

Background: Syndecan ectodomains shed by cells can enhance progression of cancer, inflammatory disease, and pathogen infection.
Results: Reducing the amount of heparan sulfate present on syndecan core proteins increases shedding of the ectodomain.
Conclusion: Heparan sulfate chains suppress syndecan shedding.
Significance: Therapeutic inhibition of heparan sulfate degradation could slow disease progression.
Keywords: Cancer, Heparan Sulfate, Proteoglycan, Proteoglycan Synthesis, Shedding, Heparanase
Abstract
Matrix metalloproteinases release intact syndecan-1 ectodomains from the cell surface giving rise to a soluble, shed form of the proteoglycan. Although it is known that shed syndecan-1 controls diverse pathophysiological responses in cancer, wound healing, inflammation, infection, and immunity, the mechanisms regulating shedding remain unclear. We have discovered that the heparan sulfate chains present on syndecan core proteins suppress shedding of the proteoglycan. Syndecan shedding is dramatically enhanced when the heparan sulfate chains are enzymatically degraded or absent from the core protein. Exogenous heparan sulfate or heparin does not inhibit shedding, indicating that heparan sulfate must be attached to the core protein to suppress shedding. Regulation of shedding by heparan sulfate occurs in multiple cell types, for both syndecan-1 and syndecan-4 and in murine and human syndecans. Mechanistically, the loss of heparan sulfate enhances the susceptibility of the core protein to proteolytic cleavage by matrix metalloproteinases. Enhanced shedding of syndecan-1 following loss of heparan sulfate is accompanied by a dramatic increase in core protein synthesis. This suggests that in response to an increase in the rate of shedding, cells attempt to maintain a significant level of syndecan-1 on the cell surface. Together these data indicate that the amount of heparan sulfate present on syndecan core proteins regulates both the rate of syndecan shedding and core protein synthesis. These findings assign new functions to heparan sulfate chains, thereby broadening our understanding of their physiological importance and implying that therapeutic inhibition of heparan sulfate degradation could impact the progression of some diseases.
Introduction
Syndecan-1 is known predominantly as a cell surface proteoglycan that regulates complex cell behaviors, such as adhesion, motility, invasion, and intracellular signaling. Syndecan-1 performs these diverse functions by acting as a co-receptor for a variety of growth factor receptors or as a binding partner to integrins (1, 2). However, syndecan-1 localization is not restricted to the cell surface. It is present in the nucleus where it regulates gene transcription (3), or it can be shed from the cell surface and become a soluble or insoluble component of the extracellular matrix (4, 5). Shed syndecan-1 retains its glycosaminoglycan chains (both heparan sulfate and chondroitin sulfate) (6) along with their bound ligands, which often include growth factors, chemokines, and other effectors that can endow the shed proteoglycans with both autocrine and paracrine signaling capacity.
Shed syndecan-1 plays important roles in the pathophysiology of many disease states, including inflammation, microbial pathogenesis, and cancer (4, 5, 7). For example, in a murine model of acute lung injury, syndecan-1 mediates the inflammatory response by facilitating the formation of a CXC chemokine gradient to enhance transepithelial migration of neutrophils (8). In pulmonary fibrosis, reactive oxygen species enhance syndecan-1 shedding, which leads to induction of neutrophil chemotaxis, impaired epithelial wound healing, and increased fibrosis (9).
Shed syndecan-1 also plays an important role in microbial pathogenesis and host defense (4). For example, Pseudomonas aeruginosa induces syndecan-1 shedding through LasA, an enzyme that activates MMP2-mediated shedding of syndecan-1 (10). The shed syndecan-1 inhibits host-derived antimicrobial peptides and thus is a component of a virulence mechanism that promotes infection (11).
In cancer, shed syndecan-1 plays an active role in driving tumor progression (5). In a model of breast cancer, syndecan-1 shed from the surface of reactive stromal fibroblasts stimulates signaling and proliferation in adjacent tumor cells (12). Syndecan-1 shed by myeloma cells enhances angiogenesis, osteolysis, growth, and spontaneous metastasis of tumor cells in vivo (13–15). Shed syndecan-1 can also be measured in the serum of patients with some cancers, including lung cancer and myeloma, where high levels of shed syndecan-1 correlate with poor outcome of the patient (16, 17).
Although the mechanisms mediating syndecan-1 shedding from the cell surface are not completely understood, shedding occurs via proteolytic cleavage of the core protein close to the cell membrane. Shedding of syndecan-1 occurs constitutively or can be accelerated in response to various agonists (e.g. growth factors, chemokines, microbial toxins, insulin, or cellular stress). These diverse stimuli set off a cascade of molecular events that include signal transduction, intracellular regulators, and, finally, protease activity at the cell surface that cleaves the core protein and releases syndecan-1 (4, 5). Intracellularly, activation of protein tyrosine kinases leads to downstream phosphorylation of the syndecan-1 cytoplasmic domain, an event that may enhance syndecan-1 shedding (18, 19). In addition, shedding agonists cause dissociation of Rab5 from the syndecan-1 cytoplasmic domain, which triggers ectodomain shedding (20). The actual cleavage of syndecan-1 from the cell surface is accomplished by various metalloproteinases, including MMPs, membrane type MMPs, and ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) (5).
We previously demonstrated that the action of heparanase, an enzyme that cleaves heparan sulfate chains, stimulates increased syndecan-1 shedding (21). We found that increased shedding is due at least in part to heparanase-mediated up-regulation of expression of MMP-9 and urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor, two enzymes known to be sheddases of syndecan-1 (22). In addition to up-regulating proteases, heparanase trims heparan sulfate chains of syndecan-1, resulting in a significantly smaller amount of heparan sulfate present on the proteoglycan. However, a role for such reduction in heparan sulfate content leading to shedding has never been tested. In the present study, we demonstrate that this reduction in heparan sulfate amount dramatically accelerates the rate of syndecan-1 shedding and that this can be blocked by inhibitors of MMPs. Thus, heparan sulfate present on syndecan-1 acts to suppress shedding of the ectodomain, possibly by interfering with MMP-mediated cleavage of the core protein. This suppression occurs in multiple cell types, in both human and murine species and for syndecan-4 as well as syndecan-1. We also find that in response to enhanced shedding there is a dramatic increase in synthesis of the core protein of syndecan-1. These data provide new insight into a previously unknown role for the heparan sulfate chains of syndecans.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
Cell lines CAG, RPMI-8226, ARH-77, and MPC-11 were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. U266 cells were grown in RPMI 1640 medium supplemented with 15% fetal bovine serum. RPMI-8226, ARH-77, and U266 cells were obtained from the American Type Culture Collection (Manassas, VA). Cell lines Panc-1 (a kind gift from Dr. Lacey McNally, University of Alabama at Birmingham), MDA-MB-231 (ATCC), and HeLa (a kind gift from Dr. Selvarangan Ponnazhagan, University of Alabama at Birmingham) were cultured in DMEM supplemented with 10% FBS. Murine mammary epithelial cell line, NMuMG, cells were grown in DMEM supplemented with 10% FBS and insulin (23). Bacterial heparinase III (Hep III) and chondroitinase ABC (Chase-ABC) were purchased from Seikagaku (Kogyo, Japan); phorbol 12-myristate 13-acetate, cycloheximide, cytochalasin, heparin, and purified bovine kidney heparan sulfate were from Sigma-Aldrich and inhibitors BB-94, U0126, bisindolylmaleimide; and genistein was from Calbiochem.
Constructs and Transfections
The full-length human syndecan-1 gene in vector pcDNA3.1(−) (Invitrogen) was used as the template to create syndecan-1 with a tobacco etch virus cleavage and C-terminal His10 tag and the mutated form of syndecan-1 with the same tag. Mutagenesis was carried out with the Change-It kit (USB/Agilent, Palo Alto, CA), following the protocols therein, because it allows the incorporation of multiple mutations in a single round of PCR. Phosphorylated insertion mutagenic oligonucleotides were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA), and the phosphorylated substitution mutagenic oligonucleotides were synthesized by Invitrogen. The phosphorylated mutagenic insertion primer for the tag was 5′-phospho-caccaaacaggaggaattctatgccGGTTCTGGTTCTGGTTCTGAAAACCTGTATTTTCAGGGCGGTTCTCATCATCACCATCACCATCACCACCATCACtgacgcaagcttaagtttaaaccgc-3′, with the complete insertion sequence capitalized. The tag is followed immediately by a termination codon. Potential glycosylation sites at amino acid positions 37, 43, 45, 47 206, 216, and 231 were replaced with alanine using the following primers in various combinations to incorporate all changes: S37AFor, 5′-phospho-gatcaagatggcGCTggggatgactc-3′; S37ARev, 5′-phospho-gagtcatccccAGCgccatcttgatc-3′; S206AFor, 5′-phospho-gcagcagagggcGCTggggagcagg-3′; S206ARev, 5′-phospho-cctgctccccAGCgccctctgctgc-3′; S216AFor, 5′-phospho-cctttgaaaccGCGggggagaatacg-3′; S216ARev, 5′-phospho-cgtattctccccCGCggtttcaaagg-3′; N231AFor, 5′-phospho-cctgaccgccggGCCcagtccccag-3′; N231ARev, 5′-phospho-ctggggactgGGCccggcggtcagg-3′, S43A/S45A/S47AFor 5′-phospho-gatgactctgacGCCttcGCCggcGCAggtgcagg-3′; and S43A/S45A/S47ARev, 5′-phospho-cctgcaccTGCgccGGCgaaGGCgtcagagtcatc-3′. Sequences were confirmed after each round of PCR by generating DNA with a miniplasmid kit (Qiagen, Santa Clarita, CA). The plasmids encoding for human wild type SDC1 (syndecan-1) sequence and the sequence for SDC1 mutated at glycosylation sites are referred to as WT hSDC1 and ΔGAG hSDC1, respectively. The murine full-length syndecan-1 gene in pcDNA3.1(−) vector and the TDM clone have been described previously (24). TDM refers to a triple deletion mutant (S43A/S45A/S47A) having all heparan sulfate attachment sites removed. The TDM clone was used as the template for additional alanine substitutions at amino acid residues 43, 207, and 217. A FLAG tag with a tobacco etch virus cleavage site was inserted toward the N terminus, after the signal sequence. Phosphorylated mutagenic oligonucleotides for substitution mutagenesis were synthesized by Invitrogen, and the FLAG tag insertion oligonucleotide was synthesized by Integrated DNA Technologies, Inc. The sequence of the phosphorylated mutagenic insertion primer, FlagNFor, was 5′-phospho-gctctgcgcgctggcgctgcgcctgGATTATAAGGATGACGATGACAAAggttctGAAAACCTGTATTTTCAGGGCcagcctgccctcccgcaaattgtgg-3′, with the FLAG and tobacco etch virus sites in capital letters. Potential glycosylation sites at amino acid positions 43, 207, and 217 were replaced with alanine (shown in capital letters) using the following primers in various combinations to incorporate all changes: mS43A For, 5′-phospho-ggatgactctgacGCCttcgcgggcgcgg-3′; mS43ARev, 5′-phospho-ccgcgcccgcgaaGGCgtcagag tcatcc-3′; mS207AFor, 5′-phospho-gcaggagagggcGCTggagaacaagac-3′; mS207ARev, 5′-phospho-tcttgttctccAGCgccctctcctgc-3′; mS217AFor, 5′-phospho-acctttgaaacaGCTggggagaacacag-3′; and mS217ARev, 5′-phospho-ctgtgttctccccAGCtgtttcaaaggt-3′. Sequences were confirmed after each round of PCR by generating DNA with a miniplasmid kit (Qiagen). DNA was confirmed by the University of Alabama, Birmingham, CFAR DNA Sequencing and Analysis Core Facility. The plasmids encoding murine wild type syndecan-1 sequence and the sequence for syndecan-1 mutated at glycosylation sites will be referred to as WT mSDC1 and ΔGAG mSDC1, respectively.
Prior to transfection of constructs, ARH-77 (2 × 106) cells were plated in 5 ml of fresh complete medium and grown for 24 h. The cells were then transfected with human plasmid or murine WT or ΔGAG SDC1 plasmid using Amaxa NucleofectorTM (Amaxa, Cologne, Germany) as per the manufacturer's instructions. The transfected cells were then selected with Geneticin (G418, Invitrogen), for 7 days. The selected cells were stained for cell surface syndecan-1 expression and sorted by fluorescence-activated cell sorting (as described below) to sort isolate cells expressing similar levels of either the WT or ΔGAG SDC1 protein. The sorted cells were grown for 7 days, and the stable expression and levels of cell surface syndecan-1 were confirmed again by flow cytometry.
Flow Cytometry and Fluorescence-activated Cell Sorting (FACS)
For FACS or flow cytometry, cells were collected by centrifugation and washed twice in phosphate-buffered saline (PBS). Cell pellets were then resuspended in anti-human syndecan-1 (clone BA-38, Cell Sciences, Canton, MA), or anti-mouse syndecan-1 monoclonal antibody (clone 281.2 (25)) diluted in PBS and incubated at 4 °C for 1 h. The cells were then washed three times in PBS and incubated with secondary antibody conjugated to Alexa Fluor 647 (Invitrogen) at 4 °C for 1 h. Following incubation, the cells were washed three times in PBS and then resuspended in PBS. For flow cytometry, after staining, cells were fixed in 4% paraformaldehyde prior to analysis. Cells stained with appropriate isotype-matched IgG antibodies served as a gating control.
Western Blotting
Cells in culture were pelleted by centrifugation and washed with PBS twice. Cell pellets were then resuspended in lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100) containing 10 μg/ml leupeptin and 1× HALT protease inhibitor mixture and incubated on ice for 30 min. Lysates were centrifuged at 12,000 × g at 4 °C for 15 min, and the supernatants were removed from the pellets. Proteins isolated from cell lines in the supernatant were quantified by a BCA protein assay reagent kit (Pierce). Equal amounts of protein were loaded onto 4–20% gradient SDS-polyacrylamide gels (Bio-Rad), transferred to a positively charged nylon membrane (Nytran SPC, Schleicher & Schuell), and probed with goat anti-human syndecan-1 (R&D Systems, Minneapolis, MN) followed by horseradish peroxidase-conjugated secondary anti-mouse antibody (GE Healthcare). The levels of actin (loading control) were probed using a β-actin primary antibody (Sigma-Aldrich). Immunoreactive bands were detected using enhanced chemiluminescence (GE Healthcare).
Preparation of Conditioned Media
In all experiments, unless mentioned otherwise, cells were seeded and incubated in serum-free RPMI medium for 1 h prior to the addition of Hep III and/or Chase-ABC. At the end of incubation, cells and cellular debris were separated by centrifuging the conditioned media at 1000 rpm for 5 min, and the supernatants were analyzed either by dot blotting or ELISA. For adherent cells (MDA-MB-231, Panc-1, HeLa, and NMuMG), equal numbers were seeded in complete growth medium in a 12-well plate. After overnight incubation, the monolayers were washed with serum-free medium at least twice prior to enzyme treatments.
Dot Blotting
Conditioned medium collected from (a) ARH-77 cells expressing either human or murine wild-type or mutated syndecan-1, (b) NMuMG cells after different treatments, and (c) HeLa cells after glycosidase treatments was processed prior to analyses by dot blotting. The conditioned media were treated with Hep III (1.25 milliunits/ml) and Chase-ABC (12.5 milliunits/ml) at 37 °C for 4 h. This enzymatic digestion ensures that all of the glycosaminoglycan chains from the shed syndecan-1 were removed prior to analyses, and therefore an increase in shedding was determined only by comparing the differences in the level of syndecan-1 core protein between different samples. After processing, the indicated amounts of the conditioned media were blotted onto nitrocellulose membrane using an immunodot apparatus (Milliblot D, Millipore, Bedford, MA). The membranes were then fixed in Tris-buffered saline containing 0.05% glutaraldehyde for 30 min at room temperature. After washes in distilled water and TBS, the membranes were blocked in TBS containing 3% nonfat dry milk, 0.5% BSA, and 0.3% Tween 20 (blocking solution) for 1 h. The membranes were incubated overnight with primary antibodies against murine or human syndecan-1 in the blocking solution. After washes in TBS containing 0.05% Tween 20 (washing solution), membranes were probed with horseradish peroxidase-conjugated secondary anti-mouse antibody (GE Healthcare), and immunoreactive bands were detected by chemiluminescence.
RNA Extraction and Real-time PCR for Human Syndecan-1
5 × 105 CAG cells were seeded in complete growth medium at 37 °C and 5% CO2. Cells were then treated with Hep III (1.25 milliunits/ml) added every 2 h for the next 8 h. Cells treated with water served as a control. At the end of the incubation, cells were washed once with PBS, and RNA was extracted using RNeasy columns (Qiagen, Valencia, CA). One microgram of total RNA was reverse transcribed to cDNA using random hexamer primers and the Maxima® First Strand cDNA synthesis kit (Fermentas, Hanover, MD) as per the manufacturer's protocol. 25 ng of diluted cDNA was mixed with 2× IQTM SYBR® green supermix (Bio-Rad) along with gene specific primers (200 nm final concentration). The primers used were 5′-TCTGACAACTTCTCCGGCTC-3′ (forward) and 5′-CCACTTCTGGCAGGACTACA-3′ (reverse) for syndecan-1 and 5′-CGGCGACGACCCATTCGAAC-3′ (forward) and 5′-GAATCGAACCCTGATTCCCCGTC-3′ (reverse) for 18S rRNA. The cycle parameters included initial denaturation at 95 °C for 3 min followed by 45 cycles of 95 °C for 10 s and 55 °C for 60 s, followed by one cycle of 95 °C for 60 s and 55 °C for 60 s. To ensure specific amplification, a melt curve was generated at the end of PCR for each sample. The PCR cycle at which the fluorescence exceeded a set threshold (CT), for each sample was determined by the iCycler software. Data were analyzed according to the comparative CT method, as described previously (26), using internal control (18S rRNA) transcript levels to normalize differences in sample loading and preparation.
Syndecan-1 ELISA
The levels of shed syndecan-1 accumulated in the conditioned media after different treatments were assessed by an ELISA using an Eli-pair kit specific for human syndecan-1 core protein (Cell Sciences). The standard curve was linear between 8 and 256 ng/ml, and all samples were diluted to concentrations within that range. All of the samples were run in duplicate.
RESULTS
Syndecan-1 Shedding Is Enhanced by Reducing Amount of Heparan Sulfate Present on Core Protein
We have previously demonstrated that syndecan-1 shedding by myeloma cells can be enhanced by increasing endogenous heparanase expression or by treating cells with exogenous heparanase or bacterial Hep III (21). These results suggest that reducing the amount of heparan sulfate present on syndecan-1 enhances shedding of the proteoglycan. To explore this further, equal numbers of CAG cells were treated with increasing amounts of Hep III enzyme to generate cells having syndecan-1 containing high, intermediate, or low levels of heparan sulfate. After treatment with enzyme for 4 h, the cells were harvested, and conditioned medium was collected. Western blots of the cell extracts revealed that with an increase in the amount of Hep III enzyme, there was a decrease in the amount of heparan sulfate present on syndecan-1. This decrease in amount of heparan sulfate is visualized on the Western blots as an increase in syndecan-1 polydispersity and a reduction in the mean molecular size of the proteoglycan (Fig. 1A; note the broadening of the syndecan-1 smear (polydispersity) following treatment with increasing amounts of Hep III and the clear emergence of a low molecular mass fraction of syndecan-1 around 80–95 kDa in cells treated with 2.5 milliunits/ml of Hep III). Although in the Western blot there appear to be much higher levels of syndecan-1 present following Hep III digestion (Fig. 1A, compare lanes 1 and 5), quantification of syndecan-1 by ELISA reveals that similar levels of syndecan-1 are present in cell extracts before and after Hep III treatment.3 We speculate that on Western blots, the high molecular mass form of syndecan-1 either transfers poorly to the membrane or is not as accessible to the anti-syndecan-1 antibody as is the low molecular mass form, thus giving the false impression that the amount of syndecan-1 in the cell fraction increases following Hep III treatment.
FIGURE 1.

Reducing the amount of heparan sulfate by enzymatic digestion enhances syndecan-1 shedding from cells. CAG cells were plated at a density of 106 cells/ml in serum-free medium for 2 h, and then the individual wells were treated with different amounts of Hep III enzyme for 4 h or with heat-inactivated Hep III enzyme as a control. At the end of incubation, the cells and conditioned media were collected and analyzed separately. A, cell lysates were analyzed by immunoblotting for syndecan-1. β-Actin was probed in the same blots to demonstrate equal loading of the total protein. B, conditioned media were analyzed for their level of shed SDC1. Data are mean ± S.E. (error bars) of three independent experiments. *, p < 0.005 versus untreated controls. C, CAG cells were treated with Hep III for 8 h, and the level of syndecan-1 was determined by real-time PCR. Control included CAG cells treated with distilled water. Transcript levels were normalized against 18S rRNA levels. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus control.
Quantification of soluble syndecan-1 in the medium conditioned by these cells revealed that with the decrease in proteoglycan size, there was a corresponding increase in syndecan-1 shedding (Fig. 1B). Inactive Hep III, lacking the ability to degrade heparan sulfate, failed to affect syndecan-1 shedding, indicating that the enzymatic activity of Hep III and the consequent decrease in heparan sulfate content are responsible for the increase in shedding (Fig. 1, A and B). In separate experiments, it was confirmed that Hep III treatment of syndecan-1 did not alter the ability to accurately quantify syndecan-1 levels by ELISA.3 In addition, Hep III-induced syndecan-1 shedding was confirmed by dot blotting using an antibody different from the one used in ELISA quantification.3
Together the results indicated that upon Hep III treatment, the amount of syndecan-1 present in the cell fraction remained constant, but there was a dramatic increase in syndecan-1 shedding. This suggested that syndecan-1 synthesis was elevated following Hep III treatment. To confirm this, we tested whether a sustained enzymatic digestion of heparan sulfates over time was accompanied by an enhanced synthesis of syndecan-1. Results revealed that there was a significant increase in syndecan-1 synthesis (Fig. 1C), thereby indicating that the amount of heparan sulfate present on the syndecan-1 core protein regulates the rate of core protein synthesis.
We next assessed whether cell lines that express either high molecular weight or low molecular weight forms of syndecan-1 differed in their rate of shedding. When an equal number of U266, RPMI-8226, and CAG cells were seeded and grown in serum-free medium for 24 h, U266 cells shed a significantly higher level of syndecan-1 than did the other two cell lines (Fig. 2A). Western blots of cell extracts revealed that syndecan-1 from U266 cells ran as a broad smear ranging from ∼100 kDa to well over 260 kDa, whereas syndecan-1 from RPMI-8226 and CAG myeloma cells was much less polydisperse and considerably larger in mean molecular mass than syndecan-1 from U266 cells (Fig. 2B). Because the polydispersity in syndecan-1 size is due predominantly to heterogeneity in the size and number of heparan sulfate chains, these results indicate that the overall heparan sulfate content in U266 cell syndecan-1 is less than that in RPMI-8226 and CAG cells. The high rate of syndecan-1 shedding by U266 cells as compared with RPMI-8226 and CAG cells was not due to a higher level of cell surface syndecan-1 on the U266 cells (Fig. 2C); nor was it due to differences in rates of proliferation, because the three cell lines grew at similar rates in serum-free medium.3 Consistent with the finding in the myeloma cell lines, we also found when assessing murine cell lines that MPC-11 myeloma cells that have a low molecular mass form of syndecan-1 shed the proteoglycan at a higher rate than do NMuMG cells that have a high molecular mass form of syndecan-1 (see Fig. 5). Together, the results from assessing different cell lines and from treating cells with Hep III (as described in Fig. 1) point to an important role for heparan sulfate in regulating shedding of syndecan-1. Retention of high levels of heparan sulfate on the core protein suppresses syndecan-1 shedding, whereas removal of heparan sulfate enhances syndecan-1 shedding.
FIGURE 2.

U266 myeloma cells shed more syndecan-1 than RPMI-8226 or CAG myeloma cells. A, 106 cells/ml of U266, RPMI-8226, or CAG myeloma cells were seeded and grown in serum-free media for 24 h. The level of shed syndecan-1 in the conditioned medium from each cell line was determined by ELISA. Data are mean ± S.E. (error bars) of three independent experiments. *, p < 0.01 versus U266 cells. B, equal protein from total cell extracts of the three myeloma cell lines was probed for syndecan-1 and β-actin by immunoblotting. C, the level of cell surface expression of syndecan-1 on each cell line was determined by staining with BA38 antibody and analyzed by flow cytometry: IgG control antibody (shaded), U266 cells (dotted line), RPMI-8226 cells (solid line), CAG cells (dashed line).
FIGURE 5.

Shedding of murine syndecan-1 is enhanced by heparan sulfate removal. A, equal numbers of murine MPC-11 myeloma cells and murine NMuMG epithelial cells were seeded in serum-free medium for 24 h, and the level of shed syndecan-1 was determined by dot blotting with monoclonal antibody 281.2. B, equal numbers of NMuMG cells were seeded in serum-free medium and treated with either Hep III (1.25 milliunits/ml) or Chase-ABC (12.5 milliunits/ml) for 24 h. Untreated monolayers served as a control. After incubation, the conditioned medium was harvested, and the level of shed syndecan-1 was determined by dot blotting. The experiments were repeated at least three times, and a representative dot blot is shown. C, ARH-77 cells were transfected with a cDNA encoding either wild-type murine syndecan-1 or mutated syndecan-1 lacking heparan sulfate chain attachment sites. Stable expression of cell surface syndecan-1 was confirmed by flow cytometry by staining with 281.2 antibody: isotype IgG control (black line), WT mSDC1 (dotted line), and ΔGAG mSDC1 (dashed line). D, equal numbers of WT mSDC1 or ΔGAG mSDC1 cells were seeded in serum-free medium for 24 h, the conditioned medium was harvested, and the level of shed syndecan-1 was determined by dot blotting. The experiments were repeated at least three times, and a representative dot blot is shown.
Heparan Sulfates Suppress Syndecan Shedding across Multiple Cell Lineages
Cell lines belonging to different lineages (for myeloma, CAG, U266, and RPMI-8226; for breast cancer, MDA-MB 231; for pancreatic cancer, Panc-1; and for cervical cancer, HeLa) were treated with Hep III, and the effect on syndecan-1 shedding was evaluated. In all cell lines, reducing the amount of heparan sulfate chains caused an up-regulation of syndecan-1 shedding by severalfold, ranging from ∼3-fold in U266 cells to ∼10-fold in HeLa cells compared with untreated cells (Fig. 3, A and B). Because syndecan-1 also bears chondroitin sulfate chains (27), we treated cells with Chase-ABC to determine if this would alter shedding. Reducing chondroitin sulfate levels impacted syndecan-1 shedding in only three (U266, CAG, and Panc-1) of the six cell lines tested (Fig. 3, A and B). Even in these three cell lines, the maximal effect seen with CAG cells (less than 2-fold increase over untreated controls) was considerably less than levels induced by Hep III treatment in the same cell line (Fig. 3A). Thus, between the two types of glycosaminoglycans present on syndecan-1, heparan sulfates are the major regulators of syndecan-1 shedding.
FIGURE 3.

Heparan sulfate suppression of syndecan-1 shedding is not restricted to myeloma cells. Shown is shedding of syndecan-1 in response to reduction of glycosaminoglycans in tumor cell lines of different origins. A, 106 cells/ml of myeloma cell lines (U266, RPMI-8226, and CAG) were seeded in serum-free medium for 6 h in the presence of either Hep III (1.25 milliunits/ml) or Chase-ABC (12.5 milliunits/ml) separately. The level of shed syndecan-1 in the conditioned medium was determined by an ELISA. Untreated cells for each cell line tested served as the control. Data are mean ± S.E. (error bars) of three independent experiments. *, p < 0.01 versus untreated control for each individual cell line tested; **, p < 0.05 versus untreated control for each individual cell line tested. B, 0.5 × 106 cells of tumor cells of different origins: MDA-MB-231 (breast cancer), Panc-1 (pancreatic cancer), and HeLa (cervical cancer) were seeded separately in serum-free medium for 24 h in the presence of either Hep III (1.25 milliunits/ml) or Chase-ABC (12.5 milliunits/ml). Untreated cells of each cell line were included as a control. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined by ELISA. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated controls. C, shedding of syndecan-4 is elevated upon reduction of the amount of heparan sulfate. 0.5 × 106 HeLa cells were seeded in serum-free medium in the presence of either Hep III (1.25 milliunits/ml) or Chase-ABC (12.5 milliunits/ml) for 16 h. Untreated cells served as a control. The designated amounts of treated conditioned medium were probed by dot blotting for shed syndecan-4. The experiment was repeated at least three times, and a representative blot is shown. The blot reflects a greater than 2-fold increase in shed syndecan-4 upon treatment with Hep III compared with untreated controls.
To test whether reduction in heparan sulfate amount elevated shedding in other members of the syndecan family, the effect of Hep III treatment on syndecan-4 was examined. Among the cell lines, HeLa cells had detectable levels of syndecan-4 and were therefore used to address this question. Similar to syndecan-1, reducing the amount of heparan sulfate, not chondroitin sulfate, elevated the rate of syndecan-4 shedding (Fig. 3C).
A Mutant Form of Human Syndecan-1 Lacking GAG Chains Is Shed at High Rate
As an independent confirmation of the effect of GAGs on syndecan-1 shedding, we stably expressed either the wild-type human syndecan-1 (WT hSDC1) or mutated form of human syndecan-1 lacking all glycosylation attachment sites including GAG attachment sites (ΔGAG hSDC1; serine to alanine mutations at residues at 37, 45, 47, 206, and 216). These constructs were expressed in ARH-77 cells, an Epstein-Barr virus-transformed human B lymphoblastoid cell line that lacks endogenous expression of syndecan-1 (21). Stable transfectants expressing similar levels of WT hSDC1 and ΔGAG hSDC1 protein on the cell surface were selected and confirmed by flow cytometric analyses (Fig. 4A). Both transfectant lines proliferated at similar rates.3 Strikingly, the cells expressing GAG-less syndecan-1 shed ∼6-fold higher levels of syndecan-1 compared to the cells expressing wild-type syndecan-1 (Fig. 4B), mirroring the effects seen with enzymatic removal of GAGs. As a control, treating ARH-77 cells expressing WT hSDC1 cells with Hep III substantially increased the rate of syndecan-1 shedding, whereas treatment of cells with Chase-ABC had much less impact (Fig. 4C). Together, these findings demonstrate that shedding following Hep III treatment is not due to an induction of shedding by the enzyme; rather it is due to the decrease in the amount of heparan sulfate present on the core protein. Moreover, the results indicate that the default state of the syndecan-1 core protein is to be shed rapidly from the cell surface and that this shedding is suppressed when sufficient heparan sulfate chains are attached to the core protein.
FIGURE 4.
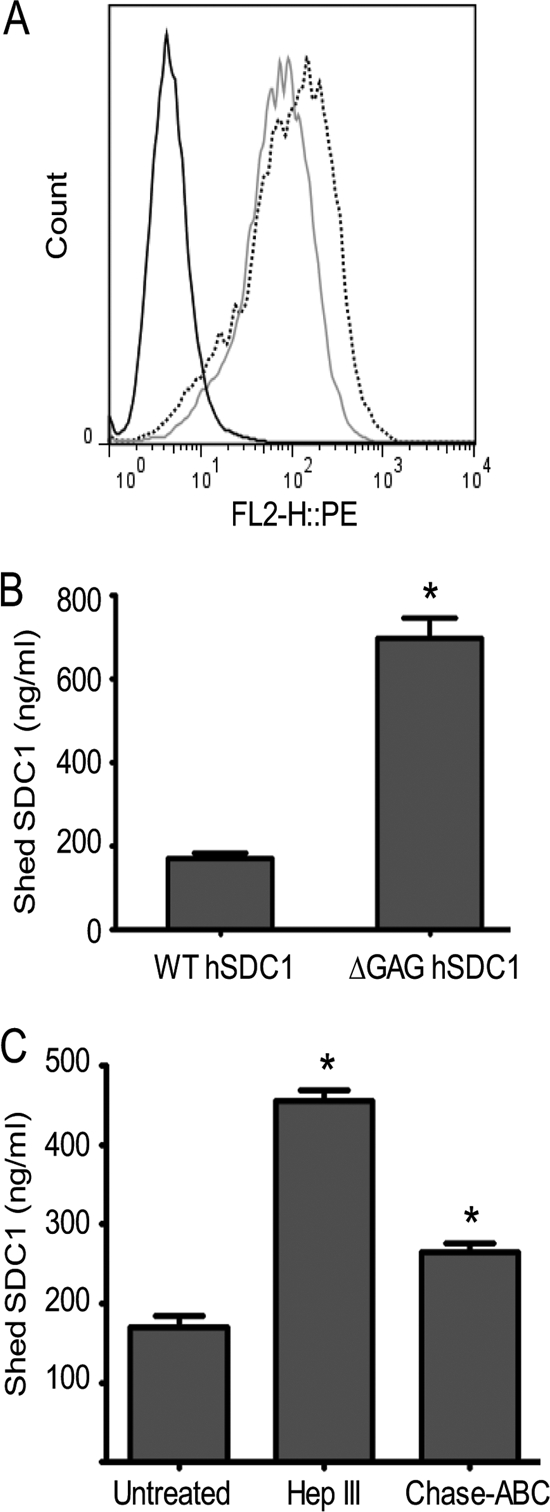
A mutated form of human syndecan-1 lacking glycosaminoglycan chains is shed at a much higher rate than wild-type syndecan-1. A, ARH-77 cells were transfected with a cDNA encoding either wild-type human syndecan-1 or mutated syndecan-1 lacking glycosaminoglycan attachment sites. Stable expression of cell surface syndecan-1 was confirmed by flow cytometry following staining with BA38 antibody: IgG control (black line), WT hSDC1 (gray line), and ΔGAG hSDC1 (dotted line). B, equal numbers of WT hSDC1 or ΔGAG hSDC1 cells were seeded in serum-free medium for 24 h. The conditioned media were harvested, and levels of syndecan-1 were assessed by ELISA. Data are mean ± S.E. (error bars) of three independent experiments. *, p < 0.0005 versus WT hSDC1 cells. C, WT hSDC1 cells were treated with either Hep III (1.25 milliunits/ml) or Chase-ABC (12.5 milliunits/ml) for 24 h. After incubation, the conditioned media were harvested, and the level of shed syndecan-1 was assessed by ELISA. Data are mean ± S.E. of three independent experiments. *, p < 0.01 versus untreated control.
Heparan Sulfate-regulated Shedding of Syndecan-1 Is Not Species-specific
In a previously published study (28), we demonstrated that syndecan-1 from a murine plasmacytoma cell line (MPC-11) had a low level of glycosaminoglycan content compared with syndecan-1 from normal murine mammary epithelial cells (NMuMG). To ascertain whether heparan sulfates regulate murine syndecan-1 shedding, we compared the rate of shed syndecan-1 between these two cell lines using dot blots probed with monoclonal antibody to murine syndecan-1. Results revealed that MPC-11 cells shed syndecan-1 at significantly higher rates compared with NMuMG cells (Fig. 5A). Additionally, the removal of heparan sulfates by Hep III treatment dramatically up-regulated shedding of murine syndecan-1 from NMuMG cells, whereas chondroitinase treatment failed to affect murine syndecan-1 shedding compared with untreated controls (Fig. 5B). This result demonstrates that heparan sulfate can regulate shedding in murine cells and, importantly, also in non-cancerous cells. As a further confirmation of the role of heparan sulfate in murine syndecan-1 shedding, we expressed either the murine wild-type syndecan-1 (WT mSDC1) or a mutated syndecan-1 lacking the GAG chains (ΔGAG mSDC1) in ARH-77 cells. The stable expression of syndecan-1 was confirmed and was found to be similar between the transfectants by flow cytometry (Fig. 5C). Similar to our observations with human syndecan-1, cells expressing ΔGAG mSDC1 shed higher levels of syndecan-1 compared with cells expressing wild-type syndecan-1 (Fig. 5D).
Heparan Sulfates Suppress MMP-mediated Shedding of Syndecan-1
Various agonists, such as phorbol ester 12-myristate 13-acetate (PMA), insulin, growth factors, and chemokines, can stimulate syndecan-1 shedding through activation of diverse molecular pathways involving extracellular signal-regulated kinase (ERK) (29), MAPKs (19), protein kinase C (PKC) (19, 29), protein-tyrosine kinases (23, 31), or Rab5 signaling (20). To determine if removal of heparan sulfate by Hep III enhances shedding by any of these pathways, CAG cells were incubated in the presence of specific inhibitors for individual pathways prior to Hep III treatment (Fig. 6A). The concentrations of inhibitors used in the current study have been reported in previously published studies (29–31). Cells treated with Hep III in the presence of DMSO served as a control. Blocking PKC activation or MEK activity had no significant effect on the level of syndecan-1 shedding (Fig. 6A). Inhibition of receptor tyrosine kinase signaling by genistein caused a modest (∼30%) but significant decrease in shedding (Fig. 6A). Blocking actin polymerization or protein synthesis failed to block the shedding induced by removal of heparan sulfate by Hep III in this short term assay.
FIGURE 6.

Heparan sulfate chains on syndecan-1 limit metalloproteinase-mediated shedding of syndecan-1. A, CAG cells were incubated in the presence of different inhibitors for 2 h in serum-free medium and then treated with Hep III (1.25 milliunits/ml) for 4 h, and the levels of shed syndecan-1 were determined by ELISA. Data are mean ± S.E. (error bars) of three independent experiments. *, p < 0.05 versus cells treated with Hep III in the presence of DMSO. The pathways inhibited and the specific inhibitors and their concentrations used were as follows: RTK signaling (genistein, 37 μm), MEK activity (U0126, 1 μm), PKC (bisindolylmaleimide, 1 μm), metalloproteinase activity (BB-94, 5 μm), actin polymerization (cytochalasin D, 10 μm), and protein synthesis (cycloheximide, 0.3 mm). B, equal numbers of WT hSDC1 or ΔGAG hSDC1 cells were seeded in serum-free medium for 24 h. Experimental groups also included ΔGAG hSDC1 cells incubated in the presence of either purified heparan sulfate, heparin, or BB-94 for 24 h. The level of shed syndecan-1 after 24 h was assessed by ELISA. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated ΔGAG hSDC1 cells. C, Panc-1 cells were seeded at equal density in serum-free medium and were treated with either Hep III (1.25 milliunits/ml) or with Hep III and DMSO for 4 h. Treatment groups also included Hep III-treated cells incubated in the presence of PMA (0.5 μm) or with PMA and BB-94 (5 μm) for the next 2 h. Cells treated with either PMA or BB-94 alone served as additional controls. Data are mean ± S.E. of three independent experiments. *, p < 0.05 versus groups treated with PMA alone. #, p < 0.01 versus groups treated with Hep III alone.
MMPs like MMP-7, MMP-9, and MMP-14 are known to regulate syndecan-1 shedding by proteolytic cleavage of the core protein (32–34). To test the possibility that heparan sulfates regulate MMP-mediated proteolysis, we utilized BB-94, a broad based MMP inhibitor. The addition of BB-94 dramatically inhibited Hep III-induced syndecan-1 shedding (∼3-fold decrease) in CAG cells (Fig. 6A). Confirming these results in another model system, the addition of BB-94 significantly reduced shed syndecan-1 from ARH-77 cells expressing GAG-less syndecan-1 (ΔGAG hSDC1) (Fig. 6B). This finding provides strong evidence that heparan sulfate present on syndecan-1 suppresses MMP-mediated shedding. Importantly, the addition of either purified heparan sulfate or heparin (100 μg/ml) failed to inhibit syndecan-1 shedding from these cells (Fig. 6B), demonstrating that not merely the presence of heparan sulfates but their localization on the syndecan-1 core protein is critical to suppress syndecan-1 shedding.
We next used a different model to verify that heparan sulfate chains of syndecan-1 inhibit core protein shedding. Panc-1 cells were treated with PMA, an agonist that has been shown to dramatically increase MMP-mediated syndecan-1 shedding (19). We hypothesized that in this model, removal of heparan sulfates by Hep III would dramatically enhance the effect of PMA on syndecan-1 shedding. As predicted, the addition of PMA to Hep III-treated cells significantly increased levels of shed syndecan-1 compared with cells treated with either Hep III alone or PMA alone (Fig. 6C). Further, this elevated shedding by combination treatment with Hep III and PMA was blocked upon the addition of BB-94 (Fig. 6C).
DISCUSSION
The importance of shed syndecan-1 in human disease has been well documented by studies showing its role in cancer progression, wound healing, inflammation, and infection (4, 5). Although the mechanisms regulating shedding are not fully understood, it is known that agonist-induced signaling up-regulates syndecan shedding via the action of MMPs. In the present work, we provide evidence for a mechanism that suppresses syndecan shedding. This occurs via the heparan sulfate chains present on syndecan-1, which act to prevent MMP cleavage of the core protein. Specifically, we found that (a) the rate of shedding from the cell surface is significantly enhanced when heparan sulfate chains of cell surface syndecan-1 are trimmed by enzymes or when the syndecan-1 core protein lacks heparan sulfate chains, (b) heparan sulfate-mediated suppression of shedding occurs in multiple cell types, for both syndecan-1 and syndecan-4 and in both human and murine cells, (c) to suppress shedding, the heparan sulfate chains must be attached to the core protein of syndecan-1 because exogenous heparan sulfate or heparin does not suppress shedding, (d) when heparan sulfate chains are removed from syndecan-1, shedding is not increased if MMP inhibitors are present, indicating that heparan sulfate chains protect the core protein from MMP-mediated proteolysis, and (e) the heparan sulfate chains on syndecan-1, by way of its regulation of shedding, also regulate syndecan-1 core protein synthesis. These findings are important because the negative regulation of syndecan-1 shedding ensures that syndecan-1 is maintained on the cell surface and that excess syndecan-1 is not released into the extracellular compartment. During disease progression, enhanced degradation of heparan sulfate chains probably shifts the balance between stimulation and suppression of syndecan-1 shedding, leading to excessive shedding and further dysregulation of tissue homeostasis.
If the amount of heparan sulfate present on syndecan-1 regulates its shedding, then it is important to understand what controls degradation of heparan sulfate chains at the cell surface. First, we know that heparanase, an enzyme that cleaves heparan sulfate chains, reduces the amount of heparan sulfate present on syndecan-1 and increases syndecan-1 shedding (21). Enhanced syndecan-1 shedding is also detected in mice expressing a heparanase transgene, indicating that the link between heparanase expression and enhanced shedding occurs in vivo (21). Heparanase expression also leads to up-regulation of MMP-9, a known sheddase of syndecan-1 (22). Thus, heparanase probably plays a dual role in promoting shedding by directly reducing the amount of heparan sulfate on syndecan-1 (thereby making it more susceptible to MMPs) and indirectly by up-regulating expression of MMP-9. It is also interesting that heparanase is known to be up-regulated in cancer, inflammation, and wound healing, all pathological states that are associated with increased syndecan-1 shedding. A second mechanism for degradation of heparan sulfate is through the action of reactive oxygen species that can chemically fragment heparan sulfate (35). This has not been studied extensively, but there is evidence that oxidative damage leads to syndecan-1 shedding, which contributes to lung fibrosis (9). Because enhanced levels of reactive oxygen species are often found in cancer and inflammatory diseases, it is possible that oxidative damage to heparan sulfates contributes significantly to the increased levels of shed syndecan-1 that help drive these diseases.
In addition to degradation of heparan sulfate, the rate of syndecan shedding may also be regulated by the length and number of heparan sulfate chains assembled onto a core protein during proteoglycan synthesis. Although the mechanisms controlling chain length and number remain unclear, the length of chains can be influenced by levels of exostosin enzymes, the glycosyltransferases responsible for heparan sulfate chain elongation. For example, the length of heparan sulfate chains was reduced when EXT1 and EXT2 (exostosin 1 and 2) genes were knocked down in HEK 293 cells. In contrast, knockdown of EXTL3 (exostosin-like 3) resulted in longer heparan sulfate chains (36). Thus, dysregulation of these enzymes during disease could indirectly lead to enhanced syndecan shedding.
Elevated shedding of syndecan in response to a reduction in heparan sulfate content was common for both human and murine syndecan-1 and for syndecan-4. This also held for the murine/human chimeric proteoglycan composed of a murine syndecan-1 core protein with attached human heparan sulfate chains that was generated when human ARH-77 cells, which are negative for syndecan-1 expression, were transfected with the cDNA for murine syndecan-1. Moreover, this shows that the intrinsic cellular mechanisms that control shedding can be present in cells that do not normally express the proteoglycan. Together these results indicate that regulation of syndecan shedding by heparan sulfate is a highly conserved molecular phenomenon and support the importance of this mechanism for control of shedding by heparan sulfate.
The precise molecular mechanism of how the heparan sulfates on syndecan-1 protect the core protein from metalloproteinases is not clear. Cell surface heparan sulfate can bind to and inhibit the activity of ADAM12 (A Disintegrin And Metalloprotease 12), a protease that mediates ectodomain shedding of pro-EGF. This inhibition by heparan sulfate occurs via its binding to the ADAM12 catalytic domain (37). However, in contrast to what we found regarding heparan sulfate inhibition of syndecan shedding, ADAM12 activity was also inhibited by exogenous heparin. We do not know if the heparan sulfates of syndecan bind to MMPs and inhibit their activity directly. It has been shown in some models that MMP-7 activity is enhanced by exogenous heparin, although it is not enhanced by heparan sulfate (38, 39). However, in our model, we found that exogenous heparin had no effect on shedding, suggesting that it is unlikely that syndecans are binding to MMPs and directly inhibiting their activity. Another possible mechanism could be related to a change in protein folding and stabilization, which can be altered by changes in glycosylation (30, 40). Removal of heparan sulfate could alter syndecan-1 core protein conformation and expose cryptic sites that are susceptible to protease cleavage. Moreover, heparan sulfate chains could simply provide a physical barrier that blocks accessibility of the protease to the core protein. Another important consideration regarding mechanism is that syndecan-1 shedding can be regulated by intracellular mechanisms. When the cytoplasmic domain of syndecan-1 associates with the protein transport regulator Rab5, shedding of syndecan-1 is inhibited (20). Removal of heparan sulfates from the core protein might disrupt formation or maintenance of the syndecan-Rab5 complex and lead to elevation of shedding.
In summary, this work defines a new role for heparan sulfate and dramatically broadens the scope of importance of this glycosaminoglycan. Although previous studies have clearly established the role of heparan sulfate in binding various ligands, our findings assign two new functions to heparan sulfate. First, heparan sulfate chains attached to the core protein retain syndecan-1 at the cell surface by inhibiting its shedding. Thus, heparan sulfate controls the location of syndecan-1, thereby impacting its function as either a cell surface molecule or as a soluble effector that can regulate distal cells. Second, heparan sulfate regulates syndecan-1 core protein synthesis. When heparan sulfate is diminished, shedding is enhanced, and the rate of core protein synthesis dramatically increases. This indicates the existence of a feedback loop in which the cell attempts to maintain a critical level of syndecan-1 on the cell surface. Last, our results indicate that therapies designed to preserve the heparan sulfate chains of syndecans or to increase heparan sulfate chain length or number could diminish syndecan shedding and help to control the progression of aggressive cancers or inflammatory diseases.
Acknowledgments
We thank Enid Keyser for technical assistance in flow cytometry and sorting and the Analytical and Preparative Cytometry Facility (supported by National Institutes of Health Grant P30 AR48311) of the Comprehensive Arthritis, Musculoskeletal, and Autoimmunity Center at the University of Alabama, Birmingham. We thank the University of Alabama, Birmingham, Center for AIDS Research DNA Sequencing Core Facility (supported by National Institutes of Health Grant P30 AI027767) for DNA sequencing.
This work was supported, in whole or in part, by National Institutes of Health Grants CA135075 and CA138340 (to R. D. S.).
V. C. Ramani, unpublished observation.
- MMP
- matrix metalloproteinase
- GAG
- glycosaminoglycan
- Hep III
- heparinase III
- PMA
- phorbol ester 12-myristate 13-acetate
- hSDC1 and mSDC1
- human and mouse SDC1 (syndecan-1), respectively
- TDM
- triple deletion mutant.
REFERENCES
- 1. Couchman J. R. (2010) Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 26, 89–114 [DOI] [PubMed] [Google Scholar]
- 2. Theocharis A. D., Skandalis S. S., Tzanakakis G. N., Karamanos N. K. (2010) Proteoglycans in health and disease. Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923 [DOI] [PubMed] [Google Scholar]
- 3. Purushothaman A., Hurst D. R., Pisano C., Mizumoto S., Sugahara K., Sanderson R. D. (2011) Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. J. Biol. Chem. 286, 30377–30383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teng Y. H., Aquino R. S., Park P. W. (2012) Molecular functions of syndecan-1 in disease. Matrix Biol. 31, 3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manon-Jensen T., Itoh Y., Couchman J. R. (2010) Proteoglycans in health and disease. The multiple roles of syndecan shedding. FEBS J. 277, 3876–3889 [DOI] [PubMed] [Google Scholar]
- 6. Jalkanen M., Rapraeger A., Saunders S., Bernfield M. (1987) Cell surface proteoglycan of mouse mammary epithelial cells is shed by cleavage of its matrix-binding ectodomain from its membrane-associated domain. J. Cell Biol. 105, 3087–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iozzo R. V., Sanderson R. D. (2011) Proteoglycans in cancer biology, tumor microenvironment, and angiogenesis. J. Cell Mol. Med. 15, 1013–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Q., Park P. W., Wilson C. L., Parks W. C. (2002) Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 111, 635–646 [DOI] [PubMed] [Google Scholar]
- 9. Kliment C. R., Englert J. M., Gochuico B. R., Yu G., Kaminski N., Rosas I., Oury T. D. (2009) Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J. Biol. Chem. 284, 3537–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park P. W., Pier G. B., Preston M. J., Goldberger O., Fitzgerald M. L., Bernfield M. (2000) Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J. Biol. Chem. 275, 3057–3064 [DOI] [PubMed] [Google Scholar]
- 11. Park P. W., Pier G. B., Hinkes M. T., Bernfield M. (2001) Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 411, 98–102 [DOI] [PubMed] [Google Scholar]
- 12. Su G., Blaine S. A., Qiao D., Friedl A. (2007) Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J. Biol. Chem. 282, 14906–14915 [DOI] [PubMed] [Google Scholar]
- 13. Yang Y., Yaccoby S., Liu W., Langford J. K., Pumphrey C. Y., Theus A., Epstein J., Sanderson R. D. (2002) Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood 100, 610–617 [DOI] [PubMed] [Google Scholar]
- 14. Purushothaman A., Uyama T., Kobayashi F., Yamada S., Sugahara K., Rapraeger A. C., Sanderson R. D. (2010) Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood 115, 2449–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Y., Ren Y., Ramani V. C., Nan L., Suva L. J., Sanderson R. D. (2010) Heparanase enhances local and systemic osteolysis in multiple myeloma by up-regulating the expression and secretion of RANKL. Cancer Res. 70, 8329–8338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joensuu H., Anttonen A., Eriksson M., Mäkitaro R., Alfthan H., Kinnula V., Leppä S. (2002) Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res. 62, 5210–5217 [PubMed] [Google Scholar]
- 17. Seidel C., Sundan A., Hjorth M., Turesson I., Dahl I. M., Abildgaard N., Waage A., Borset M. (2000) Serum syndecan-1. A new independent prognostic marker in multiple myeloma. Blood 95, 388–392 [PubMed] [Google Scholar]
- 18. Ott V. L., Rapraeger A. C. (1998) Tyrosine phosphorylation of syndecan-1 and -4 cytoplasmic domains in adherent B82 fibroblasts. J. Biol. Chem. 273, 35291–35298 [DOI] [PubMed] [Google Scholar]
- 19. Fitzgerald M. L., Wang Z., Park P. W., Murphy G., Bernfield M. (2000) Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 148, 811–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashida K., Stahl P. D., Park P. W. (2008) Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J. Biol. Chem. 283, 35435–35444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Y., Macleod V., Miao H. Q., Theus A., Zhan F., Shaughnessy J. D., Jr., Sawyer J., Li J. P., Zcharia E., Vlodavsky I., Sanderson R. D. (2007) Heparanase enhances syndecan-1 shedding. A novel mechanism for stimulation of tumor growth and metastasis. J. Biol. Chem. 282, 13326–13333 [DOI] [PubMed] [Google Scholar]
- 22. Purushothaman A., Chen L., Yang Y., Sanderson R. D. (2008) Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J. Biol. Chem. 283, 32628–32636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reiland J., Ott V. L., Lebakken C. S., Yeaman C., McCarthy J., Rapraeger A. C. (1996) Pervanadate activation of intracellular kinases leads to tyrosine phosphorylation and shedding of syndecan-1. Biochem. J. 319, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langford J. K., Stanley M. J., Cao D., Sanderson R. D. (1998) Multiple heparan sulfate chains are required for optimal syndecan-1 function. J. Biol. Chem. 273, 29965–29971 [DOI] [PubMed] [Google Scholar]
- 25. Jalkanen M., Nguyen H., Rapraeger A., Kurn N., Bernfield M. (1985) Heparan sulfate proteoglycans from mouse mammary epithelial cells. Localization on the cell surface with a monoclonal antibody. J. Cell Biol. 101, 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 27. Rapraeger A., Jalkanen M., Endo E., Koda J., Bernfield M. (1985) The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J. Biol. Chem. 260, 11046–11052 [PubMed] [Google Scholar]
- 28. Sanderson R. D., Sneed T. B., Young L. A., Sullivan G. L., Lander A. D. (1992) Adhesion of B lymphoid (MPC-11) cells to type I collagen is mediated by integral membrane proteoglycan, syndecan. J. Immunol. 148, 3902–3911 [PubMed] [Google Scholar]
- 29. Charnaux N., Brule S., Chaigneau T., Saffar L., Sutton A., Hamon M., Prost C., Lievre N., Vita C., Gattegno L. (2005) RANTES (CCL5) induces a CCR5-dependent accelerated shedding of syndecan-1 (CD138) and syndecan-4 from HeLa cells and forms complexes with the shed ectodomains of these proteoglycans as well as with those of CD44. Glycobiology 15, 119–130 [DOI] [PubMed] [Google Scholar]
- 30. Shental-Bechor D., Levy Y. (2008) Effect of glycosylation on protein folding. A close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. U.S.A. 105, 8256–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian S. V., Fitzgerald M. L., Bernfield M. (1997) Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J. Biol. Chem. 272, 14713–14720 [DOI] [PubMed] [Google Scholar]
- 32. Ding K., Lopez-Burks M., Sánchez-Duran J. A., Korc M., Lander A. D. (2005) Growth factor-induced shedding of syndecan-1 confers glypican-1 dependence on mitogenic responses of cancer cells. J. Cell Biol. 171, 729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brule S., Charnaux N., Sutton A., Ledoux D., Chaigneau T., Saffar L., Gattegno L. (2006) The shedding of syndecan-4 and syndecan-1 from HeLa cells and human primary macrophages is accelerated by SDF-1/CXCL12 and mediated by the matrix metalloproteinase-9. Glycobiology 16, 488–501 [DOI] [PubMed] [Google Scholar]
- 34. Endo K., Takino T., Miyamori H., Kinsen H., Yoshizaki T., Furukawa M., Sato H. (2003) Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 278, 40764–40770 [DOI] [PubMed] [Google Scholar]
- 35. Raats C. J., Bakker M. A., van den Born J., Berden J. H. (1997) Hydroxyl radicals depolymerize glomerular heparan sulfate in vitro and in experimental nephrotic syndrome. J. Biol. Chem. 272, 26734–26741 [DOI] [PubMed] [Google Scholar]
- 36. Busse M., Feta A., Presto J., Wilén M., Grønning M., Kjellén L., Kusche-Gullberg M. (2007) Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J. Biol. Chem. 282, 32802–32810 [DOI] [PubMed] [Google Scholar]
- 37. Sørensen H. P., Vivès R. R., Manetopoulos C., Albrechtsen R., Lydolph M. C., Jacobsen J., Couchman J. R., Wewer U. M. (2008) Heparan sulfate regulates ADAM12 through a molecular switch mechanism. J. Biol. Chem. 283, 31920–31932 [DOI] [PubMed] [Google Scholar]
- 38. Yu W. H., Woessner J. F., Jr. (2000) Heparan sulfate proteoglycans as extracellular docking molecules for matrilysin (matrix metalloproteinase 7). J. Biol. Chem. 275, 4183–4191 [DOI] [PubMed] [Google Scholar]
- 39. Ra H. J., Harju-Baker S., Zhang F., Linhardt R. J., Wilson C. L., Parks W. C. (2009) Control of promatrilysin (MMP7) activation and substrate-specific activity by sulfated glycosaminoglycans. J Biol. Chem. 284, 27924–27932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Live D. H., Kumar R. A., Beebe X., Danishefsky S. J. (1996) Conformational influences of glycosylation of a peptide. A possible model for the effect of glycosylation on the rate of protein folding. Proc. Natl. Acad. Sci. U.S.A. 93, 12759–12761 [DOI] [PMC free article] [PubMed] [Google Scholar]