrhoeae pilin antigenic variation, DNA transformation, and DNA repair. Mol. Microbiol. 30, 697–710 [DOI] [PubMed] [Google Scholar]
- 30. Stohl E. A., Seifert H. S. (2001) The recX gene potentiates homologous recombination in Neisseria gonorrhoeae. Mol. Microbiol. 40, 1301–1310 [DOI] [PubMed] [Google Scholar]
- 31. Hamilton H. L., Domínguez N. M., Schwartz K. J., Hackett K. T., Dillard J. P. (2005) Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol. Microbiol. 55, 1704–1721 [DOI] [PubMed] [Google Scholar]
- 32. Zahrl D., Wagner M., Bischof K., Bayer M., Zavecz B., Beranek A., Ruckenstuhl C., Zarfel G. E., Koraimann G. (2005) Peptidoglycan degradation by specialized lytic transglycosylases associated with type III and type IV secretion systems. Microbiology 151, 3455–3467 [DOI] [PubMed] [Google Scholar]
- 33. Dillard J. P., Hackett K. T. (2005) Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect. Immun. 73, 5697–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jayaswal R. K., Lee Y. I., Wilkinson B. J. (1990) Cloning and expression of a Staphylococcus aureus gene encoding a peptidoglycan hydrolase activity. J. Bacteriol. 172, 5783–5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bernadsky G., Beveridge T. J., Clarke A. J. (1994) Analysis of the sodium dodecyl sulfate-stable peptidoglycan autolysins of select gram-negative pathogens by using renaturing polyacrylamide gel electrophoresis. J. Bacteriol. 176, 5225–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohnishi R., Ishikawa S., Sekiguchi J. (1999) Peptidoglycan hydrolase LytF plays a role in cell separation with CwlF during vegetative growth of Bacillus subtilis. J. Bacteriol. 181, 3178–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clarke T. B., Kawai F., Park S. Y., Tame J. R., Dowson C. G., Roper D. I. (2009) Mutational analysis of the substrate specificity of Escherichia coli penicillin binding protein 4. Biochemistry 48, 2675–2683 [DOI] [PubMed] [Google Scholar]
- 38. Lloyd A. J., Gilbey A. M., Blewett A. M., De Pascale G., El Zoeiby A., Levesque R. C., Catherwood A. C., Tomasz A., Bugg T. D., Roper D. I., Dowson C. G. (2008) Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J. Biol. Chem. 283, 6402–6417 [DOI] [PubMed] [Google Scholar]
- 39. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 40. Sechman E. V., Rohrer M. S., Seifert H. S. (2005) A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol. Microbiol. 57, 468–483 [DOI] [PubMed] [Google Scholar]
- 41. Odintsov S. G., Sabala I., Marcyjaniak M., Bochtler M. (2004) Latent LytM at 1.3 Å resolution. J. Mol. Biol. 335, 775–785 [DOI] [PubMed] [Google Scholar]
- 42. Wang X., Yang X., Yang C., Wu Z., Xu H., Shen Y. (2011) Crystal structure of outer membrane protein NMB0315 from Neisseria meningitidis. PLoS One 6, e26845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stefanova M. E., Tomberg J., Davies C., Nicholas R. A., Gutheil W. G. (2004) Overexpression and enzymatic characterization of Neisseria gonorrhoeae penicillin-binding protein 4. Eur. J. Biochem. 271, 23–32 [DOI] [PubMed] [Google Scholar]
- 44. Stefanova M. E., Tomberg J., Olesky M., Höltje J. V., Gutheil W. G., Nicholas R. A. (2003) Neisseria gonorrhoeae penicillin-binding protein 3 exhibits exceptionally high carboxypeptidase and β-lactam binding activities. Biochemistry 42, 14614–14625 [DOI] [PubMed] [Google Scholar]
- 45. Fussenegger M., Kahrs A. F., Facius D., Meyer T. F. (1996) Tetrapac (tpc), a novel genotype of Neisseria gonorrhoeae affecting epithelial cell invasion, natural transformation competence, and cell separation. Mol. Microbiol. 19, 1357–1372 [DOI] [PubMed] [Google Scholar]
- 46. Garcia D. L., Dillard J. P. (2006) AmiC functions as an N-acetylmuramyl-l-alanine amidase necessary for cell separation and can promote autolysis in Neisseria gonorrhoeae. J. Bacteriol. 188, 7211–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cloud K. A., Dillard J. P. (2004) Mutation of a single lytic transglycosylase causes aberrant septation and inhibits cell separation of Neisseria gonorrhoeae. J. Bacteriol. 186, 7811–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Layec S., Gérard J., Legué V., Chapot-Chartier M. P., Courtin P., Borges F., Decaris B., Leblond-Bourget N. (2009) The CHAP domain of Cse functions as an endopeptidase that acts at mature septa to promote Streptococcus thermophilus cell separation. Mol. Microbiol. 71, 1205–1217 [DOI] [PubMed] [Google Scholar]
- 49. Stentz R., Wegmann U., Parker M., Bongaerts R., Lesaint L., Gasson M., Shearman C. (2009) CsiA is a bacterial cell wall synthesis inhibitor contributing to DNA translocation through the cell envelope. Mol. Microbiol. 72, 779–794 [DOI] [PubMed] [Google Scholar]
- 50. Camiade E., Peltier J., Bourgeois I., Couture-Tosi E., Courtin P., Antunes A., Chapot-Chartier M. P., Dupuy B., Pons J. L. (2010) Characterization of Acp, a peptidoglycan hydrolase of Clostridium perfringens with N-acetylglucosaminidase activity that is implicated in cell separation and stress-induced autolysis. J. Bacteriol. 192, 2373–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uehara T., Dinh T., Bernhardt T. G. (2009) LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J. Bacteriol. 191, 5094–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poggio S., Takacs C. N., Vollmer W., Jacobs-Wagner C. (2010) A protein critical for cell constriction in the Gram-negative bacterium Caulobacter crescentus localizes at the division site through its peptidoglycan-binding LysM domains. Mol. Microbiol. 77, 74–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goley E. D., Comolli L. R., Fero K. E., Downing K. H., Shapiro L. (2010) DipM links peptidoglycan remodeling to outer membrane organization in Caulobacter. Mol. Microbiol. 77, 56–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Möll A., Schlimpert S., Briegel A., Jensen G. J., Thanbichler M. (2010) DipM, a new factor required for peptidoglycan remodeling during cell division in Caulobacter crescentus. Mol. Microbiol. 77, 90–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nambu T., Minamino T., Macnab R. M., Kutsukake K. (1999) Peptidoglycan-hydrolyzing activity of the FlgJ protein, essential for flagellar rod formation in Salmonella typhimurium. J. Bacteriol. 181, 1555–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dijkstra A. J., Keck W. (1996) Peptidoglycan as a barrier to transenvelope transport. J. Bacteriol. 178, 5555–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dillard J. P., Seifert H. S. (2001) A variable genetic island specific for Neisseria gonorrhoeae is involved in providing DNA for natural transformation and is found more often in disseminated infection isolates. Mol. Microbiol. 41, 263–277 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.