

Background: Previous studies have failed to reconstitute an active replication complex with hepatitis C virus (HCV) RNA-dependent RNA polymerase.
Results: The replication complex from HCV was assembled, purified, and characterized.
Conclusion: A highly active replication complex can be formed with HCV polymerase that catalyzes fast and processive RNA replication.
Significance: A purified and active replication complex is essential for mechanistic studies and drug discovery.
Keywords: Enzyme Kinetics, Hepatitis C Virus, Pre-steady-state Kinetics, RNA Polymerase, Viral Polymerase, Viral Replication
Abstract
NS5B is the RNA-dependent RNA polymerase responsible for replicating hepatitis C virus (HCV) genomic RNA. Despite more than a decade of work, the formation of a highly active NS5B polymerase·RNA complex suitable for mechanistic and structural studies has remained elusive. Here, we report that through a novel way of optimizing initiation conditions, we were able to generate a productive NS5B·primer·template elongation complex stalled after formation of a 9-nucleotide primer. In contrast to previous reports of very low proportions of active NS5B, we observed that under optimized conditions up to 65% of NS5B could be converted into active elongation complexes. The elongation complex was extremely stable, allowing purification away from excess nucleotide and abortive initiation products so that the purified complex was suitable for pre-steady-state kinetic analyses of polymerase activity. Single turnover kinetic studies showed that CTP is incorporated with apparent Kd and kpol values of 39 ± 3 μm and 16 ± 1 s−1, respectively, giving a specificity constant of kpol/Kd of 0.41 μm−1 s−1. The kinetics of multiple nucleotide incorporation during processive elongation also were determined. This work establishes a novel way to generate a highly active elongation complex of the medically important NS5B polymerase for structural and functional studies.
Introduction
Hepatitis C virus (HCV)3 belongs to the Flaviviridae virus family that includes several important human and animal pathogens such as Dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), and bovine diarrhea virus (BVDV). HCV infects ∼3% of the world population and chronic infection of HCV can lead to hepatitis, liver cirrhosis, and cancer. HCV has a 9.6-kb single-stranded positive RNA genome, which encodes three structural proteins (C, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (1).
NS5B is the 68-kDa RNA-dependent RNA polymerase of HCV. Like other RNA-dependent RNA polymerases in the Flaviviridae family, NS5B catalyzes de novo RNA replication and does not require a primer to initiate replication from the 3′-end of the RNA template. RNA replication catalyzed by NS5B can be divided into three phases: de novo initiation, transition from initiation to elongation, and processive elongation. De novo initiation involves the binding of the 3′-end of the RNA to the active site of NS5B, recruiting the first two nucleotides and catalyzing the first nucleotidyl transfer reaction to form a dinucleotide (2). The dinucleotide serves as a primer for the addition of subsequent nucleotides (3, 4). Based on studies with recombinant NS5B protein, the initiation phase of RNA synthesis appears to be slow and inefficient, often leading to short abortive products (2, 3, 5). After several cycles of nucleotide incorporation, a major transition from the initiation to the elongation phase of RNA synthesis occurs, and nucleotide incorporation becomes fast and processive (6, 7). However, in previous reports, the fraction of the NS5B participating in the RNA elongation synthesis was estimated to be very low, in the range of <1% of total protein, and this has precluded detailed mechanistic analysis (8). Thus, even though the enzyme was cloned and the crystal structure was solved more than a decade ago, measurements of the kinetics, thermodynamics, and fidelity governing nucleotide incorporation are lacking, and analysis of the mechanistic basis for inhibition of HCV replication by new pharmaceuticals has been limited.
Detailed kinetic studies, combined with structural analysis, previously have provided valuable mechanistic insight to understand the function and mechanism of inhibition for many polymerases (see for example Ref. 9). Thus far, attempts to characterize HCV NS5B using steady-state kinetics have provided limited information, because of the difficulty in data interpretation. In steady-state kinetic analysis of a polymerase, the kinetic parameters kcat and Km are complex functions of the rates of multiple steps along the reaction pathway leading to the products. In contrast, pre-steady-state kinetic methods can dissect the complex reaction pathway and measure the kinetics of individual steps. These methods are especially important for characterizing the complex mechanism involving a DNA or RNA polymerase (10–12). To study the kinetics of nucleotide incorporation using pre-steady state kinetic methods, it is first necessary to obtain a stoichiometric polymerase·primer·template ternary complex. Unlike other polymerases, such a ternary complex cannot be formed productively with HCV NS5B (8, 13, 14). Crystal structures of NS5B showed that its active site is encircled by the loops extending from the “fingers” domain and is occluded by the β-loop and the C-terminal tail (15–17). These structural elements likely prevent NS5B from productively binding a primer-template duplex RNA. In support of that postulate, mutations (point mutation or deletions) in these structural elements on NS5B have increased the elongation activity of NS5B from a duplex RNA but have decreased its de novo initiation activity (18–21). HCV polymerase therefore is likely to go through a major conformational change during the transition from initiation to elongation of RNA synthesis and, in its ground state, is only able to perform de novo initiation of RNA synthesis or use very short (2–3 nucleotide) primer molecules for initiation.
In this work, a productive elongation complex containing HCV NS5B and a duplex RNA primer-template was obtained by an extension and pause reaction, without the need to mutate or delete major structural elements of NS5B. Surprisingly, we found conditions that allowed the majority (65%) of the NS5B protein to be assembled productively into active elongation complexes, thus increasing active site concentration significantly over previous reports. The elongation complex could be purified after the initial assembly reaction by a simple precipitation method, taking advantage of the fact that the elongation complex was insoluble under low salt reaction conditions. The purified elongation complex was highly active and exceptionally stable, even under high salt conditions or in the presence of heparin, suggesting a major conformational change, leading to very slow RNA dissociation from NS5B in the elongation conformation. Using this purified elongation complex, we examined the kinetics of single and multiple nucleotide incorporation catalyzed by NS5B using pre-steady-state kinetic methods. This elongation complex can now be used to study the mechanisms of replication, drug inhibition, and drug resistance of HCV involving NS5B.
EXPERIMENTAL PROCEDURES
Chemicals and Nucleic Acids
All NTPs were ultrapure grade purchased from USB Corp. (Cleveland, OH). Heparin sodium salt (195.9 USP units/mg) was from Sigma-Aldrich. MgCl2, EDTA, NaCl solutions, and Tris-Cl buffers were purchased from Ambion (Austin, TX).
The unique 20-mer RNA template (5′-AAUCUAUAACGAUUAUAUCC-3′), 5′-mono-phosphorylated pGG dinucleotide primer and non-phosphorylated OHGG dinucleotide primer were synthesized chemically by Dharmacon, Inc. (Chicago, IL). The 5′-end labeling reaction of OHGG was conducted with [γ-32P]ATP (PerkinElmer Life Sciences), and T4 polynucleotide kinase in forward reaction buffer (Invitrogen) for 1 h at 37 °C. The reaction was stopped by heating at 95 °C for 3 min. The radiolabeled pGG was mixed with cold pGG to make radiolabeled pGG stock solutions. (The original radiolabeled pGG was <1%.)
Expression and Purification of NS5B(Δ21)
N-terminal penta-His-tagged NS5BΔ21 (con1 strain, codon sequence is from GenBankTM accession no. AJ242654; 21 C-terminal amino acids were removed from the full-length NS5B protein) was cloned into a pET17b vector (Novagen) and expressed in Escherichia coli BL21(DE3) cells. Cells were cultured in TB medium at 37 °C. Isopropyl 1-thio-β-d-galactopyranoside induction was started at an A600 of 0.6 at 20 °C. After an 18-h induction, cells were collected and stored at −80 °C. Cells were resuspended in a lysis buffer (50 mm HEPES, pH 7.5, 20% (v/v) glycerol, 20 mm imidazole, 0.1% (w/v) octyl glucoside, 2 mm β-mercaptoethanol, protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN), and 300 mm NaCl). The lysate was treated with DNase for 10 min. The lysate was passed through a microfluidizer and centrifuged at 13,000 rpm for 30 min. The supernatant was applied to a Superflow nickel-nitrilotriacetic acid column (Qiagen, Valencia, CA). NS5B protein was eluted with a buffer containing 50 mm HEPES, pH 7.5, 300 mm NaCl, 20% (v/v) glycerol, 350 mm imidazole, 0.1% (w/v) octyl glucoside, and 2 mm β-mercaptoethanol. Peak fractions were pooled, diluted 1:1 with a buffer containing 20 mm HEPES, pH 7.5, 15% (v/v) glycerol, and 2 mm DTT, centrifuged at 15,000 rpm for 30 min, and finally passed through a 0.2 μm filter. The filtrated sample was applied to an SP-Sepharose column (GE Healthcare) equilibrated in buffer A (50 mm HEPES, pH 7.5, 15% (v/v) glycerol, 0.1% (w/v) octyl glucoside, and 2 mm DTT). The column was washed with five column volumes of buffer A. The protein was eluted with a gradient of 0.15 ∼ 1.0 m NaCl in buffer A. Peak fractions were pooled and loaded onto a Superdex 200 column (GE Healthcare) equilibrated in buffer B (10 mm Tris-HCl, pH 7.5, 10% (v/v) glycerol, 5 mm DTT, and 0.1% (w/v) octyl glucoside) containing 600 mm NaCl. Peak fractions were pooled, concentrated, and dialyzed to buffer B containing 200 mm NaCl. The protein was stored at −80 °C until use.
The protein concentration was measured by absorbance at 280 nm with an extinction coefficient of 170,850 cm−1 m−1. The identity of the protein was confirmed by mass spectrometry analysis of intact protein and tryptic digested products.
Extension and Pause Reaction
A typical reaction containing NS5B, pGG, 20-mer RNA template, ATP, and UTP in an optimized reaction buffer (40 mm Tris-Cl, pH 7.0, 20 mm NaCl, 5 mm DTT, and 2 mm MgCl2) was conducted at 30 °C in a Hybex microsample incubator (Scigene, Inc., Sunnyvale, CA). The concentrations of NS5B and substrates were indicated in the corresponding figure legends. Typically, a 10-μl reaction was mixed with 30 μl of a quench solution (90% formamide, 50 mm EDTA, 0.1% bromphenol blue, and 0.1% xylene cyanol) to stop the reaction at an indicated time. To test the activity of the elongation complex, 10 μl of the extension and pause reaction was mixed with 10 μl of 100 μm CTP in the optimized reaction buffer and quenched by a 60-μl quench solution after 20 s.
Purification of Elongation Complex by Centrifugation
An extension and pause reaction containing 12 μm NS5B, 10 μm radiolabeled pGG, 10 μm 20-mer, 25 μm ATP, and 12.5 μm UTP in the optimized reaction buffer (40 mm Tris-Cl, pH 7.0, 20 mm NaCl, 5 mm DTT, and 2 mm MgCl2) was run for 2 h. The 2-h reaction was spun at 16,000 × g for 5 min at room temperature using a bench top centrifuge (model 5415 D, Eppendorf). After centrifugation, the supernatant was removed, and the remaining pellet was washed twice by additional resuspension and centrifugation to remove residual contaminants. The pellet was resuspended in the optimized reaction buffer. A 5-μl aliquot from the 2-h reaction, the supernatant, or the resuspended pellet was mixed with 35 μl of quench solution. Another 5-μl aliquot from each sample was reacted with 5 μl of 100 μm CTP in the reaction buffer for 20 s and quenched with 30 μl of quench solution.
Solubility and Stability of Elongation Complex
The elongation complex in the pellet from the 2-h extension and pause reaction was obtained as described above. The pellet was resuspended in the pH7.4 reaction buffer (40 mm Tris-Cl, pH 7.4, 5 mm DTT, and 2 mm MgCl2) containing NaCl at various concentrations. After incubation for 30 min, each sample was spun at 16,000 × g for 5 min at room temperature. The supernatant and pellet were analyzed on a sequencing gel (supplemental Fig. S5A). The percentage of the soluble elongation complex was calculated as the ratio of the band intensity of the elongation complex in supernatant to the total band intensity of the elongation complex in the supernatant and the pellet.
To study the stability of the elongation complex, the pellet from the 2-h extension and pause reaction was resuspended in the pH 7.4 reaction buffer containing NaCl at various concentrations. The samples were incubated at 30 °C for 14 h. A 10-μl aliquot from each sample (0 and 14 h) was reacted with 10 μl of 100 μm CTP in the pH 7.4 reaction buffer for 20 s and quenched by 60 μl of quench solution. The samples were analyzed on a sequencing gel (supplemental Fig. S5B). The percentage of 10-mer in the sum of 10-mer and 9-mer was calculated from their respective band intensities.
To study the dissociation rate of the elongation complex, the pellet from the 2-h extension and pause reaction was resuspended in the pH 7.4 reaction buffer containing 150 mm NaCl. Heparin (0.2 mg/ml), an enzyme trap, was added at the start of the dissociation reaction at 30 °C (up to 32 h). At various times, a 10-μl aliquot was reacted with 10 μl of 100 μm CTP in the pH 7.4 reaction buffer with 150 mm NaCl for 20 s and quenched by 60 μl of quench solution to test the remaining activity of the elongation complex. The samples were analyzed on a sequencing gel (supplemental Fig. S5C). The percentage of 10-mer in the sum of 10-mer and 9-mer was calculated from their respective band intensities.
Nucleotide Incorporation Reaction Using Rapid Quench-flow Method
Reactions were conducted at 30 °C using an RQF-3 rapid quench-flow instrument (KinTek Corp., Austin, TX). The pellet containing elongation complex from a 2-h extension and pause reaction was resuspended in a buffer containing 40 mm Tris-Cl, pH 7.4, 150 mm NaCl, 5 mm DTT, and 2 mm MgCl2. The volume of this buffer was five times that of the original extension and pause reaction, and thus, the NS5B·9-mer·20-mer complex was ∼0.6 μm, assuming 100% recovery of the elongation complex during purification. In a typical quench-flow assay, each reaction was started by mixing the elongation complex with an equal volume of solution containing NTP in the same reaction buffer. Each reaction was quenched at the time interval indicated in the figures.
Product Analysis
The quenched reactions were heat-denatured for 3 min at 95 °C before electrophoresis. The samples were loaded onto a 16% denaturing polyacrylamide gel with 7 m urea (National Diagnostics, Atlanta, GA), and electrophoresis was performed at 80 watts using a Sequi-Gen GT system from Bio-Rad. Gels were dried at 80 °C for 1 h with a Model-583 gel drier (Bio-Rad). Dried gels were exposed to storage phosphor screens and visualized by a Typhoon 9400 scanner (GE Healthcare). The intensity of each band on the gel was quantified using the ImageQuant software (version 5.2, GE Healthcare). The concentration for an RNA product was calculated by multiplying the input pGG concentration by the fraction of the product band intensity in a given lane.
Data Analysis
The active site titration data were analyzed by non-linear regression using the program GraFit5 (Erithacus Software, Surrey, UK). The data were fit to a quadratic equation,
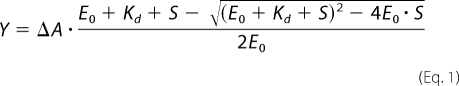 |
where Y is the measured concentration of the 10-mer, ΔA is the maximal concentration of the 10-mer, E0 is the enzyme concentration, Kd is the apparent dissociation constant, and S is the concentration of pGG. The fitted parameters were presented in the form of best-fit value ± S.E. with their respective units. The S.E. estimates were calculated by the covariance matrix during nonlinear regression using GraFit5.
The kinetics of single nucleotide (see Fig. 5) and multiple nucleotide incorporation reactions (see Fig. 6) had been analyzed using the KinTek Global Kinetic Explorer program (KinTek Corp., Austin, TX) based upon numerical integration of rate equations from an input model (22). In fitting single nucleotide incorporation data to Scheme 1, the equilibrium constant for the initial complex formation was estimated by assuming diffusion-limited nucleotide binding (kon = 100 μm−1s−1) and allowing the dissociation rate to vary during fitting to afford calculation of the equilibrium constant (Kd = koff/kon). The kinetics of multiple-nucleotide processive incorporation were fit to a model shown in Scheme 2, where each nucleotide incorporation was modeled as a single irreversible step. The processive incorporation experiment was performed three times and all three data sets were fit simultaneously to derive the rates shown in Scheme 2 and Fig. 6C. Standard error estimates were calculated from the covariance matrix during nonlinear regression using KinTek Explorer program and were verified by confidence contour analysis (22, 23). The average rate of nucleotide incorporation from the multiple nucleotide incorporation experiment was calculated using the equation,
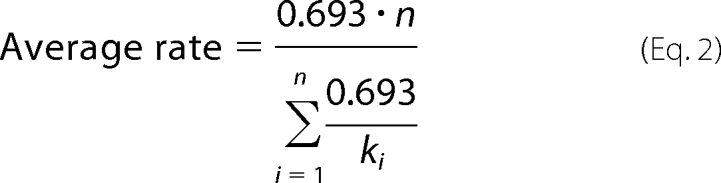 |
where ki is the rate for each nucleotide incorporation, and n represents the total number of nucleotides incorporated.
FIGURE 5.

Kd and kpol of single nucleotide incorporation catalyzed by HCV NS5B in the elongation mode. A, time courses of CTP incorporation opposite G in the template at various CTP concentrations (3.1, 6.25, 12.5, 25, 50, 100, and 200 μm) were obtained by rapid quench-flow assays. Data were fit to the mechanism in Scheme 1 using KinTek Global Kinetic Explorer. The solid lines were the fitted lines. The fitted parameters, Kd (equilibrium dissociation constant for nucleotide binding) and kpol (nucleotide incorporation rate), are 39 ± 3 μm and 16 ± 1 s−1, respectively. B, time courses of GTP incorporation opposite C in the template at various GTP concentrations (3.1, 6.25, 12.5, 25, 50, 100, and 200 μm). The Kd is 22 ± 2 μm, and the kpol is 2.1 ± 0.1 s−1. Each data set shown is the representative of three repeats, and the Kd and kpol were reported as mean ± S.D. from the repeats.
FIGURE 6.

Processive nucleotide incorporation by HCV NS5B in the elongation mode. A, a sequencing gel showing the time course of multiple nucleotide incorporation. The reaction was conducted by mixing the elongation complex, NS5B·9-mer·20-mer (0.3 μm), with ATP, UTP, GTP, and CTP (400 μm each) in the reaction buffer and quenched at indicated time intervals. B, the time courses of product formation were analyzed globally by fitting to the mechanism in Scheme 2. The data set shown was a representative from three repeats, with the solid lines indicating the best fit. The fitted rate for each step was listed in Scheme 2 and C. C, a bar graph showing rate (±S.E.) for each nucleotide incorporation during processive replication. The average of the rates from 11 steps was 4 s−1.
RESULTS
Assembly of HCV NS5B Elongation Complex by Extension and Pause Reaction
Fig. 1A shows the reaction scheme used to obtain a paused elongation complex and then to test the activity of the elongation complex using a single nucleotide incorporation assay. The dinucleotide primer (pGG), the 20-mer RNA template, and NS5B were mixed with ATP and UTP to start the RNA synthesis reaction. According to the 20-mer sequence, the reaction should pause after forming a 9-mer product because of the absence of CTP in the reaction. Fig. 1B shows that the major products of the reaction were a 3-mer and a 9-mer during the extension reaction monitored for up to 24 h. A small amount of a 10-mer product was observed in reactions with longer reaction time, presumably due to a slow misincorporation event at this position. As described below (see Fig. 3), the 3-mer was an abortive product, which accumulated in solution during the reaction.
FIGURE 1.

pGG primer extension and pause assay. A, a scheme depicting the reaction setup and the 20-mer RNA template used in all assays in this study. B, a time course of product formation during an extension and pause assay. The reaction was started by mixing 20 μm pGG, 20 μm template, 25 μm ATP, and 25 μm UTP with 12 μm HCV NS5B. An aliquot from the reaction was mixed with quench solution at each indicated time point; in the meantime, another aliquot of the reaction was reacted with 50 μm CTP and quenched after 20 s (indicated as +C). C, extension and pause assays at various concentrations of NS5B. Each reaction was started with NS5B (2, 4, 6, 8, 10, and 12 μm), 20 μm pGG, 20 μm template, 25 μm ATP, and 25 μm UTP. Aliquots from each reaction were mixed with quench solution at the time points indicated. Concentration of the elongation complex (9-mer and 10-mer) was plotted against NS5B concentration at each reaction time point. For each reaction time, the data were fit to a linear function with the slope representing the fraction of input NS5B assembled into the elongation complex.
FIGURE 3.

Purification of the elongation complex by centrifugation. An extension and pause reaction with 12 μm NS5B, 12.5 μm UTP, 25 μm ATP, and 10 μm pGG/20-mer was run for 2 h, followed by centrifugation at 16,000 × g for 5 min. Lane 1, pGG primer; lane 2, the 2-h reaction before spin; lane 3, one aliquot of the 2-h reaction was reacted with 50 μm CTP for 20 s; lanes 4 and 5, the supernatant (Sup) and its reaction with 50 μm CTP for 20 s; lanes 6 and 7, the resuspended pellet and its reaction with 50 μm CTP for 20 s.
As a first test to determine whether the 9-mer productively assembled into the elongation complex bound to NS5B, the paused reaction was mixed with CTP and allowed to react for 20 s. About 90% of the 9-mer could be rapidly extended to a 10-mer product within 20 s (see Fig. 1B). During this time interval, the enzyme catalyzed only a single turnover because both RNA dissociation (see below, Fig. 4C) and rebinding of a 9-mer primer to the NS5B (supplemental Fig. S1) were determined to be extremely slow events. These results suggested that the 10-mer formed in the reactions shown in Fig. 1B within the 20-s elongation time originated from the 9-mer stalled complex. The majority (∼90%) of the 9-mer product formed during the extension and pause reaction was therefore assembled into a productive NS5B·9-mer·20-mer elongation complex, capable of fast elongation.
FIGURE 4.

Solubility and stability of the elongation complex. A, solubility of the elongation complex at various NaCl concentrations. The pellet containing the elongation complex after centrifugation was resuspended in the reaction buffer with NaCl at various concentrations (0 ∼ 350 mm). After a 30-min incubation, the samples were spun at 16,000 × g for 5 min. The percentage of elongation complex distributed in the supernatant or the pellet was analyzed by sequencing gel. The percentage of elongation complex in the supernatant (soluble EC) was plotted against NaCl concentration. B, stability of the elongation complex at various NaCl concentrations. The pellet-containing elongation complex was resuspended in reaction buffer with NaCl at various concentrations and was incubated for 14.3 h. The activities of the elongation complex preincubation and postincubation were measured by reacting the elongation complex with 50 μm CTP for 20 s. The percentage of the 10-mer product versus NaCl concentration was shown. C, stability of the elongation complex in the reaction buffer with heparin. The pellet containing elongation complex was resuspended in the reaction buffer with 150 mm NaCl and 0.2 mg/ml heparin and incubated for various time intervals. The remaining activity of the elongation complex was measured by reacting an aliquot at each incubation time point with 50 μm CTP for 20 s. The percentage of the 10-mer product versus incubation time was shown.
We examined the effect of NS5B concentration on the kinetics of formation of the elongation complex (Fig. 1C). The sum of 9-mer and 10-mer was used to estimate the amount of the elongation complex formed. The formation of the elongation complexes followed a linear function of the input NS5B concentrations during the reaction. The slope represents the ratio of the concentration of the elongation complex relative to the input enzyme concentration, i.e. the fraction of NS5B captured in the elongation complex. During the first 5 h, the time dependence of the slope fit to a single exponential function, providing an estimated half-life for the formation of the elongation complex of 36 ± 4 min (supplemental Fig. S2). On a longer time scale (21 and 24 h), it appeared that a slower phase led to the accumulation of additional elongation complex, but was accompanied by formation of a 10-mer, presumably due to misincorporation.
Active Site Titration of HCV NS5B
To quantify the amount of NS5B that could be assembled into the elongation complex, we examined the extension and pause reactions with increasing concentrations of pGG and template RNA, in the presence of 500 μm ATP, 500 μm UTP, and 50 μm 3′-deoxy-CTP (3′-dCTP) (Fig. 2). The chain terminator, 3′-dCTP, was used as the final nucleotide to be incorporated in forming the 10-mer product to prevent subsequent mismatch incorporation. The reactions were run for 24 h (Fig. 2A). The concentration of the product (10-mer) was plotted against the input pGG concentrations, and the data were fit to a quadratic equation, yielding an apparent Kd of 3.8 ± 1.4 μm for pGG binding, and an amplitude of 7.8 ± 0.2 μm. The amplitude represents the maximum amount of elongation complexes formed at saturation (Fig. 2B). Because the input NS5B concentration was 12 μm, ∼65% of input NS5B could be assembled into a functional elongation complex.
FIGURE 2.

Active site titration of HCV NS5B. A, extension and pause reactions were started with 12 μm NS5B, 500 μm ATP, 500 μm UTP, 50 μm 3′-dCTP and pGG/20-mer at various concentrations (5, 10, 20, 40, 60, 80, 100 μm as indicated). The reactions were quenched after 24 h. B, the concentration of elongation complex (10-mer) was plotted against pGG concentration. The data were fit to a quadratic equation, yielding an apparent Kd of 3.8 ± 1.4 μm and an amplitude of 7.8 ± 0.22 μm. Each data point on the plot was the average of four independent experiments, and the error bar was S.D.
Purification of Elongation Complex
Unincorporated ATP and UTP, substrate pGG, and short abortive products such as GGA present in the extension and pause reaction could interfere with more detailed mechanistic characterization of the elongation complex; therefore, a purification procedure was established.
We conducted the extension and pause reaction using 10 μm pGG for 2 h to obtain the NS5B·9-mer·20-mer elongation complex without significant misincorporation. Under these optimized conditions, ∼3 μm of NS5B·9-mer·20-mer complex was formed. (Fig. 3, lane 2). We also noted that the reaction solution became turbid immediately after mixing NS5B with the substrates due to precipitation. A white pellet formed at the bottom of the tube after a 5-min centrifugation of the 2-h reaction mixture at 16,000 × g. Fig. 3 shows a sequencing gel analysis of total products from the 2-h reaction (Total), the products that remained in the supernatant after the centrifugation (Sup), and the products present in the resuspended pellet (Pellet). Surprisingly, most of the 3-mer product generated during the reaction was observed in the supernatant, and most of the 9-mer product was in the pellet after the centrifugation. When CTP was added for a 20-s reaction, the 9-mer in the total reaction and the 9-mer in the pellet were extended equally to 10-mer products. These data strongly suggested that the NS5B·9-mer·20-mer elongation complex precipitated during the extension and pause reaction under low salt conditions and was recovered in the pellet after centrifugation, whereas abortive reaction products such as GGA remained soluble in the supernatant. Similar to the GGA trimer RNA, the unincorporated nucleotides were also expected to be soluble and remain in the supernatant. Thus, the elongation complex resuspended from the pellet fraction was expected to be void of unincorporated nucleotides. To confirm this assumption, the purified elongation complex was incubated with CTP and GTP. From this reaction, an 11-mer product was formed as expected from the template sequence, but no products longer than 11-mer were observed (supplemental Fig. S3). In contrast, when the total 2-h extension and pause reaction was mixed for 20 s with CTP and GTP, extension products up to 20 nucleotides were observed, confirming the presence of excess UTP and ATP from the reaction buffer (supplemental Fig. S3). In conclusion, abortive products and unincorporated nucleotides could be separated from the elongation complex by precipitation of the complex in low salt buffer, centrifugation and resuspension of the pellet in a new reaction buffer.
Solubility and Stability of Elongation Complex
Next, we optimized the conditions to solubilize the elongation complex. The pellet containing the elongation complex was resuspended in reaction buffer at various NaCl concentrations. After a half-hour incubation, the samples were centrifuged again, and the fraction of the elongation complex distributed into the supernatant was measured (Fig. 4A). The elongation complex mainly remained in the white pellet at low NaCl concentration, but the majority of the complex was in the supernatant >100 mm NaCl (Fig. 4A), indicating that the elongation complex was more soluble at higher NaCl concentrations.
It is known from studies with other polymerase enzymes that high concentrations of NaCl can interfere with RNA binding and cause dissociation of protein·RNA complexes. We therefore investigated the stability of the NS5B elongation complex at increasing concentrations of NaCl. The pellet containing the elongation complex was resuspended in reaction buffers with NaCl at various concentrations and then incubated for 14 h to test for complex stability. The activity of the elongation complex before and after the 14-h incubation was measured. Surprisingly, the activity of the purified elongation complex was similar immediately after resuspension and after the 14-h incubation at all NaCl concentrations tested, up to 350 mm (Fig. 4B), suggesting exceptionally high stability as compared with previously characterized RNA binding proteins. Based on the solubility and stability of the elongation complex in different NaCl concentrations, a reaction buffer with a physiological concentration of 150 mm NaCl was used for further characterizations of the elongation complex.
We tested the stability of the elongation complex in a buffer with a polymerase enzyme trap, heparin. The elongation complex was incubated in reaction buffer with 0.2 mg/ml heparin for up to 32 h. At this concentration, heparin fully inhibited the activity of NS5B in the extension and pause reaction (data not shown). During the incubation, the remaining activity of the elongation complex was tested by incubating an aliquot of the reaction with CTP for 20 s (Fig. 4C). The activity of the elongation complex remained high during the complete heparin incubation period, suggesting a RNA dissociation half-life greater than a week. These data indicate that the dissociation rate of the HCV NS5B elongation complex was extremely slow and the elongation complex was remarkably stable in a physiologically relevant buffer.
Rapid and Processive Nucleotide Incorporation Catalyzed by HCV NS5B in Elongation Complex
Scheme 1 shows a simplified two-step mechanism of single nucleotide incorporation catalyzed by the RNA polymerase. Study of the NTP concentration dependence of the rate of nucleotide incorporation can define the apparent nucleotide dissociation equilibrium constant, Kd, and the maximal incorporation rate, kpol. We measured Kd and kpol of CTP incorporation catalyzed by HCV NS5B during elongation by rapid-quench-flow methods using the NS5B·9-mer·20-mer complex. The elongation complex was rapidly mixed with a reaction buffer containing CTP at various concentrations (Fig. 5A). The reactions were conducted under single turnover conditions because only productively bound 9-mer could be extended rapidly. The time courses of 10-mer formation were analyzed by global data fitting to the mechanism shown in Scheme 1. The Kd and kpol were 39 ± 3 μm and 16 ± 1 s−1, respectively. The calculated ratio of kpol/Kd was 0.41 μm−1 s−1, which defines kcat/Km, the specificity constant of nucleotide incorporation during processive polymerization (24). We also measured the Kd and kpol of GTP incorporation using an NS5B·10-mer·20-mer elongation complex (Fig. 5B). The Kd and kpol for GTP incorporation were 22 ± 2 μm and 2.1 ± 0.1 s−1, respectively.
 |
SCHEME 1. Simplified mechanism of nucleotide incorporation catalyzed by an RNA polymerase.
The kinetics of incorporation of multiple nucleotide were studied to estimate the rate of each nucleotide incorporation from 10-mer to 20-mer (Fig. 6) during processive synthesis. The NS5B·9-mer·20-mer elongation complex was mixed rapidly with all four nucleotides at 400 μm each. The reactions were quenched at various time intervals. The time courses of the appearance and disappearance of all intermediates during polymerization were analyzed by global data fitting to a simplified mechanism shown in Scheme 2 (Fig. 6B). The best fitted rate constant for each step is shown on Scheme 2 and was plotted in a bar graph in Fig. 6C. The rate for each step varied from 1.3 s−1 to 15 s−1, with an average of 4 s−1. Rates of the first two nucleotide incorporation reactions agree with those measured in single turnover kinetic studies (Fig. 5).
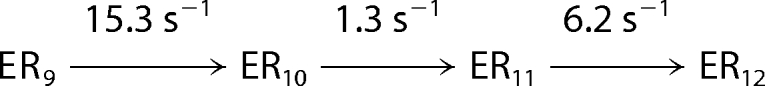 |
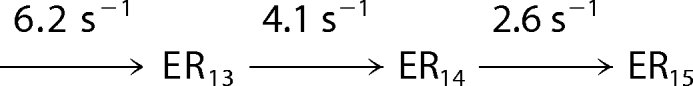 |
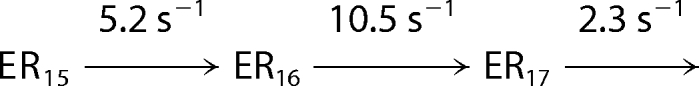 |
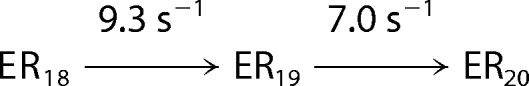 |
SCHEME 2. Simplified mechanism for processive nucleotide incorporation catalyzed by HCV NS5B.
DISCUSSION
An NS5B·primer·template ternary complex is necessary for kinetic and structural analysis of the nucleotide incorporation reaction catalyzed by HCV NS5B during elongation. In this work, we showed that such a ternary complex can be obtained effectively following an extension and pause reaction. The high yield of elongation complex from recombinant NS5B, the high activity and stability of the elongation complex were all surprising, considering previous reports of attempts to form such a complex. The kinetics of the extension and pause reaction (Fig. 1) showed that the elongation complex was formed with a half-time of ∼36 min, consistent with the initiation of RNA synthesis by HCV NS5B being rate-limiting. Active site titration demonstrated that ∼65% of the NS5B protein could form functional elongation complexes (Fig. 2) with an apparent dissociation equilibrium constant of 3.8 ± 1.4 μm for pGG binding during the initiation phase. Because misincorporation events were observed at longer reaction time (Fig. 1B), we reduced the extension and pause reaction to 2 h for most experiments, still allowing ∼25% efficiency in the assembly of the stable NS5B elongation complex (Fig. 3). We took advantage of the insolubility of the elongation complex at low NaCl concentration (Figs. 3 and 4A) to use a centrifugation step to separate the elongation complex from unincorporated nucleotides and abortive short products (Fig. 3 and supplemental Fig. S3). Interestingly, the insoluble elongation complex formed at low salt concentration was fully active in suspension, as indicated by the efficient elongation of stalled 9-mer to extended 10-mer product (Fig. 3, lanes 3 and 7). Thus, the precipitation occurred without affecting the enzyme activity.
In contrast to the elongation reaction, the nucleotide incorporation reactions during the initiation and transition phases to form a stable elongation complex appear to be quite complex. First, NS5B must bind to the 3′-end of the template, followed by the binding of pGG in a base pair with two cytosines on the 3′-end of the template. Reaction with the next nucleotide to form a trimer is slow (3) and is not sufficient to form a stable elongation complex; rather, the trimer accumulates in solution (Fig. 3). The transition from the initiation to the elongation phase of the reaction can be defined by observation of accumulated intermediates. The amount of each intermediate seen to accumulate depends on the relative rate of its forward reaction versus its rate of dissociation from the enzyme. Accordingly, inspection of the data in Figs. 1 and 2 suggests that the transition from initiation to elongation occurs after 6-mer formation because of the accumulation of intermediates 3 to 6 nucleotides in length. More detailed studies are underway to establish the kinetics of initiation to elongation transition.
De novo RNA initiation in vivo is likely to be a highly regulated process, which may involve additional nonstructural viral proteins and/or host factors. It is reasonable to assume that initiation is relatively fast and efficient in vivo, and it will be of interest to identify factors that regulate this process. Here, we circumvented the normal initiation reaction by starting RNA synthesis with a pGG dinucleotide and allowing reaction for several hours at low salt buffer. By adding only two of the four nucleotides required for full replication, those NS5B molecules that succeed in completing the relatively inefficient transition process accumulate at a stalled complex after the formation of a primer 9 nucleotides in length.
Several features of our extension and pause reaction conditions contributed to the higher activity of HCV NS5B observed in our assays. One was the use of pGG instead of mononucleotides to initiate the reaction. Previous reports showed that the enzyme can initiate from pGG more efficiently by skipping the slow reaction of first nucleotidyl transfer (4). This strategy had been previously used to attempt to obtain an elongation complex, but the amount of the complex was very low under those reaction conditions (14). The most important optimization in our assay was the choice of NaCl concentration (supplemental Fig. S4). We found that lower NaCl concentrations substantially increased the RNA synthesis initiation efficiency and subsequently the amount of the elongation complex formed in agreement with several previous reports (25, 26). It has been reported that NaCl could control the reversible oligomerization of NS5B (3) and that NS5B oligomerization increased initiation activity of NS5B (27–29). While investigating the impact of salt on reaction conditions, we observed that more NS5B precipitate was formed at lower NaCl concentrations but that NS5B was soluble in higher NaCl concentration, suggesting that the precipitation process was reversible (Fig. 4A). Based on the correlation between precipitation and oligomerization as discussed above, it is reasonable to propose that the NS5B precipitate could contain one form of the NS5B oligomers reported by several groups (3, 8, 30, 31). The exact interactions between NS5B molecules in the oligomers are not clear. If at least one molecule of NS5B served as accessory function for one active NS5B in a functional dimer as in HIV reverse transcriptase, no more than 50% NS5B would be assembled into the elongation complex. Our data indicate that more than half of the NS5B (∼65%) was assembled into the elongation complex, arguing against the existence of a dimeric complex with one active site. However, slight errors in protein concentration determination or activity determination are possible; therefore, the formation of functional dimers in the elongation complex cannot be fully excluded by the current data.
We found that the productive NS5B·primer·template elongation complex was extraordinarily stable, with a half-life for dissociation of approximately 1 week. Our data also showed that binding or rebinding of primer/template RNA from solution is exceedingly slow (supplemental Fig. S1). These results were consistent with previous reports indicating that the formation of a productive complex of NS5B with duplex RNA was very slow with a half-life of ∼30 h (13), even though the initial binding to RNA to NS5B was fast and diffusion limited (3). The inability of NS5B to bind productively to primer/template RNA from solution is also consistent with the NS5B crystal structure, which shows a steric hindrance of a β-loop and a C-terminal tail close to the polymerase active site interfering of duplex RNA binding (15). These data all suggest that a large conformational rearrangement in the NS5B protein must occur to form a productive complex with RNA. Once duplex RNA is formed via our extension and pause reaction, the NS5B protein has likely completed this conformational change to the elongation complex, such that the RNA duplex becomes locked at the active site in a mode that is kinetically very stable. Because of the very slow on and off rates for RNA from the elongation complex, we are unable to estimate the affinity constant. However, the complex is stable enough to allow complete replication of the HCV genome without dissociation.
A general minimal mechanism for nucleotide incorporation catalyzed by an RNA or DNA polymerase is summarized in Scheme 1 (32). This mechanism includes ground state binding of the incoming nucleotide followed by phosphotidyl transfer reaction to elongate the primer by one base. The dissociation equilibrium constant for nucleotide binding, Kd, and the rate of incorporation, kpol, can be measured readily by transient kinetic methods (12) under single turnover conditions. The direct measurement of the Kd and kpol for incorporation of CTP and GTP showed that these two incorporation events had similar Kd (39 μm versus 22 μm) but about one magnitude change in the incorporation rate kpol (16 s−1 versus 2.1 s−1) (Fig. 5). These values are comparable with the kinetic parameters observed for the incorporation by HIV reverse transcriptase where Kd ∼ 4 μm and kpol ∼ 30 s−1 (33, 34). Compared with Kd measurements of other RNA-dependent RNA polymerases such as poliovirus RNA polymerase (130 μm) and dengue RNA polymerase (275 μm), the Kd for HCV NS5B was much lower, suggesting its active site was tighter and more interactions between the nucleotide and the active site were formed (11, 35). The slow rate of GTP incorporation was not due to a slow step after CTP incorporation and preceding GTP binding such as pyrophosphate release because the rate was measured directly from a preformed elongation complex containing the 10-mer (Fig. 5B). A conformational change preceding chemistry had been demonstrated to play a critical role in several DNA polymerases (24, 36). The rate of nucleotide incorporation thus could be the combination of the rate of the conformational change step and the rate of chemistry. For HCV RNA polymerase, it was not clear whether a similar conformational change step existed. Further investigation is needed using the NS5B elongation complex to resolve these steps.
The processive nucleotide incorporation assay allowed simultaneous quantification of the kinetics of multiple nucleotide incorporation events (Fig. 6). Interestingly, identical nucleotides were incorporated at different rates during elongation, which may be explained by the nearest neighboring and sequence context effects (37). Slight differences in the alignment of all elements at or near the active site might change the catalytic rate significantly. The average rate of nucleotide incorporation was ∼4 s−1 based on our measurement on a 20-mer template. This rate was close to the estimated rates of 200 min−1 ∼ 700 min−1 for NS5B replicating longer RNA templates in vitro (6, 7).
The assembly of a stable and pure NS5B·primer·template ternary complex in this work has removed one major hurdle that has hampered the study of HCV replication enzymology for a long time. This complex will be an essential reagent to study the molecular mechanisms of drug inhibition and drug resistance of HCV NS5B. The methods of assembly and purification developed for HCV NS5B may also apply to other RNA-dependent RNA polymerases that catalyze de novo RNA synthesis. The high yield and the high kinetic stability of the NS5B·primer·template complex also suggest that crystallization of the elongation complex may now be within reach as well.
Supplementary Material
Acknowledgment
We thank Lena Liang for the preparation of HCV NS5BΔ21 protein stock.
Kenneth A. Johnson is president of KinTek Corp., which provided the RQF-3 rapid chemical quench-flow instrument and the KinTek Explorer data fitting software used in this study.

This article contains supplemental Figs. S1–S6.
- HCV
- hepatitis C virus.
REFERENCES
- 1. Tellinghuisen T. L. (2011) An overview of the hepatitis C virus life cycle in Hepatitis C: Antiviral Drug Discovery and Development (Tan S.-L., He Y., eds), pp. 1–48, Caister Academic Press, Norfolk, UK [Google Scholar]
- 2. Dutartre H., Boretto J., Guillemot J. C., Canard B. (2005) A relaxed discrimination of 2′-O-methyl-GTP relative to GTP between de novo and Elongative RNA synthesis by the hepatitis C RNA-dependent RNA polymerase NS5B. J. Biol. Chem. 280, 6359–6368 [DOI] [PubMed] [Google Scholar]
- 3. Cramer J., Jaeger J., Restle T. (2006) Biochemical and pre-steady-state kinetic characterization of the hepatitis C virus RNA polymerase (NS5BΔ21, HC-J4). Biochemistry 45, 3610–3619 [DOI] [PubMed] [Google Scholar]
- 4. Zhong W., Ferrari E., Lesburg C. A., Maag D., Ghosh S. K., Cameron C. E., Lau J. Y., Hong Z. (2000) Template/primer requirements and single nucleotide incorporation by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 74, 9134–9143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selisko B., Dutartre H., Guillemot J. C., Debarnot C., Benarroch D., Khromykh A., Desprès P., Egloff M. P., Canard B. (2006) Comparative mechanistic studies of de novo RNA synthesis by flavivirus RNA-dependent RNA polymerases. Virology 351, 145–158 [DOI] [PubMed] [Google Scholar]
- 6. Lohmann V., Roos A., Körner F., Koch J. O., Bartenschlager R. (1998) Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology 249, 108–118 [DOI] [PubMed] [Google Scholar]
- 7. Tomei L., Vitale R. L., Incitti I., Serafini S., Altamura S., Vitelli A., De Francesco R. (2000) Biochemical characterization of a hepatitis C virus RNA-dependent RNA polymerase mutant lacking the C-terminal hydrophobic sequence. J. Gen. Virol. 81, 759–767 [DOI] [PubMed] [Google Scholar]
- 8. Carroll S. S., Sardana V., Yang Z., Jacobs A. R., Mizenko C., Hall D., Hill L., Zugay-Murphy J., Kuo L. C. (2000) Only a small fraction of purified hepatitis C RNA-dependent RNA polymerase is catalytically competent: implications for viral replication and in vitro assays. Biochemistry 39, 8243–8249 [DOI] [PubMed] [Google Scholar]
- 9. Spence R. A., Kati W. M., Anderson K. S., Johnson K. A. (1995) Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Science 267, 988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel S. S., Wong I., Johnson K. A. (1991) Pre-steady-state kinetic analysis of processive DNA replication, including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 [DOI] [PubMed] [Google Scholar]
- 11. Arnold J. J., Cameron C. E. (2004) Poliovirus RNA-dependent RNA polymerase (3Dpol): Pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry 43, 5126–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson K. A. (1992) Transient-state kinetic analysis of enzyme reaction pathways. The Enzyme XX, 1–61 [Google Scholar]
- 13. Liu Y., Jiang W. W., Pratt J., Rockway T., Harris K., Vasavanonda S., Tripathi R., Pithawalla R., Kati W. M. (2006) Mechanistic study of HCV polymerase inhibitors at individual steps of the polymerization reaction. Biochemistry 45, 11312–11323 [DOI] [PubMed] [Google Scholar]
- 14. Deval J., Powdrill M. H., D'Abramo C. M., Cellai L., Götte M. (2007) Pyrophosphorolytic excision of nonobligate chain terminators by hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 51, 2920–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lesburg C. A., Cable M. B., Ferrari E., Hong Z., Mannarino A. F., Weber P. C. (1999) Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6, 937–943 [DOI] [PubMed] [Google Scholar]
- 16. Bressanelli S., Tomei L., Roussel A., Incitti I., Vitale R. L., Mathieu M., De Francesco R., Rey F. A. (1999) Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 96, 13034–13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biswal B. K., Cherney M. M., Wang M., Chan L., Yannopoulos C. G., Bilimoria D., Nicolas O., Bedard J., James M. N. (2005) Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol. Chem. 280, 18202–18210 [DOI] [PubMed] [Google Scholar]
- 18. Hong Z., Cameron C. E., Walker M. P., Castro C., Yao N., Lau J. Y., Zhong W. (2001) A novel mechanism to ensure terminal initiation by hepatitis C virus NS5B polymerase. Virology 285, 6–11 [DOI] [PubMed] [Google Scholar]
- 19. Maag D., Castro C., Hong Z., Cameron C. E. (2001) Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 276, 46094–46098 [DOI] [PubMed] [Google Scholar]
- 20. Chinnaswamy S., Yarbrough I., Palaninathan S., Kumar C. T., Vijayaraghavan V., Demeler B., Lemon S. M., Sacchettini J. C., Kao C. C. (2008) A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J. Biol. Chem. 283, 20535–20546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lévêque V. J., Johnson R. B., Parsons S., Ren J., Xie C., Zhang F., Wang Q. M. (2003) Identification of a C-terminal regulatory motif in hepatitis C virus RNA-dependent RNA polymerase: Structural and biochemical analysis. J. Virol. 77, 9020–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson K. A., Simpson Z. B., Blom T. (2009) Global kinetic explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 23. Johnson K. A., Simpson Z. B., Blom T. (2009) FitSpace explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 24. Tsai Y. C., Johnson K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhong W., Uss A. S., Ferrari E., Lau J. Y., Hong Z. (2000) De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 74, 2017–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bellón-Echeverría I., López-Jiménez A. J., Clemente-Casares P., Mas A. (2010) Monitoring hepatitis C virus (HCV) RNA-dependent RNA polymerase oligomerization by a FRET-based in vitro system. Antiviral Research 87, 57–66 [DOI] [PubMed] [Google Scholar]
- 27. Wang Q. M., Hockman M. A., Staschke K., Johnson R. B., Case K. A., Lu J., Parsons S., Zhang F., Rathnachalam R., Kirkegaard K., Colacino J. M. (2002) Oligomerization and cooperative RNA synthesis activity of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 76, 3865–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin W., Luo H., Nomura T., Hayashi N., Yamashita T., Murakami S. (2002) Oligomeric interaction of hepatitis C virus NS5B is critical for catalytic activity of RNA-dependent RNA polymerase. J. Biol. Chem. 277, 2132–2137 [DOI] [PubMed] [Google Scholar]
- 29. Qin W., Yamashita T., Shirota Y., Lin Y., Wei W., Murakami S. (2001) Mutational analysis of the structure and functions of hepatitis C virus RNA-dependent RNA polymerase. Hepatology 33, 728–737 [DOI] [PubMed] [Google Scholar]
- 30. Chinnaswamy S., Murali A., Li P., Fujisaki K., Kao C. C. (2010) Regulation of de novo-initiated RNA synthesis in hepatitis C virus RNA-dependent RNA polymerase by intermolecular interactions. J. Virol. 84, 5923–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Labonté P., Axelrod V., Agarwal A., Aulabaugh A., Amin A., Mak P. (2002) Modulation of hepatitis C virus RNA-dependent RNA polymerase activity by structure-based site-directed mutagenesis. J. Biol. Chem. 277, 38838–38846 [DOI] [PubMed] [Google Scholar]
- 32. Johnson K. A. (1993) Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713 [DOI] [PubMed] [Google Scholar]
- 33. Kati W. M., Johnson K. A., Jerva L. F., Anderson K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 34. Kellinger M. W., Johnson K. A. (2010) Nucleotide-dependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 107, 7734–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin Z., Deval J., Johnson K. A., Swinney D. C. (2011) Characterization of the elongation complex of dengue virus RNA polymerase: Assembly, kinetics of nucleotide incorporation, and fidelity. J. Biol. Chem. 286, 2067–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin Z., Johnson K. A. (2011) Role of a GAG hinge in the nucleotide-induced conformational change governing nucleotide specificity by T7 DNA polymerase. J. Biol. Chem. 286, 1312–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mendelman L. V., Boosalis M. S., Petruska J., Goodman M. F. (1989) Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem. 264, 14415–14423 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.