

Background: M-channels are potassium channels that are activated by phosphatidylinositol 4,5-bisphosphate, but their response to other phospholipids is unknown.
Results: M-channel proteins were activated by phosphoinositides and lipid phosphates but not by inositol phosphates.
Conclusion: Minimum activation requirements are an acyl chain and one or more phosphate groups.
Significance: M-channels control cell excitability, so their regulation by membrane constituents is important for biology.
Keywords: Biophysics, Ion Channels, Membrane Lipids, Phosphoinositides, Phospholipid, M-channels, Potassium Channels
Abstract
M-channels are voltage-gated potassium channels that regulate cell excitability. They are heterotetrameric assemblies of Kv7.2 and Kv7.3 subunits. Their opening requires the presence of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). However, the specificity of PI(4,5)P2 as a binding and activating ligand is unknown. Here, we tested the ability of different phosphoinositides and lipid phosphates to activate or bind to M-channel proteins. Activation of functional channels was measured in membrane patches isolated from cells coexpressing Kv7.2 and Kv7.3 subunits. Channels were activated to similar extents (maximum open probability of ∼0.8 at 0 mV) by 0.1–300 μm dioctanoyl homologs of the three endogenous phosphoinositides, PI(4)P, PI(4,5)P2, and PI(3,4,5)P3, with sensitivity increasing with increasing numbers of phosphates. Non-acylated inositol phosphates had no effect up to 100 μm. Channels were also activated with increasing efficacy by 1–300 μm concentrations of the monoacyl monophosphates fingolimod phosphate, sphingosine 1-phosphate, and lysophosphatidic acid but not by phosphate-free fingolimod or sphingosine or by phosphate-masked phosphatidylcholine or phosphatidylglycerol. An overlay assay confirmed that a fusion protein containing the full-length C terminus of Kv7.2 could bind to a broad range of phosphoinositides and phospholipids. A mutated Kv7.2 C-terminal construct with reduced sensitivity to PI(4,5)P showed significantly less binding to most polyphosphoinositides. We concluded that M-channels bind to, and are activated by, a wide range of lipid phosphates, with a minimum requirement for an acyl chain and a phosphate headgroup. In this, they more closely resemble inwardly rectifying Kir6.2 potassium channels than the more PI(4,5)P2-specific Kir2 channels. Notwithstanding, the data also support the view that the main endogenous activator of M-channels is PI(4,5)P2.
Introduction
The activity of many membrane ion channels is regulated by the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)2 (1–3). One such channel is the M-type potassium channel. This is a voltage-gated potassium channel composed of Kv7 family subunits, primarily Kv7.2 and Kv7.3 (4). Although gated by membrane voltage, the channels require PI(4,5)P2 to enter in and stabilize the open state (5–7). They are particularly interesting because their sensitivity to PI(4,5)P2 is set at such a level that they rapidly close when the endogenous membrane content of PI(4,5)P2 is reduced by a neurotransmitter such as acetylcholine that activates phospholipase C and so stimulates PI(4,5)P2 hydrolysis (2, 3, 8). The loss of outward potassium current then initiates a marked increase in the excitability of many neurons, with profound physiological consequences (9).
Ion channels (and other proteins) vary in the specificity of their interaction with PI(4,5)P2 vis-à-vis other phosphoinositides or other membrane phospholipids (3). Kv7.2/7.3 channels appear to show a rather specific dependence on PI(4,5)P2 in that activity is rapidly and substantially reduced when the 5′-phosphate is cleaved by an inositol-5-phosphatase (10). Furthermore, in experiments in which the individual channel subunits were expressed and then isolated membrane patches were exposed to the water-soluble PI(4,5)P2 analog diC8PI(4,5)P2, the two subunits showed a very large (∼100-fold) difference in the concentrations of diC8PI(4,5)P2 required to activate them (7). This was attributable to variations within a small cluster of basic amino acids in a region of the C terminus of the subunit protein, suggesting a specific PI(4,5)P2 interaction site (11).
However, there are also some indications that Kv7.2/7.3 channels may be activated by other phosphoinositides (6, 7). Furthermore, a C-terminal fusion protein of the homologous cardiac Kv7.1 channel was noted to bind to a variety of phosphoinositides and phospholipids in a protein-lipid overlay assay (12). This raised the question of how broad might be the range of phosphoinositides and lipid phosphates that can interact with Kv7.2/7.3 channels.
In these experiments, we have tried to answer this question by (a) testing a range of phosphoinositides and their analogs and derivatives for their ability to activate Kv7.2/7.3 channels when applied to an isolated membrane patch in which they are expressed and (b) using a lipid overlay assay to observe the binding of a Kv7.2 C-terminal fusion protein to a number of membrane phospholipids. From this, we have been able to draw some conclusions regarding the minimum requirements for M-channel activation by membrane phospholipids and lipid phosphates.
EXPERIMENTAL PROCEDURES
Kv7.2 and Kv7.3 Constructs
Electrophysiological experiments were undertaken using CHO cells stably cotransfected with the full-length human Kv7.2 and Kv7.3 M-channel subunits (designated Kv7.2/7.3 cells) (13). For in vitro binding tests, the full-length C terminus of Kv7.2 (Kv7.2C; amino acids 318–845; NCBI accession number NM_004518) was cloned into the pMAL-c2X vector (New England Biolabs) to create a C-terminal fusion with maltose-binding protein (MBP). MBP-Kv7.2C was expressed and purified as described previously (12). Experiments were repeated using the full-length C terminus of the mutant channel Kv7.2(K452E/R459E/R461E) (called Kv7.2C-EEE), which is ∼2-fold less sensitive to PI(4,5)P2 than the wild-type channel when expressed as a homomer (11).
Cell Culture
Kv7.2/7.3-CHO cells were incubated in α-minimal essential medium (Invitrogen) supplemented with 10% fetal calf serum, 1% l-glutamine, 1% penicillin/streptomycin, 0.2 mg/ml hygromycin, and 0.4 mg/ml neomycin. The cell line was maintained in a humidified incubator gassed with 5% CO2 and 95% air. Cells were passaged every 2–3 days at a ratio of 1:10. Ca2+- and Mg2+-free phosphate-buffered Hanks' balanced saline solution was used to detach the cells. This was subsequently followed by centrifugation at 800 × g and resuspension in the supplemented medium described above. For subsequent electrophysiological experimentation, cells were settled in specially designed siliconized chambers (volume of ∼200 μl) within plastic Petri dishes. They were incubated for at least 24 h before being mounted on the stage of an inverted microscope equipped with phase-contrast optics and continuously superfused at ∼5 ml/min.
Electrophysiological Recordings
Single M-channel activity was recorded using patch electrodes in membrane patches excised from Kv7.2/7.3-CHO cells in inside-out configuration at a controlled room temperature (22 ± 0.5 °C). Pipette voltage was set at 0 mV. For current recording, we used an Axopatch 200A amplifier and Digidata 1440 A/D interface (Axon Instruments, Forster City, CA) and a pipette holder optimized for low-noise recordings (G23 Instruments, University College London). All recordings were filtered with an 8-pole Bessel filter at 2 kHz and digitized at 5 kHz. The pipette resistance when filled with the pipette solution was ∼5–10 megohms. Recorded channel currents were judged to be through a single class of heteromeric Kv7.2/7.3 channels because they had a constant single current amplitude of 0.52 ± 0.01 pA at 0 mV (n = 27 patches). In parallel studies on these cells, a channel slope conductance of 9.2 ± 0.1 picosiemens (n = 6) was determined in cell-attached mode, in agreement with previous data (7, 14).
Bath and pipette solutions contained 144 mm NaCl, 2.5 mm KCl, 0.5 mm MgCl2, 2 mm CaCl2, 10 mm d-glucose, and 5 mm HEPES; the pH was adjusted to 7.4 with Trizma (Tris base). Bath solutions in the inside-out studies contained 165 mm KCl, 5 mm HEPES, and 10 mm EGTA; the pH was adjusted to 7.2 with NaOH. Non-hydrolyzable ATPγS (0.1 μm) was constantly present in the bath solutions in inside-out studies to inhibit endogenous production of PI(4,5)P2 by phosphatidylinositol-phosphate kinases, which possibly remained associated with the patch after excision. All compounds studied were applied to the isolated inside-out membrane patches in incremental concentrations using a fast microperfusion system (delivery time of <1 s). All applied phospholipids were reversible. The effects of water-soluble phosphoinositides recovered within 100–500 ms on washout. A longer recovery time (up to 10 s) was needed to wash out compounds dissolved in methanol, which itself slightly depressed M-channel activity.
Data Analysis
Single channel current amplitudes were determined individually in each patch using a Gaussian all-point amplitude distribution. Because in these experiments we were concerned only with open probability (Po) values, not open-shut time distributions, we included patches with several channels in the analysis. The number of channels (N) in multichannel patches was determined in two ways: first, by observing the number of incremental current steps attained as Po rose to 0.6–0.8; and second, by using variance analysis. In the latter, current variance (Var(Im)) was fitted to the mean current (Im) using the parabolic function shown in Equation 1,
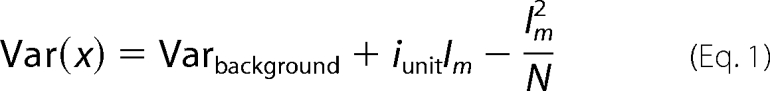 |
with the unit current (iunit) constrained to the value estimated from the point amplitude distribution. Values for N were accepted if the two estimates agreed to the nearest whole number. Patch NPo was measured over the last 20 s of each concentration of phospholipid, when activity was stable as judged from stability plots, and then corrected for the estimated number of channels in each patch to give channel Po as a function of phospholipid concentration.
The concentrations for half-activation (EC50) were calculated from concentration-response curves of the Po using two-component Hill equations (15) (Equation 2).
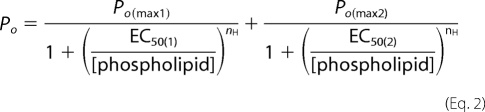 |
When fitting, the Hill coefficient (nH) was constrained to be the same for both components. Two-component curves were found to fit the Po/[phospholipid] concentration-response curves of heteromeric M-channels significantly better than one-component curves. Statistical comparisons were performed using one-way analysis of variance, and the difference was considered as significant at the level of p < 0.05. Data were analyzed using Clampfit Version 10.2, WinEDR, Microsoft Office Excel 2003 and MicroCal Origin Version 6.0 software.
Protein-Lipid Overlay Assay
Purified MBP-Kv7.2C was incubated with PIP Strips (Invitrogen) at a concentration of 5 μg/ml as described previously (16, 17). Rabbit anti-MBP antiserum (New England Biolabs) and ECL detection were used to detect binding of MBP-Kv7.2C to the relevant phospholipids. Densitometric analysis was carried out using Scion Image. Experiments were repeated using mutant Kv7.2C-EEE, which is ∼2-fold less sensitive to PI(4,5)P2 than the wild-type channel when expressed as a homomer (11).
Chemicals
All compounds for solutions, ATPγS, and dihydroxyacetone phosphate were purchased from Sigma-Aldrich. Phosphatidylinositol mono-, bis-, and trisphosphates (diC8PI(4)P, diC8PI(4,5)P2 and diC8PI(3,4,5)P3), inositol 1,4,5-trisphosphate (I(1,4,5)P3), sphingosine 1-phosphate (S-1-P), d-erythro-sphingosine, and biotinylated phosphatidylglycerol (PG) and phosphatidylcholine (PC) were purchased from Echelon Inc. Fingolimod phosphate (FTY720-P), fingolimod (FTY720), and 1-oleoyl lysophosphatidic acid (LPA) were purchased from Cayman Chemical Co. I(4,5)P2/triethanolamine salt was provided by Prof. B. V. L. Potter. Stock solutions of S-1-P, d-erythro-sphingosine, fingolimod phosphate, fingolimod, LPA, PG, and PC were dissolved in methanol (maximum concentration applied to patch of 12%).
RESULTS
Electrophysiological Assays
Phosphoinositides
PI(4,5)P2 is normally synthesized from PI(5)P by PI5K and then can be further phosphorylated to PI(3,4,5)P3 by PI3K (Fig. 1) (18). We therefore tested the effect of incremental concentrations of the diC8 analogs of these three phosphoinositides on the activity of Kv7.2/7.3 channels in excised membrane patches held at a constant pipette voltage of 0 mV. All three produced a strikingly similar activation of the channels (Fig. 2, A–C). As previously noted for diC8PI(4,5)P2 (15), incremental concentrations between 0.1 and 300 μm generated a biphasic concentration-Po curve, resolvable into “high-affinity” and “low-affinity” components; the former maximized at Po = 0.19 (0.10 for diC8PI(4)P), whereas the latter carried Po up to an extrapolated value approaching ∼0.8 (Fig. 2D). The three phosphoinositides had very similar EC50 values (EC50(1) = 1.1–1.7 μm) for the high-affinity component, whereas the low-affinity EC50 (EC50(2)) varied inversely with the number of phosphates (Table 1). As a result, diC8PI(4)P produced a slightly lower observed Po (0.61 ± 0.10, n = 9) at the maximum applied concentration of 300 μm compared with diC8PI(4,5)P2 (0.79 ± 0.05, n = 11) or diC8PI(3,4,5)P3 (0.80 ± 0.04, n = 11) (Table 1). Thus, diC8 homologs of all three endogenous phosphoinositides strongly activated the M-channels, with a potency that increased with increasing numbers of phosphates.
FIGURE 1.

Structures and phosphorylation of phosphatidylinositols. Shown are sequential reactions for the endogenous phosphorylation of PI(4)P to PI(4,5)P2 and PI(3,4,5)P3 by PI5K and PI3K.
FIGURE 2.

M-type channel activity stimulated by diC8 phosphatidylinositides. A–C, exemplar traces of M-channel recordings showing effects of sequentially increasing concentrations of diC8PI(4)P, diC8PI(4,5)P2, and diC8PI(3,4,5)P3 applied to the inner leaflet of excised inside-out patches from CHO cells stably expressing the M-channel subunits Kv7.2 and Kv7.3 (holding potential of 0 mV). C, closed state; O1–O5, open state currents for one to five channels. D, mean ± S.E. for Po of M-channels plotted against 0.1–300 μm phosphatidylinositols. ●, diC8PI(4)P (n = 6–24); ○, diC8PI(4,5)P2 (n = 10–27); ■, diC8PI(3,4,5)P3 (n = 7–18). The data points were fitted to a two-component Hill equation (see “Experimental Procedures”) with the parameters listed in Table 1.
TABLE 1.
Activation of Kv7.2/7.3 M-channels by phosphoinositides
Data are from least-squares curve fits (see “Experimental Procedures” and Fig. 2).
| PI(4)P | PI(4,5)P2 | PI(3,4,5)P3 | |
|---|---|---|---|
| High-affinity component | |||
| EC50(1) (μm) | 1.7 ± 0.3 | 1.1 ± 0.1 | 1.1 ± 0.2 |
| Po(max1) | 0.10 ± 0.007a | 0.19 ± 0.007 | 0.19 ± 0.017 |
| Low-affinity component | |||
| EC50(2) (μm) | 98.6 ± 4.5 | 49.6 ± 1.9 | 35.4 ± 2.4 |
| Po(max2) | 0.57 ± 0.022 | 0.61 ± 0.012 | 0.64 ± 0.025 |
| Combined | |||
| Po(max) | 0.61 ± 0.10 | 0.79 ± 0.05 | 0.80 ± 0.04 |
| nH | 1.9 ± 0.2 | 2.0 ± 0.1 | 1.5 ± 0.1 |
| nb | 6–24 | 10–27 | 7–18 |
a The value significantly differs from the Po(max1) of PI(4,5)P2 (p < 0.03).
b Number of patches at each concentration.
Inositol Phosphates
Studies on other potassium channels indicate that the inositol phosphate headgroup of PI(4,5)P2 projects from the inner leaflet of the lipid bilayer into the cytoplasm, where it docks onto a receptive site (or sites) in the cytoplasmic domain of the channel (19, 20). The above results suggest that the charges on the headgroup are one factor governing the ability of phosphoinositides to activate M-channels. We therefore wondered whether the inositol phosphates themselves might activate the Kv7.2/7.3 channels or, conversely, compete with PI(4,5)P2 to inhibit its action. These are not unreasonable possibilities because (a) the PI(4,5)P2-binding pleckstrin homology domain of the membrane enzyme phospholipase C also binds I(1,4,5)P3 with high affinity (21, 22), and (b) I(1,4,5)P3 has been reported to antagonize the effect of PI(4,5)P2 on some transient receptor potential cation channels (23). Accordingly, we applied incremental concentrations of I(1,4,5)P3 to excised Kv7.2/7.3-containing patches as illustrated in Fig. 3A. No channel activation occurred up to 300 μm in any of six experiments, whereas in each case, 100 μm PI(4,5)P2 increased Po to an average value of 0.54 ± 0.09 (n = 6). Furthermore, 100 μm I(1,4,5)P3 did not inhibit the effect of PI(4,5)P2 when applied either before or during the application of 100 μm PI(4,5)P2, so the mean Po remained unaffected (0.58 ± 0.08, n = 5) (Fig. 3B). I(4,5)P2 (n = 5) or a small water-soluble monophosphate, dihydroxyacetone phosphate (n = 8), also had no effect at concentrations of 100 μm (data not shown). Thus, water-soluble analogs of the phosphoinositide headgroups do not activate M-channels.
FIGURE 3.

I(1,4,5)P3 does not stimulate or inhibit M-channel activity. A, exemplar trace of sequential increases in I(1,4,5)P3 concentrations showing no stimulation of M-channel activity applied to the inner leaflet of excised inside-out patches from CHO cells stably expressing M-type Kv7.2/7.3 channels (holding potential of 0 mV). C, closed state; O, open state current. B, superimposition of 100 μm PI(4,5)P2 does not reduce channel activity stimulated by 100 μm diC8PI(4,5)P2 (holding potential of 0 mV).
Other Phospholipids
The above results indicate that, although the docking of the inositol headgroups of the phosphoinositides onto the cytoplasmic domain of the channel may be responsible for channel opening, attachment of the phosphate to a lipophilic moiety is necessary for (or facilitates) this effect. This accords with recent structural information regarding inwardly rectifying Kir channels, in which the acyl chain of PI(4,5)P2 interacts nonselectivity with the transmembrane domain of Kir2 (20). To assess what the minimum lipophilic acyl phosphate requirement for M-channel activation might be, we tested the effect of several other phospholipids and their congeners (Fig. 4): S-1-P, its analog fingolimod phosphate, and LPA, whose aliphatic structure corresponds in principle to PI(4,5)P2.
FIGURE 4.

Structures of some lipid phosphates tested.
S-1-P activated the channels at concentrations of 3 μm upward to a mean Po of 0.16 ± 0.03 (n = 9) at 100 μm (Fig. 5, A and B). Fingolimod phosphate also activated the channels over the same concentration range but only to a lower mean Po of 0.02 ± 0.006 (n = 8) (Fig. 5C). Concentration-Po curves (Fig. 5D) could be resolved into two (high- and low-affinity) components like those generated by the phosphoinositides (Table 2). In both cases, incorporating two components gave a significantly better fit than a single component. This suggests that they acted mechanistically, rather like the phosphoinositides.
FIGURE 5.

M-channel activity stimulated by S-1-P and fingolimod phosphate. A–C, exemplar traces of M-channel recordings showing the effects of sequential increases in S-1-P (A and B) and fingolimod phosphate (FTY720-P; C) concentrations applied to the inner leaflet of excised inside-out patches from CHO cells stably expressing M-type Kv7.2/7.3 channels (holding potential of 0 mV). C, closed state; O1–O5, open state currents for one to five channels. D, mean ± S.E. for M-channel Po plotted against lipid phosphate concentration. – – –, diC8PI(4,5)P2; ▴, S-1-P; ▾, fingolimod phosphate (including inset); △ and ▿, 100 μm diC8PI(4,5)P2 (reference points for S-1-P and fingolimod phosphate, respectively). The data points were fitted to a two-component Hill equation (see “Experimental Procedures”) with the parameters listed in Table 2.
TABLE 2.
Activation of Kv7.2/7.3 M-channels by non-inositol lipid monophosphates
Data are from least-squares curve fits (see Figs. 5 and 6). FTY720-P, fingolimod phosphate.
| S-1-P | FTY720-P | LPA | |
|---|---|---|---|
| High-affinity component | |||
| EC50(1) (μm) | 3.2 ± 0.6 | 0.5 ± 0.001 | 1.5 ± 0.4 |
| Po(max1) | 0.072 ± 0.006a | 0.007 ± 0.000001a | 0.204 ± 0.027 |
| Low-affinity component | |||
| EC50(2) (μm) | 157.3 ± 40.8 | 63.2 ± 0.2 | 40.0 ± 9.2 |
| Po(max2) | 0.26 ± 0.043b | 0.021 ± 0.00006b | 0.626 ± 0.075 |
| Combined | |||
| Po(max) | 0.26 ± 0.05b | 0.02 ± 0.006b | 0.68 ± 0.09 |
| nH | 1.5 ± 0.2 | 1.8 ± 0.0042 | 1.3 ± 0.2 |
| nc | 6–15 | 8–11 | 4–12 |
a Values significantly differ from the Po(max1) of PI(4,5)P2 (p < 0.02).
b Values significantly differ from the Po(max2) and combined Po(max) of PI(4,5)P2 (p < 0.000006 and p < 0.0000000001 for S-1-P and fingolimod phosphate, respectively).
c Number of patches at each concentration.
Somewhat surprisingly, LPA proved as effective as PI(4,5)P2 as a channel activator, driving Po to a value of 0.68 ± 0.09 (n = 6) at 100 μm (Fig. 6, A and B). Again, the concentration-Po curve could be resolved into two components, although less clearly demarcated than with PI(4,5)P2 (Fig. 6C and Table 2). Hence, the property of activating M-channels is not totally restricted to diacylphosphoinositides but extends to some monoacyl phosphates.
FIGURE 6.

M-type channel activity stimulated by LPA. A and B, exemplar traces of M-channel recordings showing effects of sequential increases in LPA concentrations applied to the inner leaflet of excised inside-out patches from CHO cells stably expressing M-type Kv7.2/7.3 channels (holding potential of 0 mV). C, closed state; O1–O5, open state currents for one to five channels. C, mean ± S.E. for Po of M-channels plotted against LPA concentration. – – –, diC8PI(4,5)P2 (from Fig. 2); ♦, LPA; ♢, 100 μm diC8PI(4,5)P2 (reference point for LPA). The data points were fitted to a two-component Hill equation (see “Experimental Procedures”) with the parameters listed in Table 2.
Phosphate-free Acyl Compounds
As noted, the acyl groups of phosphoinositides interact with the transmembrane domain of the Kir2 channels (20). The question then arises whether such an interaction might itself be capable of activating the M-channel in the absence of a phosphate headgroup. To answer this, we tested phosphate-free d-erythro-sphingosine, its analog fingolimod, and the biotinylated phospholipids PG and PC, in which the phosphates are masked with glycerol and choline groups, respectively. However, neither of the non-phosphorylated precursors of S-1-P and fingolimod phosphate (d-erythro-sphingosine and fingolimod, respectively) nor biotinylated PG or PC was able to activate the Kv7.2/7.3 channels when tested at 100 μm (n = 6–13) (Fig. 7). In each test, channels were strongly activated by 100 μm PI(4,5)P2. Thus, the charged phosphate headgroup seems an absolute requirement for phospholipid activation of M-channels.
FIGURE 7.

d-erythro-Sphingosine, fingolimod, and biotinylated PG and PC are unable to stimulate M-type channel activity. A–C, applications of 100 μm d-erythro-sphingosine (S) (A), fingolimod (FTY720) (B), and biotinylated PG and PC (C) to three separate patches did not activate M-channels. In each patch, channels were activated by 10 μm diC8PI(4,5)P2 (holding potential of 0 mV). C, closed state; O, open state current.
Protein-Lipid Overlay Assays
A fusion protein between MBP and the full-length C terminus of the Kv7.2 channel proteins (MBP-Kv7.2C, 100 kDa) was expressed and purified (Fig. 8A). The full-length protein is visible at 100 kDa; in addition, there is a clear degradation product of ∼43 kDa, which most likely corresponds to MBP alone. When the purified protein was incubated with the PIP Strips, the overlay assay revealed a broad association with all phosphoinositides, including phosphatidylinositol mono-, bis-, and trisphosphates (Fig. 8, B and C). There was also some association with phosphatidylserine (12, 24, 25). Binding to PI, LPA, and S-1-P was low or non-significant compared with the background. As reported previously (12), when expressed alone, MBP did not show any binding to the PIP Strips, indicating that the observed binding came directly from the Kv7.2C portion of the protein. These results are similar to those obtained with a fusion protein containing the C terminus of Kv7.1 (12) and confirm a wide range of potential phospholipid modulators of Kv7 channel function.
FIGURE 8.

Purification and lipid binding of MBP-Kv7.2C. A, 10% SDS-PAGE of typical MBP-Kv7.2C protein purification. The soluble fraction (S) and 2 μg of the eluted (E) protein were loaded as indicated. The protein size was estimated using Bio-Rad prestained markers (M) of known molecular mass (shown in kilodaltons). The band representing the expected size of the protein is boxed. B, PIP Strips were incubated overnight with 5 μg/ml MBP-Kv7.2C or MBP-Kv7.2C-EEE (11) protein. Recognition of binding was obtained by incubation with rabbit anti-MBP antibody (1:1000 dilution), followed by anti-rabbit antibody as contained in the ECL kit (GE Healthcare). C, densitometric measurements (A.U, arbitrary units) of the binding of MBP-Kv7.2C (WT; dark gray bars) and MBP-Kv7.2C-EEE (EEE; light gray bars) to phospholipids. Error bars are S.E. for the number of strips shown in parentheses. * and **, MBP-Kv7.2C-EEE binding was significantly less than that of MBP-Kv7.2C at p < 0.05 and p < 0.01, respectively (two-tailed t test for unequal numbers). LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine; PA, phosphatidic acid; PS, phosphatidylserine.
Mutant Kv7.2C-EEE showed significantly reduced binding to most of the phosphoinositides and also to phosphatidic acid (Fig. 8, B and C). No significant change in the low-level binding to LPA and S-1-P could be detected.
DISCUSSION
The first point emerging from these experiments is that M-channel subunits show a rather broad spectrum of interactions with phosphoinositides (and some other phospholipids). This is evident both from the activation of Kv7.2/Kv7.3 heteromers by intracellularly applied phospholipids and from phospholipid binding of the purified, solubilized Kv7.2C fusion protein. The apparent order of interactions revealed by these two approaches appears to diverge quite appreciably, but this is not surprising. The fusion protein contains only the C terminus (albeit full-length), so it is devoid of other cytoplasmic domains of the channel to which the phospholipids might potentially bind (26) and also of the transmembrane domains with which the lipophilic moieties of the phospholipids might interact (20). The overlay assays do not provide any information about relative affinities. Conversely, binding does not necessarily lead to channel activation; even where it does, the link between agonist binding and channel opening may be complex (27).
The second point concerns the activation by the phosphoinositides. We found that the channels can be equally well activated (i.e. to near-comparable maximum Po values) by the mono-, di-, and triphosphates diC8PI(4)P, diC8PI(4,5)P2, and diC8PI(3,4,5)P3. This accords with previous observations that diC8PI(4,5)P2 and diC8PI(3,4,5)P3 are equally efficacious (6) and that the channels are also activated by diC8PI(3,4)P2 (6, 7). It seems likely that they all interact with the same domain of the channel because a Kv7.2C fusion protein that contained a group of mutations that reduce the sensitivity of the functional channel to PI(4,5)P2 by 2-fold (11) showed less binding to all of the polyphosphoinositides. Notwithstanding, the channels showed a quantitative difference in their response to the different phosphoinositides, with sensitivity increasing with increasing numbers of phosphates. Interestingly, this applied only to the low-affinity component of the concentration-response curve, with no significant differences between the EC50 values for the high-affinity component; instead, diC8PI(4)P appeared to show a lower efficacy on this component of channel response. One possibility is that these two components relate to the contributions made by binding to the two subunits, Kv7.3 and Kv7.2, the former having a higher affinity than the latter (7). If this is the case, then the differences in the amino acid sequences in the C terminus that are responsible for the different sensitivities of the two subunits to diC8PI(4,5)P2 (11) also affect the relative activities of the different phosphoinositides at the two sites.
In contrast to the phosphoinositides, neither of the free inositol phosphates, I(1,4,5)P3 and I(4,5)P2, activated the M-channels or inhibited their response to PI(4,5)P2 at concentrations up to 300 μm. Thus, the presence of the lipophilic diacyl chain appears essential to orient the polar headgroup in position with respect to the channel to activate it. It has recently been shown that this lipophilic chain forms a nonspecific association with the transmembrane domains of the Kir channel (20) to facilitate the interaction of the polar headgroup with the binding sites in the cytoplasmic domain. The natural PI(4,5)P2 contains 16–20-carbon diacyl chains (28, 29). The 8-carbon derivative is less lipophilic and hence more convenient experimentally because it more rapidly enters and leaves the membrane and thus is more rapid in both onset and offset (30). A 4-carbon analog proved ineffective on Kir channels (30), presumably because it does not insert into the membrane so readily.
In this study, we also found that the M-channels could be activated by the monoacyl lipid monophosphates LPA, S-1-P, and fingolimod phosphate. Although in other circumstances lysophospholipids are known to act on specific G protein-coupled transmembrane receptors when released into the extracellular fluid (31), in our experiments, they presumably interacted directly with the inner (cytoplasmic) face of the M-channels. This view is strengthened by the fact that the effect of LPA quite strikingly replicated that of diC8PI(4,5)P2 both in efficacy and in its concentration dependence (Fig. 6). S-1-P and fingolimod phosphate were appreciably less efficacious, although they were effective over a similar concentration range to diC8PI(4,5)P2. We cannot be sure that these acted on the same C-terminal domain as the phosphoinositides because their binding to the fusion protein was weak and not obviously affected by the mutation that altered phosphoinositide binding and PI(4,5)P2 activation. However, the presence of high- and low-affinity components to their concentration-activation curves suggests a similar subunit-dependent activation mechanism if our hypothesis for the two components is correct.
Hence, M-channels can be activated by a range of lipid phosphates. From our limited survey, the minimum requirements would appear to be a phosphate group attached to an appropriate-length acyl chain, with a potency and efficacy that (in phosphoinositides) increases with increasing numbers of phosphates. Neither acyl compounds without the headgroups nor the inositol phosphates without the acyl chains activated the channels, so presumably, the acyl chains facilitate activation by interacting with the transmembrane domains of the channel (20).
How does this compare with other phosphoinositide-sensitive ion channels? The most thoroughly studied channels in terms of phosphoinositide selectivity are the inwardly rectifying Kir channels (26). Kir2 channels are activated most strongly by PI(4,5)P2, although Kir2.2 and Kir2.3 are also activated by PI(3,4,5)P3; Kir3 channels are activated by both, plus PI(3,4)P2, and show evidence of a wider range of interactions with phosphatidylinositol monophosphates in fusion protein binding studies (16). In contrast, Kir6.2 (KATP) channels do not discriminate between PI(4,5)P2, PI(3,4)P2, and PI(3,4,5)P3 and can also be activated by other negatively charged lipids such as long-chain acetyl coenzyme A (32, 33). The M-channel appears to more closely resemble the Kir6.2 channel in this respect rather than the more PI(4,5)P2-specific Kir2 channels.
Other phospholipid-sensitive membrane proteins show very variable ligand selectivities. The pleckstrin homology domain of phospholipase Cδ is selective for PI(4,5)P2 but binds I(1,4,5)P3 even more tightly (21). In contrast, the membrane-located transcription factor tubby binds PI(3,4)P2, PI(4,5)P2, and PI(3,4,5)P3 but not the inositol phosphates (34). Very many proteins bind phospholipids, with a wide range of substrate specificities that are associated with distinct differences in the structures and flexibility of their binding sites (35). Indeed, interaction of basic proteins or regions thereof with abundant membrane phospholipids may occur simply through electrostatic attraction rather than specific binding (36).
A plausible binding site in the Kv7.2 and Kv7.3 subunits of the M-channel, in which electrostatic interactions are reinforced by strong hydrogen bonding, has been derived from previous mutational studies coupled with homology modeling (11). It would be interesting to know whether this has sufficient flexibility to accommodate the range of phospholipids that we have studied.
An additional question arises regarding the physiological significance of these observations. The first major consideration is how the concentrations of phospholipid applied to the inner face of the membrane relate to those the channel may encounter in the normal cell membrane. PI(4,5)P2 comprises ∼1% of the phospholipids in the cell membrane (37). It has been estimated that the concentration of membrane PI(4,5)P2, if dissolved in the cytoplasm of a cell with a radius of 10 μm, would yield a cytoplasmic concentration of 10 μm (29). Thus, the lower concentrations of PI(4,5)P2 (giving a Po of up to ∼0.2) are likely to encompass this physiological range. PI(4)P is also present in cell membranes at ∼78% of PI(4,5)P2 (38). However, its lower potency and considerably weaker effect at low (<10 μm) concentrations (Po1 = 0.1 for PI(4)P versus 0.2 for PI(4,5)P2) (Fig. 2) might explain why it is unable to maintain channel activity when the 5-phosphate is selectively cleaved from PI(4,5)P2 (10). Furthermore, the high potency of PI(3,4,5)P3 cannot compensate for the fact that its maximum concentration is only about one-hundredth of that of PI(4,5)P2 (39); the same applies to PI(3,4)P2, which also activates M-channels (6, 7). Thus, within the phosphoinositides, our results agree with the conclusions of others in suggesting that PI(4,5)P2 is the specific physiological regulator (2, 40). The insensitivity of the channels to I(1,4,5)P3 at up to 300 μm also implies that this will not affect M-channel activity at the cytoplasmic concentrations likely to be generated following receptor-mediated hydrolysis of PI(4,5)P2 (up to ∼16 μm in neuroblastoma cells (41)).
Activation by LPA and S-1-P raises new possibilities. Although derived from abundant membrane lipids (phosphatidic acid and sphingosine), they are not retained in the membrane at high concentrations but are instead released into the extracellular solution, where they act primarily on specific G protein-coupled receptors (31). However, their ability to activate M-channels when applied to their inside face raises the interesting question as to whether they might have additional, more direct effects on these and other PI(4,5)P2-regulated membrane proteins when present in the cytoplasm.
In conclusion, our experiments reveal that the M-channel can be activated by a wide range of lipid phosphates, with a minimum requirement for an acyl chain of sufficient length and one or more phosphate headgroups; and within the phosphoinositides, that potency increases with increasing numbers of phosphates. Notwithstanding, they also accord with the view that the phosphoinositide PI(4,5)P2 is the primary endogenous phospholipid regulator.
Acknowledgments
We thank Dr. Mark S. Shapiro (Department of Physiology, University of Texas Health Science Center, San Antonio, TX) for the kind gift of the mutated Kv7.2 cDNA and Professor B. V. L. Potter (Medicinal Chemistry, Department of Pharmacy and Pharmacology, and Sterix Ltd., University of Bath, Bath, United Kingdom) for the gift of I(4,5)P2.
This work was supported by Grant 085419 from the Wellcome Trust and Grant RG/10/10/28447 from the British Heart Foundation.
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- diC8
- dioctanoyl
- Kv7.2C
- Kv7.2 C terminus
- MBP
- maltose-binding protein
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- I(1,4,5)P3
- inositol 1,4,5-trisphosphate
- S-1-P
- sphingosine 1-phosphate
- PG
- phosphatidylglycerol
- PC
- phosphatidylcholine
- LPA
- 1-oleoyl lysophosphatidic acid.
REFERENCES
- 1. Hilgemann D. W., Feng S., Nasuhoglu C. (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, re19. [DOI] [PubMed] [Google Scholar]
- 2. Gamper N., Shapiro M. S. (2007) Regulation of ion transport proteins by membrane phosphoinositides. Nat. Rev. Neurosci. 8, 921–934 [DOI] [PubMed] [Google Scholar]
- 3. Suh B. C., Hille B. (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 37, 175–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang H. S., Pan Z., Shi W., Brown B. S., Wymore R. S., Cohen I. S., Dixon J. E., McKinnon D. (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893 [DOI] [PubMed] [Google Scholar]
- 5. Suh B. C., Hille B. (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520 [DOI] [PubMed] [Google Scholar]
- 6. Zhang H., Craciun L. C., Mirshahi T., Rohács T., Lopes C. M., Jin T., Logothetis D. E. (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975 [DOI] [PubMed] [Google Scholar]
- 7. Li Y., Gamper N., Hilgemann D. W., Shapiro M. S. (2005) Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delmas P., Brown D. A. (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862 [DOI] [PubMed] [Google Scholar]
- 9. Brown D. A., Passmore G. M. (2009) Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suh B. C., Inoue T., Meyer T., Hille B. (2006) Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314, 1454–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hernandez C. C., Zaika O., Shapiro M. S. (2008) A carboxyl-terminal interhelix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas A. M., Harmer S. C., Khambra T., Tinker A. (2011) Characterization of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem. 286, 2088–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Main M. J., Cryan J. E., Dupere J. R., Cox B., Clare J. J., Burbidge S. A. (2000) Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 58, 253–262 [DOI] [PubMed] [Google Scholar]
- 14. Selyanko A. A., Hadley J. K., Brown D. A. (2001) Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J. Physiol. 534, 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Telezhkin V., Brown D. A., Gibb A. J. (2010) Stoichiometry of potassium M-channel activity by PIP2. Program 340.1, Poster F20, 2010 Neuroscience Meeting Planner, Society for Neuroscience, San Diego, CA [Google Scholar]
- 16. Thomas A. M., Brown S. G., Leaney J. L., Tinker A. (2006) Differential phosphoinositide binding to components of the G protein-gated K+ channel. J. Membr. Biol. 211, 43–53 [DOI] [PubMed] [Google Scholar]
- 17. Thomas A. M., Tinker A. (2008) Determination of phosphoinositide binding to K+ channel subunits using a protein-lipid overlay assay. Methods Mol. Biol. 491, 103–111 [DOI] [PubMed] [Google Scholar]
- 18. Osborne S. L., Meunier F. A., Schiavo G. (2001) Phosphoinositides as key regulators of synaptic function. Neuron 32, 9–12 [DOI] [PubMed] [Google Scholar]
- 19. Stansfeld P. J., Hopkinson R., Ashcroft F. M., Sansom M. S. (2009) PIP2-binding site in Kir channels: definition by multiscale biomolecular simulations. Biochemistry 48, 10926–10933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hansen S. B., Tao X., MacKinnon R. (2011) Structural basis of PIP2 activation of the classical inward-rectifier K+ channel Kir2.2. Nature 477, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia P., Gupta R., Shah S., Morris A. J., Rudge S. A., Scarlata S., Petrova V., McLaughlin S., Rebecchi M. J. (1995) The pleckstrin homology domain of phospholipase Cδ1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34, 16228–16234 [DOI] [PubMed] [Google Scholar]
- 22. Lemmon M. A., Ferguson K. M., O'Brien R., Sigler P. B., Schlessinger J. (1995) Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. U.S.A. 92, 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ju M., Shi J., Saleh S. N., Albert A. P., Large W. A. (2010) Ins(1,4,5)P3 interacts with PIP2 to regulate activation of TRPC6/C7 channels by diacylglycerol in native vascular myocytes. J. Physiol. 588, 1419–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manna D., Bhardwaj N., Vora M. S., Stahelin R. V., Lu H., Cho W. (2008) Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKCα C2 domain. J. Biol. Chem. 283, 26047–26058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yeung T., Gilbert G. E., Shi J., Silvius J., Kapus A., Grinstein S. (2008) Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 [DOI] [PubMed] [Google Scholar]
- 26. Logothetis D. E., Jin T., Lupyan D., Rosenhouse-Dantsker A. (2007) Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflugers Arch. 455, 83–95 [DOI] [PubMed] [Google Scholar]
- 27. Colquhoun D. (1998) Binding, gating, affinity, and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allan D., Cockcroft S. (1983) The fatty acid composition of 1,2-diacylglycerol and polyphosphoinositides from human erythrocyte membranes. Biochem. J. 213, 555–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McLaughlin S., Wang J., Gambhir A., Murray D. (2002) PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 [DOI] [PubMed] [Google Scholar]
- 30. Rohács T., Chen J., Prestwich G. D., Logothetis D. E. (1999) Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J. Biol. Chem. 274, 36065–36072 [DOI] [PubMed] [Google Scholar]
- 31. Ishii I., Fukushima N., Ye X., Chun J. (2004) Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73, 321–354 [DOI] [PubMed] [Google Scholar]
- 32. Rohács T., Lopes C. M., Jin T., Ramdya P. P., Molnár Z., Logothetis D. E. (2003) Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc. Natl. Acad. Sci. U.S.A. 100, 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tucker S. J., Baukrowitz T. (2008) How highly charged anionic lipids bind and regulate ion channels. J. Gen. Physiol. 131, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Santagata S., Boggon T. J., Baird C. L., Gomez C. A., Zhao J., Shan W. S., Myszka D. G., Shapiro L. (2001) G protein signaling through tubby proteins. Science 292, 2041–2050 [DOI] [PubMed] [Google Scholar]
- 35. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 36. McLaughlin S., Murray D. (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 [DOI] [PubMed] [Google Scholar]
- 37. Stephens L., McGregor A., Hawkins P. (2000) in Biology of Phosphoinositides (Cockcroft S., ed) pp. 32–130, Oxford University Press, Oxford [Google Scholar]
- 38. Willars G. B., Nahorski S. R., Challiss R. A. (1998) Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J. Biol. Chem. 273, 5037–5046 [DOI] [PubMed] [Google Scholar]
- 39. Stephens L. R., Hughes K. T., Irvine R. F. (1991) Pathway of phosphatidylinositol (3,4,5)-trisphosphate synthesis in activated neutrophils. Nature 351, 33–39 [DOI] [PubMed] [Google Scholar]
- 40. Suh B. C., Hille B. (2007) Regulation of KCNQ channels by manipulation of phosphoinositides. J. Physiol. 582, 911–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu C., Watras J., Loew L. M. (2003) Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell Biol. 161, 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]