

Abstract
At high concentrations, glutamate (Glu) exerts potent neurotoxic properties, leading to irreversible brain damages found in numerous neurological disorders. The accepted notion that Glu homeostasis in brain interstitial fluid is maintained primarily through the activity of Glu transporters present on glial cells does not take into account the possible contribution of endothelial cells constituting the blood–brain barrier (BBB) to this process. Here, we present evidence for the presence of the Glu transporters, excitatory amino-acid transporters (EAATs) 1 to 3, in porcine brain endothelial cells (PBECs) and show their participation in Glu uptake into PBECs. Moreover, transport of Glu across three in vitro models of the BBB is investigated for the first time, and evidence for Glu transport across the BBB in both directions is presented. Our results provide evidence that the BBB can function in the efflux mode to selectively remove Glu, via specific transporters, from the abluminal side (brain) into the luminal compartment (blood). Furthermore, we found that glial cells lining the BBB have an active role in the efflux process by taking up Glu and releasing it, through hemichannels, anion channels, and possibly the reversal of its EAATs, in close proximity to ECs, which in turn take up Glu and release it to the blood.
Keywords: astrocytes, blood–brain barrier, coculture, excitatory amino-acid transporters, glutamate, in vitro model
Introduction
The amino-acid -glutamate (Glu) serves as the primary excitatory neurotransmitter in the mammalian central nervous system. Under physiological conditions, it is well accepted that the Glu released in the synaptic cleft is being cleared by the action of a family of Na+-dependent high-affinity Glu transporters, also known as excitatory amino-acid transporter (EAAT), located on the plasma membrane of neurons and glial cells. (Beart and O'Shea, 2007). However, this widely accepted view on the homeostasis of Glu in brain fluids does not take into consideration the possible contribution of the blood–brain barrier (BBB).
The BBB is an essential barrier for the normal function of the central nervous system (Ballabh et al, 2004). It is formed by brain endothelial cells (ECs) lining the cerebral microvasculature, together with perivascular elements such as closely associated astrocytic end-feet processes, perivascular neurons, and pericytes (Cecchelli et al, 2007). The brain EC membrane is asymmetric and composed of a luminal membrane (L, facing the blood) and an abluminal membrane (A, facing the brain) characterized by either qualitative or quantitative difference in expression of selected membrane components, including specific influx and efflux transporters (Cornford and Hyman, 2005).
A facilitative Na+-independent saturable transport of Glu has been described to be present exclusively on the luminal membrane of EC (Benrabh and Lefauconnier, 1996; Lee et al, 1998), and a high-affinity Na+-dependent Glu uptake was found to exist in rat brain microvessels (Hutchison et al, 1985) and restricted to the abluminal membrane of EC (O'Kane et al, 1999). O'Kane et al (1999) described the existence of three members of the high-affinity Na+-dependent Glu transporters, EAATs 1 to 3, on the abluminal side of brain capillary EC using membrane vesicles, and suggested that the BBB has a role in the removal of brain Glu. Indeed, in vivo studies indicate that a naturally occurring brain-to-blood Glu efflux does exist (Al-Sarraf et al, 2000; Berl et al, 1961; Davson et al, 1982; Gottlieb et al, 2003; Hosoya et al, 1999).
The notion that the homeostasis of Glu in the brain extracellular space is maintained not only by the glial and neuronal transporters but also by those present on the brain capillary ECs is supported by the fact that the brain is highly vascularized and nearly every neuron in the brain has its own capillary located at an average distance of 8 to 20 μm (Spencer and Verma, 2007). Such a distance is reached, for instance, by Glu diffusing from the synaptic cleft after long-term potentiation (Carter and Regehr, 2000).
The purpose of the present study was to determine and verify the existence of Glu transporters, and evaluate for the first time the Glu transport activity across the BBB using functional in vitro models of the BBB based on porcine brain endothelial cells (PBECs) cultured alone or together with rat glial cells in noncontact or contact cocultures (Figure 1). The fact that there are some similarities between the porcine and human vascular physiology (Hughes, 1986) makes this porcine model highly relevant and suitable for studying the BBB. Surprisingly, we found evidence for the participation of the glial cells that are in contact with the endothelial cells in the removal of Glu from the brain to the blood. The data obtained herein strengthen the concept that the BBB contributes to brain Glu homeostasis and detoxification.
Figure 1.

In vitro models of blood–brain barrier (BBB): schematic drawings of the three (A–C) in vitro BBB culture systems studied.
Materials and methods
Materials
Dulbecco-modified Eagle's medium was purchased from Gibco (Carlsbad, CA, USA). Trypsin EDTA (solution B), fetal calf serum, gentamicin, glutamine, newborn calf serum, and penicillin/streptomycin were obtained from Biological Industries (Beit-Haemek, Israel). [U-14C]-sucrose (633 mCi/mmol) and L-[U-3H]-glutamic acid (55 Ci/mmol) were purchased from Amersham Biosciences (Uppsala, Sweden). For the immunocytochemistry and Western blot analysis, we used rabbit polyclonal anti-EAATs 1, 2, and 3 (Santa Cruz, CA, USA). Biotin-labeled anti-rabbit (Jackson ImmunoResearch, West Grove, PA, USA), Cy3-conjugated streptavidin (Jackson ImmunoResearch), and Alexa Fluor 546-conjugated goat anti-mouse (Molecular Probes, Eugene, OR, USA) were used as secondary antibodies. Dihydrokainic acid (DHK) and -threo-β-benzyloxyaspartate (-TBOA) were purchased from Tocris (Ellisville, MO, USA). All other materials used were from Sigma-Aldrich (Rehovot, Israel).
All procedures involving animals were reviewed and approved by the Weizmann Institutional Animals Care Committee.
Primary Cell Cultures
Primary cultures of ECs were established from freshly collected porcine brain as described previously (Franke et al, 2000). Primary cultures of mixed glial cells were isolated from the brain of neonatal (P1 to P2) Wistar rats (Harlan, Rehovot, Israel) as described previously (Cohen-Kashi Malina et al, 2009).
Immunofluorescence Staining and Flow Cytometry Analysis
Porcine brain endothelial cell monolayers, reaching confluence, were harvested using trypsin and then subjected to single immunofluorescence staining using polyclonal rabbit anti-EAATs 1 to 3 (1:50, 100 μL/tube, 30 minutes on ice), followed by FITC-conjugated anti-rabbit immunoglobulin G (1:100, 100 μL/tube, 30 minutes on ice). For antibody isotype control staining, normal rabbit immunoglobulin Gs and similar secondary antibodies were used. Cells stained with the secondary antibody only served as additional negative controls. After secondary staining with the FITC-labeled antibody, 10,000 cells were analyzed by FACSort and the percentage of positive cells, after subtraction of the parallel region in the isotype control staining, was calculated. A shift in fluorescence was detected when cells were probed with anti-EAAT antibodies.
Glutamate Uptake
For Glu uptake experiments, PBECs were cultured on 24-well plates at a density 125,000 cells/well precoated with rat-tail collagen (27 μg/mL). In such a configuration, the endothelial cells grow with their abluminal side facing the collagen-coated plate (which mimics the basement membrane), while exposing to the cell culture medium their luminal side (Zhang and Liu, 1999). At the day of the experiment (day in vitro 3), the culture medium was removed and the cells were preincubated at 37°C for 10 minutes with 250 μL of warm (37°C) Tris-buffer solution (TB) containing 5 mmol/L Tris base, 140 mmol/L NaCl, 2.5 mmol/L KCl, 1.2 mmol/L K2HPO4, 1.2 mmol/L CaCl2, 1.2 mmol/L MgCl2, 10 mmol/L -glucose, and 10 mmol/L HEPES, pH 7.4.
When studying sodium-independent effects, a TB–choline solution was prepared in which NaCl was substituted by an equimolar concentration of choline chloride. At the end of the 10-minute preincubation time, the buffer was removed and fresh TB–choline containing 91 nmol/L of [3H]-Glu was added for a duration of 25 minutes.
For studying the concentration dependence of Glu uptake, several concentrations of unlabeled Glu were added. Uptake was terminated by washing the cells three times with ice-cold TB–choline. The cells were then lysed with 0.5N NaOH/0.05% SDS solution and 100 μL of the lysate was analyzed for radioactivity in 4 mL of scintillation liquid (AquaSafe, Zinsser Analytic, Frankfurt, Germany), and disintegration per minute (DPMs) were counted in a β-counter. Protein concentrations were determined by BCA protein assay kit (Pierce, Rockford, IL, USA). Na+-dependent uptake was defined as the difference in radioactivity accumulated in Na+-containing buffer and in choline-containing buffer.
In Vitro Blood–Brain Barrier Models
For the in vitro BBB models, 250,000 PBECs were seeded on a transwell insert precoated with rat-tail collagen (27 μg/mL). The cells were grown to confluence for 3 days in ‘plating medium' (10% newborn calf serum, 2 mmol/L -glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 0.1 mg/mL gentamicin in Earl's medium 199). The medium was then replaced to a serum-free medium named ‘assay medium' (2 mmol/L -glutamine, penicillin/streptomycin, 0.1 mg/mL gentamicin, and 550 nm hydrocortisone in Dulbecco's Modified Eagle's Medium (DME)/Ham's F12 medium) for an additional 24 hours. The ‘assay medium' contains 50 μmol/L of glutamic acid.
For the noncontact and contact cocultures of PBECs with rat brain glial cells (Figure 1), mixed glial cells were seeded as described previously (Cohen-Kashi Malina et al, 2009).
Permeability Studies
For transport assays, the transwell inserts were transferred to a new 12-well plate and incubated for 30 minutes with fresh assay medium (1.5 mL) at the abluminal side. To avoid barrier damage, the ‘assay medium' at the luminal side was not changed. At the end of the preincubation period, to measure the luminal-to-abluminal (L-to-A) or abluminal-to-luminal (A-to-L) flux, 2.5 μCi of [3H]-radiolabeled Glu and 1.5 μCi of [U-14C]-radiolabeled sucrose or mannitol were added to the luminal or abluminal side of the inserts, respectively. During the transport assay, the cells were incubated at 37°C, and every 10 minutes for a total duration of 40 minutes, samples of 100 μL from the luminal side or 200 μL from the abluminal side were collected into 4 mL of scintillation liquid, and DPMs were counted in a β-counter. When the pharmacology of Glu transport was studied, 100 μmol/L Glu was added to the assay medium. For calculation of permeabilities, blank filters with no PBECs seeded on them were used. As blank filters for the contact coculture model, filters with only glial cells at the bottom of the insert were used. The total permeability coefficient (Pt) was derived from the slope of the calculated clearance (Cl) curve according to the following equations:
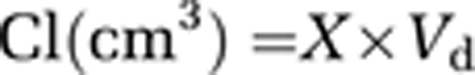 |
where X is the fraction of total compound transferred from donor to acceptor compartment at each time point and Vd is the volume of the donor chamber.
Division of the calculated slope by time (Δt) and the filter surface area (A) leads to the total permeability (Pt in cm/second):
To correct for the contribution of filter and substrate, P is also determined for the cell-free system (Pf). The filter-independent permeability for only the cell layer (Pe) was calculated as follows:
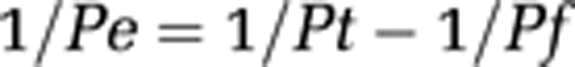 |
Immunocytochemistry
The purity of the primary cultures was verified using specific endothelial and glial cell markers, Von Willebrand factor, and glial fibrillary acidic protein, respectively, as described previously (Cohen-Kashi Malina et al, 2009). For the immunostaining of EAATs 1 to 3, PBECs were grown (250,000 cells/24-well) on rat-tail collagen-coated slides (27 μg/mL, 180 μl/slide in a 24-well plate, dried for 24 hours). The cells were then fixed with 4% paraformaldehyde in water for 10 minutes and exposed to blocking solution made of 20% normal horse serum/0.1% Triton X-100/phosphate-buffered saline for 1 hour. The PBECs were then incubated with the respective polyclonal antibodies at 1:50 concentrations (ON, 4°C), washed with phosphate-buffered saline, and stained with biotin goat anti-rabbit antibody (1:100 in phosphate-buffered saline, 1.5 hours), followed by incubation with Cy-3-conjugated streptavidin (0.5 hour). All steps were performed at room temperature. After mounting (Aqua-Poly/Mount, Polysciences, Eppelheim, Germany), the slides were observed and photographed using Nikon (Tokyo, Japan) Eclipse E600 fluorescent microscope.
Western Blot and On-Cell Western Blot Analysis
For the detection of EAATs 1, 2, and 3, whole-cell lysates from PBECs grown on 24-well plates were extracted using lysis buffer (2.5 mmol/L Tris/HCl, pH 7.4, 0.25% SDS, 1 μmol/L dithiothreitol, 1 μmol/L MgCl, 1 μmol/L ethylene glycol tetraacetic acid, and 1% antiprotease cocktail (Sigma, Rehovot, Israel)) and loaded on 10% SDS-polyacrylamide gel electrophoresis. Proteins on the gels were transferred by electroblotting to 0.2 mm nitrocellulose membranes. The nitrocellulose membranes were blocked for 1 hour in 5% nonfat milk in TB saline containing 0.1% Tween 20 (TTBS). After washing four times with TTBS, the blots were incubated over night (ON) at 4°C with the appropriate first antibodies (1:200) in TTBS containing 1% blocking agent. The blots were than washed with TTBS, incubated with anti-rabbit horseradish peroxidase-conjugated immunoglobulin G for 1 hour (1:2000), washed again, and processed for immunoreactivity using Supersignal chemiluminescence detection kit (Pierce).
For detection of the Glu transporters on the surface membrane of ECs, we performed an on-cell Western blot, which was analyzed in an Odyssey apparatus according to the protocol supplied by the manufacture (LI-COR Biosciences, Lincoln, NE, USA). In short, PBECs were seeded on 96-well plates. At day 3 after seeding, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 minutes and exposed to a blocking solution made of 5% nonfat milk in TTBS. Then, the cells were incubated with the antibodies at concentrations of 1:50 (ON, 4°C). After several washing steps, the cells were probed with IRdye 800CW goat anti-rabbit (LI-COR Biosciences) at a concentration of 1:1000 (1 hour room temperature). As a control, cells were probed only with the secondary antibody.
Results
Porcine Brain Endothelial Cells Express the Glutamate Transporters Excitatory Amino-Acid Transporters 1 to 3
To confirm the existence of the Glu transporters EAATs 1 to 3 in PBECs, Western blot analysis was performed on pure PBEC cultures grown as monolayers and devoid of any glial fibrillary acidic protein-positive immunoreactivity (data not shown). As can be seen in Figure 2A, immunoreactive bands for EAAT1, EAAT2, and EAAT3 were observed in PBECs at their expected molecular weights. The expression of EAATs 1 to 3 on PBECs was further verified by immunocytochemical staining of fixed PBECs. A clear positive staining over background for the three transporters could be detected (Figure 2B), although the EEAT1 signal is weak and the bulk of the signals is intracellular.
Figure 2.

Expression of excitatory amino-acid transporters (EAATs) 1 to 3 in porcine brain endothelial cells (PBECs). (A) Western blot analysis showing EAATs 1 to 3 immunoreactivity in PBEC lysates. (B) Immunocytochemistry of glutamate transporters in PBECs. PBEC cultures were stained using a rabbit polyclonal antibody to EAATs 1, 2, and 3, and nuclei were counterstained with Hoechst. (C) On-cell Western blot analysis showing the expression of EAATs 1 to 3 in PBECs. (D) Fluorescence-activated cell sorting analysis of surface membrane expression of EAATs 1 to 3 in PBECs. Fluorescence profiles were recorded in a fluorescence-activated cell sorter. A shift in fluorescence was detectable when cells were probed with anti-EAAT antibodies. “The color reproduction of this figure is available on the Journal of Cerebral Blood Flow and Metabolism online.”
Because EAATs 1 to 3 are membranous proteins, we also examined their expression on the endothelial plasma membrane by on-cell Western blot and EAAT-specific immunofluorescence staining and flow cytometry. Figures 2C and 2D show that PBECs indeed express on their plasma membrane the three transporters.
Glutamate Uptake into Porcine Brain Endothelial Cells
We next investigated the kinetic properties of Glu uptake by monolayers of cultured PBECs. One should mention that the previous studies were restricted to membrane vesicles derived from bovine brain ECs (Lee et al, 1998; O'Kane et al, 1999) or based on in situ brain perfusion techniques (Benrabh and Lefauconnier, 1996).
We found that the uptake of Glu by PBECs was specific, because it was reduced significantly in the presence of excess of unlabeled Glu (1 mmol/L, 55%, P<0.001; Figure 3A). In addition, -aspartate (1 mmol/L), which serves as an additional substrate of the EAATs (Nicholls and Attwell, 1990), inhibited the uptake of labeled Glu by PBECs (25%, P<0.01).
Figure 3.

[3H]-glutamate (Glu) uptake by porcine brain endothelial cells (PBECs). (A) Uptake of 91 nmol/L [3H]-Glu in the presence of excess unlabeled Glu, -aspartate (-Asp), and serine. Each bar corresponds to mean±s.e.m. of three experiments performed in triplicates. **P<0.01 and ***P<0.001 compared with uptake of [3H]-Glu (Student's t-test). (B) The PBECs were incubated with 91 nmol/L [3H]-Glu and increasing concentration of unlabeled Glu in the presence or absence of sodium. Each point corresponds to the average from at least five determinations performed in triplicates±s.e.m. (C) Effect of Glu transporter inhibitors on Glu uptake by PBECs. Uptake of 100 μmol/L Glu was measured in the absence (control) or presence of the indicated concentrations of inhibitors. Each point represents the mean±s.e.m. of at least six determinations from at least two distinct experiments. *P<0.05 compared with control (Student's t-test).
Because Glu can also be transported by the ACST2 transporter (Utsunomiya-Tate et al, 1996), which is a Na+-dependent transport system for -alanine, -serine, and -cysteine and is present at the BBB (Palacin et al, 1998), the uptake of Glu by PBECs was examined, as well as in the presence of saturating -serine (1 mmol/L), a substrate of the ACST2 transporter. We found that the uptake of Glu was reduced in the presence of 1 mmol/L -serine (29%, P<0.001), suggesting that, at least, part of the observed Na+-dependent Glu uptake in PBECs takes place via ACST2 transporters.
The concentration dependence of Glu uptake by PBECs was then studied using a range of cold Glu concentration (Figure 3B). To distinguish the contributions of putative Na+-dependent and Na+-independent Glu transports, the Glu uptake experiments were performed either in the presence of NaCl or of an equimolar concentration of choline chloride. Nonlinear regression analysis was performed, using the PRISM software (GraphPad), to yield Km and Vmax values. We found that in the presence of NaCl (total uptake), the Km and Vmax values were 367±15 μmol/L and 656±21 pmol/mg protein/minute, respectively. The Km and Vmax values for the Na+-independent Glu uptake were 260±6 μmol/L and 355±6 pmol/mg protein/minute, respectively. The Na+-dependent Glu uptake was obtained by subtracting the uptake measured in the presence of choline chloride from the total uptake. This procedure revealed Km and Vmax values of 94±15 μmol/L and 82±13 pmol/mg protein/minute, respectively. These results suggest that two saturable specific uptake mechanisms coexist in PBECs.
Pharmacology of Glutamate Uptake
To further characterize the Glu uptake in PBECs and determine whether the Na+-dependent Glu uptake is mediated by the EAATs present on PBECs, the uptake of Glu was measured in the absence or presence of specific EAAT inhibitors, DHK and -TBOA. Dihydrokainic acid is a selective, nontransported inhibitor of EAAT2, and -TBOA is a nonspecific potent inhibitor of EAAT1, 2, and 3 (Shimamoto et al, 1998). We found that 0.5 mmol/L DHK (at ∼20-fold increased concentration of its reported Ki (dissociation constant for inhibitor binding); Arriza et al, 1994) did not inhibit the Glu uptake into PBECs, whereas 300 μmol/L -TBOA (at 4-fold increased concentration of its reported Ki; Shimamoto et al, 1998) caused a significant inhibition of the total Glu uptake (36% inhibition, P<0.05). No statistically significant inhibition of Glu uptake was observed when increased concentration of DHK (5 mmol/L) was applied (data not shown). When DHK and -TBOA were applied together, a similar inhibition as for -TBOA alone was achieved (41% Figure 3C). These results suggest the Na+-dependent Glu uptake in PBECs is mediated, at least in part, by EAAT1 and/or EAAT3, whereas the contribution of EAAT2 (the homologs of the glial transporter GLT1) is apparently negligible.
Glutamate Transport Across the Blood–Brain Barrier
The Glu transport across PBECs forming an in vitro model of the BBB was studied not only to establish the ability of the BBB to transport Glu but also to determine the sidedness of this transport. Hence, the transport of [3H]-Glu was measured in the A-to-L direction, thereby mimicking its efflux from the brain into the blood, as well as that in the L-to-A direction, mimicking its influx from the blood into the brain. In view of the pivotal importance of the interactions of glia cells with the brain endothelial cells when studying solute transport across the BBB, the permeability of [3H]-Glu was measured and compared in three in vitro models of the BBB monoculture, noncontact coculture, and a contact coculture (Table 1). The permeability of [14C]-sucrose, as a marker for the paracellular transport across the BBB, was measured in parallel to that of [3H]-Glu.
Table 1. Pe value of [3H]-Glu across PBECs in the three BBB in vitro models.
| In vitro model |
A-to-L Pe value (cm/second × 10−6) |
A-to-L TEER (Ω/cm2) |
L-to-A Pe value (cm/second × 10−6) |
L-to-A TEER (Ω/cm2) | ||
|---|---|---|---|---|---|---|
| Glu | Sucrose | Glu | Sucrose | |||
| Monoculture | 2.84±0.41 | 1.09±0.3 | 306.6±83.4 | 1.87±0.25 | 0.56±0.13 | 296.5±41.5 |
| Noncontact coculture | 1.75±0.07 | 0.44±0.04 | 855.5±208.4 | 1.36±0.25 | 0.26±0.09 | 491.1±14.4 |
| Contact coculture | 3.31±1.29 | 0.38±0.03 | 1132.6±337 | 1.38±0.21 | 0.19±0.03 | 943.6±93.9 |
A-to-L, abluminal-to-luminal; BBB, blood–brain barrier; Glu, glutamate; L-to-A, luminal-to-abluminal; PBEC, porcine brain endothelial cell; Pe, permeability; TEER, transendothelial electrical resistance.
The permeability of [3H]-Glu was significantly higher at the A-to-L direction compared with the L-to-A direction in the three models tested (Table 1, P<0.05, Student's t-test). In addition, we found that the permeability of [3H]-Glu was significantly higher than the corresponding measured permeability of [14C]-sucrose in both directions (Table 1, P<0.0001, Student's t-test).
To rule out the possibility that the observed difference in the Pe values for Glu and sucrose resulted from their difference in size (molecular weight of 342 versus 147 Da for sucrose and Glu, respectively), we measured the A-to-L permeability of [14C]-mannitol, a marker for paracellular transport at the BBB, which has a molecular weight similar to that of Glu (182 Da). The Pe value for [14C]-mannitol across the PBECs in the monoculture model was found to be similar to that of sucrose and significantly smaller than the measured Pe value for Glu (Pe=0.71±0.24 × 10−6 cm/second, P<0.01, Student's t-test).
When the Pe values of [3H]-Glu in the A-to-L direction were compared in the three in vitro models of the BBB, we observed that its Pe value was smaller when PBECs were in noncontact coculture with glial cells as compared with the monoculture model (Table 1, P<0.05, one-way analysis of variance repeated measures test and Bonferroni post-test analysis). As expected, however, there was also an accompanying decrease in the Pe value of sucrose (Cohen-Kashi Malina et al, 2009).
Because the Pe value represents the overall permeability and is composed of the individual contributions of the paracellular and transcellular permeabilities, one can explain the decrease in the overall Glu permeability as a result of the glial cell-induced tightening of the barrier, which decreases the paracellular permeability of Glu. Conversely, when PBECs were cocultured in contact with glial cells, the decrease of the paracellular permeability was reflected by a further reduction in the sucrose Pe value. Surprisingly, the Pe value of Glu significantly increased in comparison with the noncontact coculture (P<0.05; two-way analysis of variance and Tukey post-test analysis). These results indicate that the contact of the PBECs with glial cells caused not only a tightening of the barrier (as reflected by a decrease in the paracellular permeability of both sucrose and Glu) but also caused an increase in the transcellular permeability of Glu. In support of the notion that the presence of glial cells causes a tightening of the barrier and impedes the paracellular transit, the decrease in the permeability measured in the L-to-A direction for sucrose in the contact coculture model of the BBB was also accompanied by a decrease of the Pe value for Glu.
Because the results presented above clearly show that the contact of PBECs with glial cells had a beneficial effect on the A-to-L transcellular transport of Glu, we proceeded to explore the mechanism(s) responsible for the observed beneficial effect of the culture conditions on this transport.
The Pharmacology of Glutamate Transport
The A-to-L permeability of [3H]-Glu across PBECs in the contact coculture model was measured again in the presence of excess of -aspartic acid (0.5 mmol/L) and of unlabeled Glu at 0.1 and 5 mmol/L. In the presence of 0.5 mmol/L -aspartate, the Pe value for [3H]-Glu was reduced by 15% (Figure 4A), whereas 5 mmol/L of unlabeled Glu caused the [3H]-Glu Pe value to decrease significantly by 50% (Figure 4A, P<0.01, Student's t-test). This indicates that the Glu efflux is at least in part transporter-mediated. In addition, the EAATs present on the glial cells and/or the endothelial cells contribute significantly to the A-to-L transport of Glu across the contact coculture in vitro BBB model, as the presence of 300 μmol/L -TBOA reduced the [3 H]-Glu Pe value by 50% (Figure 4B). No inhibitory effect of -TBOA on Glu permeability was observed when control experiments were performed on monoculture or noncontact coculture models of the BBB (data not shown). As in the monolayer experiments, no inhibitory effect on Glu permeability was observed in the presence of 300 μmol/L DHK.
Figure 4.

Permeability (Pe) value of [3H]-glutamate (Glu) across porcine brain endothelial cell monolayer in contact with glial cells. (A) The Pe of [3H]-Glu was measured in the presence or absence of 0.1 and 5 mmol/L of unlabeled Glu and 0.5 mmol/L of -aspartic acid (-Asp). The Pe values are presented as means±s.d. (n=3). **P<0.01. (B) The Pe of 100 μmol/L Glu and [14C]-sucrose was measured in cells in the presence 300 μmol/L -threo-β-benzyloxyaspartate (-TBOA) or dihydrokainic acid (DHK). The Pe values are presented as means±s.d. (n≥3). *P<0.05 compared with control (Student's t-test).
Glutamate Transport Across the Blood–Brain Barrier does not Involve a Glutamine–Glutamate Cycle Between Astrocytes and Endothelial Cells
One of the plausible mechanisms for Glu efflux from the brain extracellular fluids into the blood may involve a glutamine–Glu cycle (Teichberg et al, 2009). In such mechanism, the uptake of Glu into astrocytes would lead to its conversion into glutamine, which would then diffuse within the cell and be released at the astrocytic end-feet. Released glutamine would then be pumped into endothelial cells via glutamine transporters (Lee et al, 1998) and converted back into Glu that would diffuse through the luminal membrane into the blood by a facilitated transport.
To investigate the involvement of such mechanism in the observed increased A-to-L permeability of Glu in the contact coculture model of the BBB, the Pe value of 100 μmol/L Glu was measured again in the presence of methionine sulfoximine (MSO), an inhibitor of the glutamine synthetase responsible for the conversion in astrocytes of Glu to glutamine (Hertz et al, 1999), and 6-diazo-5-oxo--norleucine (DON), an irreversible inhibitor of glutaminase, the endothelial enzyme that converts glutamine into Glu. Both drugs were added to the cells during the preincubation period (30 minutes, see Materials and methods) and were present during the permeability assays as well.
The presence of 3 mmol/L of MSO (Isaacks et al, 1999) caused an increase of the [3H]-Glu Pe value from 4.89±1.5 to 10.37±4.58 × 10−6 cm/second (Figure 5A, P<0.05, Student's t-test). Additionally, 1 mmol/L DON (Jayakumar et al, 2004) did not alter the permeability of Glu in a significant way. These results clearly rule out the involvement of a glutamine–Glu cycle between the brain endothelial cells and the neighboring astrocytes, because the measured permeability coefficients would be expected to decrease as a result of the inhibition of the cycle by MSO and/or DON. The addition of 3 mmol/L MSO or 1 mmol/L DON had no effect on the permeability of Glu measured in a monoculture or a noncontact coculture model of the BBB (data not shown).
Figure 5.

Abluminal-to-luminal permeability (Pe) of 100 μmol/L glutamate and [14C]-sucrose across porcine brain endothelial cell monolayers in contact with glial cells in the presence of 3 mmol/L methionine sulfoximine (MSO) or 1 mmol/L 6-diazo-5-oxo--norleucine (DON; A) or 300 μmol/L suramin, 200 μmol/L 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), 1 μmol/L thapsigargin (TG), and 100 μmol/L carbenoxolone (CBX; B). The Pe values are presented as means±s.d. (n≥3). *P<0.05, **P<0.01, and ***P<0.001.
Glutamate Transport Across the Blood–Brain Barrier Involves Glutamate Release from Astrocytes Through Hemichannels
Another option for the observed beneficial effect of astrocytes on A-to-L Glu permeability could be that the astrocytes take up Glu from the abluminal compartment and release it at their end-feet in close proximity to the Glu transporters present on the endothelial cells. Such dedicated process concentrating Glu at the astrocyte/EC contact should contribute to an increase of the permeability of Glu.
Several Glu release mechanisms exist in astrocytes and include exocytosis, hemichannels, anion transporters, or purinergic P2X receptors (Haydon and Carmignoto, 2006). Examining the possible contribution of these factors to the A-to-L [3H]-Glu Pe, we found that suramin (300 μmol/L), a broad-spectrum antagonist of purinergic receptors, exerted no effect on the measured Glu permeability (Figure 5B). This indicates that the ATP-dependent Glu release by astrocytes is not involved in the A-to-L transport of Glu across the contact coculture in vitro BBB. The nonselective blocker of anion transporters, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (200 μmol/L; Rutledge et al, 1998), caused a small (∼20%), but not statistically significant, decrease in Glu permeability. No further decrease of A-to-L [3H]-Glu Pe value was observed when 1 mmol/L 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid was applied (data not shown).
To investigate the role of Ca2+-dependent exocytotic Glu release from astrocytes, 1 μmol/L of thapsigargin, a blocker of the sarcoendoplasmic reticulum Ca2+–ATPase, was added to the abluminal side, and the permeabilities of Glu and sucrose were measured. Both Glu and sucrose permeabilities increased significantly (P<0.001) in the presence of 1 μmol/L thapsigargin compared with control (Figure 5B). The increased permeability of both compounds is likely because of the effect of thapsigargin on the integrity of the EC barrier, because the formation of TJs in EC relies on intact intracellular Ca2+ stores (Huber et al, 2001). In line with this finding, the addition of 1 μmol/L thapsigargin to a monoculture or noncontact coculture in vitro model of the BBB caused a significant increase in the permeabilities of Glu and sucrose as well (data not shown).
Because astrocytes can release Glu through the functional unpaired connexons (hemichannels) present on their cell surface, the permeabilities of Glu and sucrose were measured in the presence of 100 μmol/L carbenoxolone, a commonly used gap junction blocker. We found that although the permeability to sucrose increased significantly (P<0.01), the permeability of Glu remained the same (Figure 5B).
These results indicate that despite the loosening of the barrier (as reflected by the increase paracellular permeability to sucrose), the transcellular transport of Glu probably decreased as a result of the blockage of astrocytic hemichannels. In support of this notion, 100 μmol/L carbenoxolone caused a significant increase in the permeabilities of both Glu and sucrose when added to monocultures or noncontact cocultures in vitro BBB (data not shown).
Discussion
Glutamate homeostasis in the brain extracellular fluids is of importance, because the maintenance of a very low Glu concentration is crucial for the proper physiological functioning of the brain. High Glu concentrations, as those found in numerous brain insults, cause ultimately the excitotoxic death of the exposed neurons and entails irreversible neurological deficits. In the studies of Glu homeostasis in the brain, an almost exclusive emphasis has been put on the role of glial cells (mainly, astrocytes) and of their Na+-dependent high-affinity Glu transporters (Danbolt, 2001). However, the existence of a brain-to-blood Glu efflux that is modulated by the blood Glu levels (Gottlieb et al, 2003), together with the fact that brain capillary cells carry Glu transporters EAATs 1 to 3 on their antiluminal side, strongly suggests that the brain vasculature and its fine blood capillaries also have a role in Glu homeostasis and brain Glu detoxification.
In this paper, we provide evidence for the existence of immunoreactivities toward the EAATs 1 to 3 proteins in PBECs. This demonstration was based on Western blot, immunocytochemistry, and fluorescence-activated cell sorting analysis. We further characterized the Glu uptake in monolayers of endothelial cells, and analyzed the Glu transport properties of endothelial cells grown on filters either in contact with astrocytes or in noncontact but in the presence of astrocytes at the bottom of the culture well. These studies clearly indicate the ability of the brain endothelial cells to specifically transport Glu and emphasize the role of astrocytes in this process.
Glutamate Uptake by Monolayers of Brain Capillary Endothelial Cells
In this work, we show that Glu transporters present on monolayers of PBECs are clearly functional, because they display sensitivity to specific Glu transport blockers. The Glu uptake occurs, at least in part, through a Na+-dependent mechanism to which the EAATs have a clear contribution as the uptake is inhibited by -aspartate, a well-known substrate of the EAATs (Nicholls and Attwell, 1990), and by the wide-spectrum Glu transporter inhibitor, -TBOA.
In contrast to -TBOA, DHK, a selective inhibitor of EAAT2, fails to inhibit the Glu uptake by PBECs, although the latter express EAAT2 on their cell surface (Figure 2), suggesting that EAAT2 under our experimental conditions has none or a very minor contribution to the Na+-dependent Glu uptake. This finding appears to be in contradiction with the findings of O'Kane et al (1999) that reported that the relative activities of the EAATs in membrane vesicles isolated from bovine brain capillaries were in a ratio of 1:3:6 for EAATs 1, 2, and 3 respectively. The observed differences could be because of differences among animal species, the used methodology (membranal vesicles versus endothelial cells in monolayers) or the experimental conditions. In this respect, it is known that the membranal EAAT3 and EAAT4 transporters, for instance, can interact with the intracellular proteins GTRAPs 3 to 18 and GTRAP41 and 48, respectively (Jackson et al, 2001; Lin et al, 2001), and that the latter can modulate their activity. Because one of the splice variants of EAAT2 (GLT1b) possesses a PDZ domain, which is a known structural motif that serves as a protein-interacting module (Gonzalez-Gonzalez et al, 2008), it could well be that EAAT2, although present at the PBEC cell surface membrane, is not functionally active or has a limited Glu uptake ability because of the lack of an associated activating intracellular protein or the presence of a putative inhibitory protein.
The Na+-dependent Glu uptake by PBECs cannot be solely attributed to EAATs 1 to 3, because it was inhibited by -serine, a substrate of the ASCT2 transporter. The ASCT2 transporter is a Na+-dependent transport system for -alanine, -serine, and -cysteine (Palacin et al, 1998), which was found to be expressed at the BBB (Tayarani et al, 1987) and be able to transport Glu (Utsunomiya-Tate et al, 1996). Our results suggest that such a transporter is present on PBECs and contributes to the overall Na+-dependent Glu uptake by PBECs, although its exact relative contribution awaits further investigations.
The kinetic properties of Glu uptake were measured and two distinct saturable and specific transport system were revealed; a Na+-dependent component with Km and Vmax values of 94±15 μmol/L and 82±13 pmol/mg protein/minute, respectively, and a Na+-independent component with Km and Vmax values of 260±6 μmol/L and 355±6 pmol/mg protein/minute, respectively. The Na+-dependent and independent components are likely to represent the ability of PBECs' abluminal and luminal membranes, respectively, to take up Glu, because Na+-dependent transport systems were identified exclusively on the abluminal membrane of ECs (Hawkins et al, 2006), whereas facilitative transport systems mainly on the luminal membrane of ECs.
The measured Km for the Na+-dependent Glu uptake is in agreement with reported Km values in the literature (1–100 μmol/L), which vary greatly depending on transporter subtype and the assay system (Danbolt, 2001). A possible reason for the apparent dominant contribution of the Na+-independent transport to the overall measured uptake, as reflected by approximately four times higher capacity (Vmax) for Glu, can be because in the monolayer configuration, the endothelial cells grow with their abluminal side facing the collagen-coated plate, whereas their luminal side is exposed to the cell culture medium (Zhang and Liu, 1999). Thus, Glu has an open access to the luminal side, whereas it has to diffuse under the cell layer to be taken up by the abluminal Na+-dependent Glu transporters. This also accounts for the fact that none of the specific Glu uptake inhibitors is able to block Glu uptake by more than 50%.
Glutamate Efflux Through Brain Capillary Endothelial Cells
Three in vitro models of the BBB were used for the in vitro investigations of the Glu transport across the BBB: an endothelial monoculture, a noncontact coculture of endothelial cells with astrocyte, and a contact co culture between endothelial cells and astrocytes (Figure 1). Using these in vitro BBB models, evidence for the ability of [3H]-Glu to cross the BBB in both directions was presented, where the A-to-L efflux, that is, brain-to-blood transport was significantly higher than the L-to-A influx, that is, blood-to-brain transport. These results strengthen the concept that the BBB has an important role in the homeostasis of Glu in brain fluids by removing the excess Glu from the brain into the blood.
In an apparent paradox, we have observed that the A-to-L transport of Glu in the contact coculture model was significantly larger than that taking place in the other configurations. Despite the decrease in Pe value of sucrose, because the model acquires properties close to those of the BBB in vivo, the Pe value of Glu increased significantly in the contact coculture as compared with the noncontact culture. This suggests that the observed increase in Glu Pe value most probably results from an increased transport of Glu through the transcellular pathway, whereas the paracellular pathway becomes restricted. In support of that notion, we have noticed that the expected decrease in the permeability measured in the L-to-A direction for sucrose was also accompanied by a decrease of the Pe value of Glu in the contact coculture.
The smaller A-to-L Pe measured in the noncontact coculture model compared with the monoculture model (1.75±0.07 × 10−6 and 2.84±0.41 × 10−6 cm/second, respectively) indicates that the measured permeability of Glu in the monoculture configuration is likely to reflect a paracellular rather than transcellular transport.
The contribution of the glial cells/astrocytes to the transcellular transport of Glu is expected to result from the close proximity or even from the contact between ECs and glial cells, because glial cells in the noncontact culture did not contribute to an improvement of Glu transport. This close proximity may generate the appropriate conditions for at least three mechanisms to occur: (1) the first mechanism calls for the putative involvement of a glutamine–Glu cycle between the astrocytes and ECs, where Glu is taken up by astrocytes, converted via the glutamine synthetase into glutamine, and released as glutamine from the astrocytic end-feet. This extracellular glutamine would then pumped into ECs via their antiluminal glutamine transporters and converted back, via a glutaminase, into Glu. The latter would then diffuse through the luminal membrane into the blood by a facilitated transport (pathway 2, Figure 6). (2) The second mechanism involves the possible Glu concentrative activity of astrocytes and their Glu release activity. In this scheme, the astrocytic Glu transporters would take up and concentrate Glu from the growth medium and release it by one of the known astrocyte Glu release mechanisms (Haydon and Carmignoto, 2006) in close proximity to the abluminal membrane of ECs. The increase of extracellular Glu taking place at this glutamatergic ‘synapse' should facilitate its transport into ECs via the abluminal Glu transporters. (3) The third mechanism may involve a glia-mediated increased expression of Glu transporters on ECs.
Figure 6.

Glutamate (Glu) transport into neurons, astrocytes, and endothelial cells at the blood–brain barrier. Proposed pathway for the removal of Glu from the brain to the blood. Modified from Hawkins (2009). Gln, glutamine. “The color reproduction of this figure is available on the Journal of Cerebral Blood Flow and Metabolism online.”
We analyzed these respective mechanisms one by one and found no contribution of a glutamine–Glu cycle between the astrocytes and ECs in the A-to-L transport of Glu. In fact, on administration of MSO, which prevents the conversion of Glu to glutamine in astrocyte, an increased permeability to Glu was measured. This could possibly be accounted by the fact that in the presence of MSO, the astrocytic intracellular concentration of Glu is likely to increase, causing larger amounts of Glu to be released at the Glu synapse and be taken up by the ECs' Glu transporters.
A more modest increase in the permeability of Glu was also measured when DON, the irreversible inhibitor of the endothelial glutaminase, was applied. The larger Pe value of Glu could possibly result from the expected decrease in the ECs' intracellular Glu concentration that amplifies the Glu concentration gradient across the endothelial abluminal membrane and facilitates an increased transport of Glu across the in vitro BBB.
Examining the contributions of putative astrocytic Glu release mechanisms, we found that ATP or Ca+2-dependent mechanisms are not involved in the observed increase permeability of Glu. Our results using carbenoxolone, a commonly used gap junction blocker, suggests, however, the contribution of functional connexon unpaired hemichannels, present on the astrocyte surface to the overall transport of Glu across the BBB. Because carbenoxolone was also reported to inhibit Glu release through volume-regulated anion channels at similar concentrations at which it blocked hemichannels (Ye et al, 2009), and as a small decrease in Glu Pe was observed in the presence of the volume-regulated anion channel blocker, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid, it might be that Glu release through anion channels also contributes to the efflux of Glu across the BBB. Larger number of experiments are needed, however, to confirm the observed trend. One cannot rule out, however, that the small effects of some of the tested inhibitors are because of a restricted access to their target sites in the in vitro BBB models.
The transport of Glu in the contact coculture model was inhibited by the presence of -TBOA. The latter, however, had no effect in the mono or the noncontact coculture models. This was possibly expected, because the transport of Glu in the mono and noncontact coculture system appears to result mostly from a nonspecific paracellular diffusion of Glu. The inhibitory effect of -TBOA in the contact coculture model, however, does not help to reveal the cellular location of the blocked EAATs, because -TBOA blocks both astrocytic and EC EAATs.
A possible explanation for the lack of inhibitory effect of DHK on Glu permeability in the contact coculture could be because of a selective loss of GLT1 that has been reported to occur on in vitro culture of astroglia (Gegelashvili and Schousboe, 1997; Kondo et al, 1995). In addition, it is know that the expression of GLT1 is highly regulated by neuronal factors (Gegelashvili et al, 1997) and axonal interactions (Yang et al, 2009) that are clearly missing in our culture model.
As to the possibility of a glia-mediated increased expression of Glu transporters on ECs, we have not been able to properly examine it, because we failed to obtain EC lysates devoid of glial contamination in the contact coculture model as indicated by a clear immunoreactivity toward the astrocytic marker glial fibrillary acidic protein (data not shown). Thus, we cannot rule out the possibility that the glial cells influenced the expression levels of EAATs on ECs.
In summary, we suggest that glial cells have an active role in the efflux of Glu from the brain extracellular spaces into the blood, because they are able to take up Glu and release it in close proximity to the ECs through connexon hemichannels and volume-regulated anion channels and possibly by the reversal of its Glu transporter activity. The ECs would take up the Glu by their antiluminal Na+-dependent transporters and release it into the blood by the facilitative transporters (Figure 6) present on the EC luminal side.
Acknowledgments
We thank Professor H. Galla (Munster, Germany) and his lab members for introducing us to his in vitro model of the BBB.
The authors declare no conflict of interest.
Footnotes
This work was supported by a research grant from Irwin Green Alzheimer's Research Fund and by the Nella and Leon Benoziyo Center for Neurological Diseases. VIT is the incumbent of the Louis and Florence Katz-Cohen Chair of Neuropharmacology.
References
- Al-Sarraf H, Preston JE, Segal MB. Acidic amino acid clearance from CSF in the neonatal versus adult rat using ventriculo-cisternal perfusion. J Neurochem. 2000;74:770–776. doi: 10.1046/j.1471-4159.2000.740770.x. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Beart PM, O′Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benrabh H, Lefauconnier JM. Glutamate is transported across the rat blood-brain barrier by a sodium-independent system. Neurosci Lett. 1996;210:9–12. doi: 10.1016/0304-3940(96)12635-5. [DOI] [PubMed] [Google Scholar]
- Berl S, Lajtha A, Waelsch H. Amino Acid and protein metabolism-VI cerebral compartments of glutamic acid metabolism. J Neurochem. 1961;7:186–197. doi: 10.1111/j.1471-4159.1959.tb12638.x. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6:650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- Cohen-Kashi Malina K, Cooper I, Teichberg VI. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. 2009;1284:12–21. doi: 10.1016/j.brainres.2009.05.072. [DOI] [PubMed] [Google Scholar]
- Cornford EM, Hyman S. Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx. 2005;2:27–43. doi: 10.1602/neurorx.2.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davson H, Hollingsworth JG, Carey MB, Fenstermacher JD. Ventriculo-cisternal perfusion of twelve amino acids in the rabbit. J Neurobiol. 1982;13:293–318. doi: 10.1002/neu.480130402. [DOI] [PubMed] [Google Scholar]
- Franke H, Galla H, Beuckmann CT. Primary cultures of brain microvessel endothelial cells: a valid and flexible model to study drug transport through the blood-brain barrier in vitro. Brain Res Brain Res Protoc. 2000;5:248–256. doi: 10.1016/s1385-299x(00)00020-9. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol. 1997;52:6–15. doi: 10.1124/mol.52.1.6. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gonzalez IM, Garcia-Tardon N, Cubelos B, Gimenez C, Zafra F. The glutamate transporter GLT1b interacts with the scaffold protein PSD-95. J Neurochem. 2008;105:1834–1848. doi: 10.1111/j.1471-4159.2008.05281.x. [DOI] [PubMed] [Google Scholar]
- Gottlieb M, Wang Y, Teichberg VI.2003Blood-mediated scavenging of cerebrospinal fluid glutamate J Neurochem 87119–126.(write to the Help Desk NCBI ∣ NLM ∣ NIH Department of Health & Human Services Privacy Statement ∣ Freedom of Information Act ∣ Disclaimer) [DOI] [PubMed] [Google Scholar]
- Hawkins RA. The blood-brain barrier and glutamate. Am J Clin Nutr. 2009;90:8675–8745. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RA, O′Kane RL, Simpson IA, Vina JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999;57:417–428. [PubMed] [Google Scholar]
- Hosoya K, Sugawara M, Asaba H, Terasaki T. Blood-brain barrier produces significant efflux of L-aspartic acid but not D-aspartic acid: in vivo evidence using the brain efflux index method. J Neurochem. 1999;73:1206–1211. doi: 10.1046/j.1471-4159.1999.0731206.x. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- Hughes HC. Swine in cardiovascular research. Lab Anim Sci. 1986;36:348–350. [PubMed] [Google Scholar]
- Hutchison HT, Eisenberg HM, Haber B. High-affinity transport of glutamate in rat brain microvessels. Exp Neurol. 1985;87:260–269. doi: 10.1016/0014-4886(85)90216-x. [DOI] [PubMed] [Google Scholar]
- Isaacks RE, Bender , Kim CY, Shi YF, Norenberg MD. Effect of ammonia and methionine sulfoximine on myo-inositol transport in cultured astrocytes. Neurochem Res. 1999;24:51–59. doi: 10.1023/a:1020928029845. [DOI] [PubMed] [Google Scholar]
- Jackson M, Song W, Liu MY, Jin L, Dykes-Hoberg M, Lin CI, Bowers WJ, Federoff HJ, Sternweis PC, Rothstein JD. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature. 2001;410:89–93. doi: 10.1038/35065091. [DOI] [PubMed] [Google Scholar]
- Jayakumar AR, Rama Rao KV, Schousboe A, Norenberg MD. Glutamine-induced free radical production in cultured astrocytes. Glia. 2004;46:296–301. doi: 10.1002/glia.20003. [DOI] [PubMed] [Google Scholar]
- Kondo K, Hashimoto H, Kitanaka J, Sawada M, Suzumura A, Marunouchi T, Baba A. Expression of glutamate transporters in cultured glial cells. Neurosci Lett. 1995;188:140–142. doi: 10.1016/0304-3940(95)11408-o. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Hawkins RA, Vina JR, Peterson DR. Glutamine transport by the blood-brain barrier: a possible mechanism for nitrogen removal. Am J Physiol. 1998;274:C1101–C1107. doi: 10.1152/ajpcell.1998.274.4.C1101. [DOI] [PubMed] [Google Scholar]
- Lin CI, Orlov I, Ruggiero AM, Dykes-Hoberg M, Lee A, Jackson M, Rothstein JD. Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3–18. Nature. 2001;410:84–88. doi: 10.1038/35065084. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- O′Kane RL, Martinez-Lopez I, DeJoseph MR, Vina JR, Hawkins RA. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J Biol Chem. 1999;274:31891–31895. doi: 10.1074/jbc.274.45.31891. [DOI] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev. 1998;78:969–1054. doi: 10.1152/physrev.1998.78.4.969. [DOI] [PubMed] [Google Scholar]
- Rutledge EM, Aschner M, Kimelberg HK. Pharmacological characterization of swelling-induced D-[3H]aspartate release from primary astrocyte cultures. Am J Physiol. 1998;274:C1511–C1520. doi: 10.1152/ajpcell.1998.274.6.C1511. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Spencer BJ, Verma IM. Targeted delivery of proteins across the blood-brain barrier. Proc Natl Acad Sci USA. 2007;104:7594–7599. doi: 10.1073/pnas.0702170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayarani I, Lefauconnier JM, Roux F, Bourre JM. Evidence for an alanine, serine, and cysteine system of transport in isolated brain capillaries. J Cereb Blood Flow Metab. 1987;7:585–591. doi: 10.1038/jcbfm.1987.109. [DOI] [PubMed] [Google Scholar]
- Teichberg VI, Cohen-Kashi-Malina K, Cooper I, Zlotnik A. Homeostasis of glutamate in brain fluids: an accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience. 2009;158:301–308. doi: 10.1016/j.neuroscience.2008.02.075. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem. 1996;271:14883–14890. doi: 10.1074/jbc.271.25.14883. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Oberheim N, Kettenmann H, Ransom BR. Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia. 2009;57:258–269. doi: 10.1002/glia.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu G. A novel method to determine the localization of high and low-affinity GABA transporters to the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res Brain Res Protoc. 1999;4:288–294. doi: 10.1016/s1385-299x(99)00031-8. [DOI] [PubMed] [Google Scholar]