

Abstract
The hallmark of acute lung injury (ALI) is the influx of proinflammatory cytokines into lung tissue and alveolar permeability that ultimately leads to pulmonary edema. However, the mechanisms involved in inflammatory cytokine production and alveolar permeability are unclear. Recent studies suggest that excessive production of ceramide has clinical relevance as a mediator of pulmonary edema and ALI. Our earlier studies indicate that the activation of inflammasome promotes the processing and secretion of proinflammatory cytokines and causes alveolar permeability in ALI. However, the role of ceramide in inflammasome activation and the underlying mechanism in relation to alveolar permeability is not known. We hypothesized that ceramide activates the inflammasome and causes inflammatory cytokine production and alveolar epithelial permeability. To test this hypothesis, we analyzed the lung ceramide levels during hyperoxic acute lung injury in mice. The effect of ceramide on activation of inflammasome and production of inflammatory cytokine was assessed in primary mouse alveolar macrophages and THP-1 cells. Alveolar transepithelial permeability was determined in alveolar epithelial type-II cells (AT-II) and THP-1 co-cultures. Our results reveal that ceramide causes inflammasome activation, induction of caspase-1, IL-1β cleavage and release of proinflammatory cytokines. In addition, ceramide further induces alveolar epithelial permeability. Short hairpin RNA silencing of inflammasome components abrogated ceramide-induced secretion of proinflammatory cytokines in vitro. Inflammasome silencing abolishes ceramide induced alveolar epithelial permeability in AT-II. Collectively, our results demonstrate for the first time that ceramide-induced secretion of proinflammatory cytokines and alveolar epithelial permeability occurs though inflammasome activation.
Keywords: Macrophage, Inflammasome, ceramide, reactive oxygen species, acute lung injury, IL-1β
Introduction
The most severe form of acute lung injury (ALI) is acute respiratory distress syndrome (ARDS) which is a major cause for admission to critical care units (Matthay and Idell, 2010). Patients often die by drowning as a result of fluid leaking into the lungs from damaged capillaries (Barnes, 2004, Goggel, et al., 2004). IL-1β is an early cytokine in acute lung injury patients and induces alveolar permeability and causes production of other cytokines such as IL-6 and TNF-α (Ganter, et al., 2008).
Recently, we reported that caspase-1-mediated processing of IL-1β is mediated by the inflammasome (Kolliputi, et al., 2010). Our previous report indicate that inflammasome mediated IL-1β is responsible for alveolar epithelial permeability (Kolliputi, et al., 2010). In our earlier studies we have also shown that inflammasome inhibition blocks hyperoxia induced alveolar permeability and cytokine production (Kolliputi, et al., 2010).
Recently sphingolipids have been recognized as signaling molecules involved in a number of important cellular functions (Ogretmen, et al., 2001, Milhas, et al., 2010). Ceramide has long been recognized as a signaling molecule in the inflammatory response in ALI (Barnes, 2004, Goggel, et al., 2004). Ceramide is also known to induce the expression of multiple inflammatory proteins that amplify the inflammatory response (Goldkorn and Filosto, 2010). Ceramide, released from sphingomyelin is hydrolysed by sphingomyelinase (aSMase) or through de novo synthesis, which in turn, acts as a messenger for apoptotic signaling triggered by various stresses, including oxidative stress, acid, ionizing radiation, and extracellular stimuli such as proinflammatory cytokines and lipopolysaccharide (LPS)(Yao, et al., 1995)(Mathias, et al., 1998). A recent report indicated that suppression of aSMase improves pulmonary function through reduction of ceramide generation in a piglet model of surfactant deficiency-induced ALI (von Bismarck, et al., 2008). In response to various forms of stimuli, ceramide induces apoptosis via activation of caspases and by inducing clustering of death receptors in the cell membrane (Carpinteiro, et al., 2008). Increased alveolar permeability has been ascribed to ceramide-induced activation and cytokine production (Mathias, et al., 1998). However, the role of inflammasome in relation to ceramide-induced activation and inflammatory cytokine production leading to alveolar permeability remains unknown.
In the present study, we studied hyperoxic acute lung injury, in which we demonstrate an increase in the intracellular ceramide levels. Furthermore, we investigated the role of ceramide in inflammasome activation and cytokine release. The mechanisms for ceramide-induced cellular damage was investigated by utilizing a co-culture model of THP-1 cells in combination with AT-II cells, in which we demonstrate the link for the role of NALP-3 inflammasome in relation to alveolar permeability and ALI.
Materials and methods
Reagents
Cell culture growth media, buffers, and fetal bovine serum (FBS) used was purchased from Life Technologies, Inc. (Grand Island, NY). C6-ceramide was obtained from Matreya Inc. (Pleasant Gap, PA). Adenosine triphosphate (ATP) (25 mCi/ml) was retrieved from ICN Biomedical (Costa Mesa, CA). Dithiothreitol (DTT), 2-mercaptoethanol, and all other reagents were purchased from Sigma Chemical Co. (St. Louis, MO). Microscope slides, methanol, chloroform, and all other solvents originated from Fisher Scientific (Houston, TX). NALP-3 antibody was obtained from Enzo Life Sciences, Inc. (Farmingdale, NY) and Cleaved Caspase-1 antibody was purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Isolation of mouse lung macrophages
Alveolar macrophages were isolated as mentioned earlier (Kolliputi, et al., 2010) (Lavnikova, et al., 1993). Briefly, lungs were perfused at 37°C with buffer A (1% NaCl, 0.1% glucose, 25 mM HEPES, 5 mM KCl, 0.1 mg/ml streptomycin sulfate, 0.07 mg/ml penicillin G, 0.05 mg/ml EGTA, 5 mM Na2HPO4, 5 mM NaH2PO4, pH 7.4) followed by five fluid lavages using buffer B (buffer A plus 5 mM MgSO4 and 3 mM CaCl2). Digestion of the remaining lung tissue was achieved by treatment with 7-8 ml elastase (3 U/ml in buffer A) for 15 min at 37°C. After lung tissue was homogenized, the resultant lysate was mixed with 100 μg/ml DNase I, and incubated for 5 min at 37°C under light rotation. The cells were passed through a 160-μm and 37-μm nylon mesh once, and then filtered through a 15-μm nylon mesh twice. The culture dish was scraped, detaching macrophages, which were then collected and centrifuged at 200 × g for 10 min. The resultant pellet was resuspended with 10 ml buffer C (RPMI 1640 medium containing 25 mM HEPES and 1% FBS) at a final concentration of 15 × 106 cells/ml as mentioned earlier (Lavnikova, et al., 1993). Animal procedures were conducted in accordance to institutional standards and with official IACUC approval.
THP-1 culture and cytokine analysis
THP-1 cell cultures were grown following predetermined protocols (Dostert, et al., 2008). Observation of inflammasome activation followed the standard two step activation of THP-1 cells as mentioned earlier (Kahlenberg and Dubyak, 2004, (Dostert, et al., 2008). THP-1 supernatants were then tested for cytokine levels. Quantification of cytokines by ELISA: IL-1β, TNF-α, and IL-6 cytokines found in extracellular culture media were measured using commercial ELISA kits (Pierce, Rockford, IL) according to manufacturer instructions.
Measurement of transepithelial albumin flux
Alveolar type II (ATII) cells were isolated as mentioned earlier (Ganter, et al., 2008)(Barker, et al., 2006) and cultured in 24-well transwell plates until the monolayers achieved 100% confluence. THP-1 cells (1.5 × 105) were inserted to the ATII monolayers, cultured along with ATII monolayers for 3 h, and then treated with ceramide. Some experiments used THP-1 cells transfected with control short hairpin RNA (shRNA) or NALP-3 shRNA which were then added to the monolayer of ATII cells. Transepithelial albumin flux across each monolayer was tested by the addition of 0.05 μCi [125I]-labeled human serum albumin along all upper compartments followed by incubation for 1 h, after which the contents from each lower compartment was collected and counted using a Wizard γ-counter (Perkin-Elmer, Waltham, MA) as mentioned earlier(Ganter, et al., 2008, Kolliputi, et al., 2010).
Determination of Lung Ceramide Levels by Diacylglycerol Kinase Assay
Ceramide quantification used a diacylglycerol (DAG) kinase assay as mentioned earlier (Taga, et al., 1997). Cells were extracted using methanol: chloroform: 1 N HCl (100:100:1, vol/vol/vol). The organic phase lipids were dried under vacuum and resuspended in 100 μl of [P32]-ATP reaction mixture followed by incubation at room temperature for 1 h. The reactions were halted by lipid extraction using 1 ml methanol: chloroform: 1 N HCl, 170 μl of buffered saline solution, and 30 μl of 0.1 M EDTA. The lower organic phase was dried under vacuum, and the lipids were identified by thin layer chromatography (TLC) using chloroform: methanol: acetic acid (65:15:5, vol/vol/vol) solvent. Addition of [32P] into ceramide was quantified using liquid scintillation counting after removing the spots from the plates. Comparison of the standard curve using C16-ceramide as substrate allowed for the determination of ceramide concentration. Six independent experiments were performed with at least five mice per group.
Statistical analysis
Triplicates, if not more, of each experiment were conducted. Granted relevance, all appropriate data was expressed as mean ± SEM. Data sets were analyzed using one-way and two-way ANOVA tests, with individual group means being compared using the Student unpaired t test.
Results
Hyperoxia induced ceramide levels in the lung are correlated to alveolar protein leak in BAL fluid and pulmonary edema: To induce acute lung injury, C57BL/6 mice were exposed to hyperoxia (100% O2)(Bhandari, et al., 2006, Bhandari, et al., 2007). Hyperoxic acute lung injury is a well established animal model to study human ALI and ARDS (Bhandari, et al., 2006, Bhandari, et al., 2007, Corne, et al., 2000, He, et al., 2005). In animal models, hyperoxia-induced acute lung injury mimics human ALI (Bhandari, et al., 2006, Zhang, et al., 2009). To test the hypothesis that the ceramide pathway contributes to hyperoxia-induced pulmonary edema, we analyzed lung ceramide levels at various time points of hyperoxia exposure. We found that exposure to hyperoxia (100% O2) causes time dependent increase in lung ceramide levels (Fig.1A). The increase in ceramide levels at 24, 48 and 72 hour time point after hyperoxia was highly significant (p<0.001) compared with control (normoxia). To assess the functional damage in terms of lung edema, we monitored the wet to dry ratio of the lung tissue. As shown in Fig.1B, hyperoxia caused a time dependent increase in edematous lung injury. The edematous lung damage caused by hyperoxia at 24, 48 and 72 hours was highly significant (p<0.001) compared with controls. These results clearly indicate that hyperoxia causes increase in ceramide levels and lung edema. We also monitored the total protein leak caused by hyperoxia. As shown in Fig. 1C, hyperoxia caused a significant increase in alveolar leak at 24, 48 and 72 hours compared with controls. The increase in total protein leak is indicative of lung damage and suggests that with increased exposure to hyperoxia the ceramide levels increases leading to lung edema and total protein leak. Furthermore, we assessed the hyperoxia induced lung damage by monitoring the infiltration of macrophages. As shown in Fig. 1D, up on exposure to hyperoxia for 24, 48 and 72 hours, a statistically significant increase in macrophage infiltration compared with control was observed. Taken together these results suggest that alveolar protein leak and pulmonary edema in hyperoxia-induced acute lung injury may be due to accumulation of ceramide levels in the lung tissue and is accompanied by an enhanced infiltration of macrophages.
Figure1. Hyperoxia induced lung injury causes increase of intracellular ceramide levels.
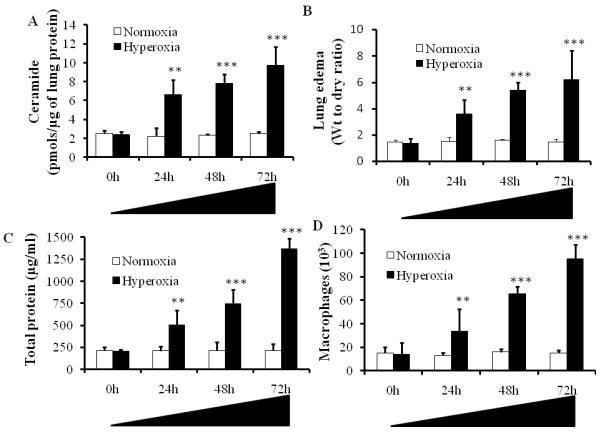
Ceramide concentrations were analyzed in homogenized lung tissues at different stages of hyperoxia-induced acute lung injury. C57BL/6 mice were exposed to hyperoxia (100% O2) for 0, 24, 48 and 72h time points and lung tissues were isolated and analyzed for ceramide levels by the DAG kinase assay. Asterisks indicate significant differences compared to the respective time points of normoxia mice at 0h (n=6) (mean s.d.; **P < 0.01, ***P < 0.001). The noted experiments are representative of a minimum of three similar evaluations.
Dose Response of Mouse Alveolar Epithelial Cells Exposed to Ceramide
Experiments were first conducted to define the effects of ceramide exposure on mouse alveolar epithelial cells cultures. Mouse alveolar epithelial cells were isolated as described before (Ganter, et al., 2008, Barker, et al., 2006) and were grown until confluence was reached and then exposed to varying concentrations of ceramide (1, 10, 20, 30, 40 and 50μM) for 12 h. Cell survival was assessed based on Trypan blue exclusion assay. We observed a cell survival ranging from 82-98% after 30 μM treatment (Fig. 2A). Thus, this concentration of ceramide was used for all subsequent experiments to provide a maximum dynamic range for quantifying protective versus harmful inflammatory responses.
Fig 2. Effect of ceramide on mouse alveolar epithelial and macrophages.

(A) Dose-response curve showing survival of alveolar epithelial type-II cells (AT-II) after exposure to varying concentrations of ceramide ranging from 10 μM to 50 μM for 12 h. Cell viability was analyzed by using trypan blue exclusion. Mouse alveolar macrophages were isolated and treated with 30 μM of ceramide for the indicated time points and levels of (B) IL-1β, (C) IL-6 and (D) TNF-α were analyzed by ELISA. Asterisks indicate significant differences compared to the respective time points of control and ceramide treatment (mean s.d.; **P < 0.01). The noted experiments are representative of a minimum of three similar evaluations.
IL-1β is the early cytokine produced in ceramide treated alveolar macrophages
Cytokine release and macrophage activation are an early event in hyperoxia induced lung injury. However, the mechanism of ceramide induced cytokine release by macrophages is not known. To determine the effect of ceramide on macrophages, we isolated mouse lung macrophages and treated with ceramide, and monitored IL-1β, TNF-α and IL-6 at various time points after treatment. As shown in Fig. 2B, our results indicate that IL-1β reaches significantly high levels at 12h after ceramide treatment and maintains a steady state until 36 hours. The increase in IL-1β is statistically significant (p<0.01) compared with control treatment. High levels of TNF-α and IL-6 were observed at 24h after ceramide treatment. However, the 12 hour time point the levels of these cytokines did not change (control vs. ceramide). These results suggest that IL-1β is a key early cytokine produced by macrophages, while IL-6 and TNF-α are secreted at later time points (Fig. 2B-D).
Ceramide induces inflammasome activation
Our recent studies suggest that caspase-1 activation and IL-1β cleavage in acute lung injury is mediated by inflammasome activation (Kolliputi, et al., 2010). However the role of ceramide in inflammasome activation is not known. To determine the effect of ceramide on inflammasome activation, we evaluated caspase-1 activation in THP-1 cells treated with ceramide (30μM) and analyzed its expression by Western blot analysis, using monoclonal anti caspase-1 antibody (Cell Signaling Technology, Inc, Danvers, MA). We found that exposure to ceramide leads to enhanced caspase-1 cleavage (Fig. 3A) compared with controls. To understand the mechanism for ceramide induced release of cytokines from the macrophages, we probed for NALP-3 which is an important component of inflammasome and triggers the inflammatory response. Macrophages were treated with ceramide with or without NALP-3 shRNA. As shown in Fig. 3A, ceramide increased the cleaved caspase-1 in control group (scramble shRNA). However, silencing the NALP-3 gene abrogated ceramide-induced caspase-1 cleavage (Fig. 3A) compared with untreated controls. These results suggest that ceramide induced caspase-1 cleavage through NALP-3 inflammasome.
Fig 3. Inflammasome silence blunted ceramide induced cytokine production in THP-1.

(A)Inflammasome activation was assessed by analyzing caspase-1 cleavage using Western blot analysis. shRNA silencing of NALP-3 abrogated ceramide-induced caspase-1 cleavage (upper panel) as well as secretion of IL-1β in ceramide treated (30 μM) THP-1 cells (lower panel, graph). ATII cells were grown in 24-well transwell plates until the cellular monolayers reached confluence. THP-1 cells were added to the appropriate ATII monolayers, cocultured for 6 h, and exposed to ceramide for 12 h. In some experiments, THP-1 cells were transfected with control shRNA or NALP-3 shRNA and then added to a monolayer of ATII cells. The media contents were collected and (B) IL-1β, (C) TNF-α and (D) IL-6 proinflammatory cytokines were analyzed in extracellular media by ELISA. Asterisks indicate significant differences compared to the AT-II (***P < 0.001) and compared to control shRNA (mean s.d.; **P < 0.01). The noted experiments are representative of a minimum of three similar evaluations.
Inflammasome silencing blunted ceramide induced cytokine production by THP-1 cells
Based on our Western blot analysis we identified the role of NALP-3 in ceramide induced inflammatory response. To test the possible in vivo effects, we employed a co-culture model in which ATII and THP-1 interactions were assessed. As shown in Fig. 3B, ceramide treatment caused a significant increase in IL-1β in the control shRNA group and silencing NALP-3 gene in THP-1 cells, caused a significant decrease in ceramide induced IL-1β release. These results suggest that ceramide induced IL-1β release is regulated via NALP-3 mediated mechanism. We also monitored IL-6 and TNF-α in control shRNA treated THP-1 cells and in NALP-3 silenced group. We found that the ceramide induced increase in TNF- α and IL-6 release was attenuated by NALP-3 silencing (Fig.3C-D). Taken together, these results suggest that inflammasome mediates ceramide-induced cytokine production in THP-1.
Inflammasome silencing inhibited ceramide induced transepithelial albumin flux in alveolar epithelial type II cells
We then asked the question if ceramide-mediated epithelial permeability occurs through inflammasome activation? To determine whether, ceramide contributes to epithelial cell permeability, we studied epithelial permeability in THP-1 and AT-II co-culture.
As shown in Fig. 4, epithelial permeability was observed in co-cultures of AT-II and control sh RNA transfected THP-1 cells after ceramide treatment. However, silencing of the NALP-3 gene in the THP-1 macrophages significantly decreased the trans-epithelial albumin permeability. Taken together these results clearly suggest that inflammasome mediates ceramide-induced transepithelial albumin flux in alveolar epithelial type II cells.
Fig 4. Inflammasome silencing inhibited ceramide induced transepithelial albumin flux in alveolar epithelial type II cells.

(A) ATII cells were grown in 24-well transwell plates until the cellular monolayers reached confluence. THP-1 cells were added to the appropriate ATII monolayers, cocultured for 6 h, and exposed to ceramide for 12 h. In some experiments, THP-1 cells were transfected with control shRNA or NALP-3 shRNA and then added to a monolayer of ATII cells. Transepithelial albumin flux across each monolayer was determined by the addition of [125I]-labeled human serum albumin to each upper compartment. The contents from the lower compartment were collected and counted in a Wizard γ-counter. *p<0.01 relative to AT-II cells; #p < 0.05, relative to control shRNA. (B) Effect of inflammasome silencing on TER in AT-II and THP-1 cells. ATII cells were grown in 24-well transwell plates until the cellular monolayers reached confluence. THP-1 cells were added to the appropriate ATII monolayers, cocultured for 6 h, and exposed to ceramide for 12 h. In some experiments, THP-1 cells were transfected with control shRNA or NALP-3 shRNA and then added to a monolayer of ATII cells. TER was measure by an epithelial voltammeter. *p<0.01 relative to AT-II cells; **p < 0.05, relative to NALP-3 shRNA. The noted experiments are representative of a minimum of three similar evaluations.
Inflammasome silencing in THP-1 cells blocks the ceramide suppressed TER in alveolar epithelial type II cells and THP-1 co culture
Since epithelial permeability is known to influence monolayer bioelectric properties (Mairbaurl, et al., 2002), we monitored Trans epithelial resistance (TER) by using a voltammeter as described earlier (Mairbaurl, et al., 2002).This allowed us to perform the functional bioelectric measurements. Alveolar epithelial cells grown in monolayer or with THP-1 were transfected with control shRNA or NALP-3 shRNA and TER was measured in the presence and absence of ceramide. Data from these studies suggest that monolayer of alveolar epithelial cells exposed to ceramide suppresses TER to lesser extent. However, ceramide treated co-cultures of THP-1 with epithelial cells showed significant suppression in the TER. Interestingly, we found ceramide suppression of TER was blocked by silencing NALP-3 under co-cultured conditions. These results suggest that inflammasome mediates ceramide-suppressed TER occurred though inflammation activation (Fig. 4B).
Discussion
In the present study we investigated the mechanisms for hyperoxia induced lung injury with a specific focus on the role of ceramide mediated triggering of inflammasome and its downstream targets. In this study, we evaluated the injured lung tissue from hyperoxic mice and measured the levels of ceramide. The results presented in this study show that hyperoxia induced an increase in lung ceramide levels. The increased levels of ceramide are consistent with damaging lung function as evidenced from the functional components such as alveolar protein leak and pulmonary edema in vivo. Based on our findings in the mouse model of acute lung injury, we investigated the specific link between inflammasome formation and IL-1β processing induced by ceramide. To test the specific role of ceramide, we utilized an in vitro model and found that ceramide increased alveolar epithelial protein permeability. These studies established that ceramide causes lung injury under cell culture conditions and in mouse model. To establish the specific mechanism, we evaluated the role of NALP-3 an important component of inflammasome, a multi protein complex involved in triggering acute lung injury response. By using shRNA based technology for silencing the NALP-3 component within the inflammasome complex, we abrogated ceramide-induced secretion of proinflammatory cytokines and abolished alveolar epithelial protein permeability. These results suggest that ceramide plays a key role in acute lung injury, suggesting the role of proinflammatory cytokines and alveolar epithelial protein permeability mediated by inflammasome activation.
Our studies are the first showing the NALP-3inflammasome formation; we have also established that ceramide causes cleavage of IL-1β and is directly associated with alveolar epithelial protein permeability. Recent reports suggest that IL-1β causes both alveolar epithelial and vascular endothelial permeability (Ganter, et al., 2008) which are important events in alveolar edema, and a key component of lung injury. Activated ceramide is known to induce cytokine production, resulting in alveolar permeability and edema (Barnes, 2004, Goggel, et al., 2004, Uhlig, et al., 2005). Based on our results it is highly likely that ceramide induces inflammasome activation triggered by IL-1β cleavage leading to increased alveolar permeability and cytokine production resulting in pulmonary edema and damage. Our data supports this hypothesis, and our results indicate that the inhibition of the inflammasome by using shRNA abrogates ceramide-induced secretion of IL-1β and ameliorates lung epithelial permeability. Thus, our results suggest that inflammasome activation is required for IL-1β cleavage and epithelial permeability in response to ceramide. The findings reported in this study have potential clinical relevance. The transient blockade of the inflammasome can provide new causal therapies for pulmonary edema resulting from ALI, which is a major cause for morbidity and mortality in critically ill patients that are currently largely untreatable.
After establishing the role of NALP-3 inflammasome in ceramide mediated hyperoxic lung we next evaluated the downstream mediators that participate in lung inflammation. Our results suggest the role of activated caspase-1 specifically regulating NALP-3 inflammasome, which in turn releases IL-1β and initiates several downstream events that may amplify inflammatory responses by releasing other cytokines such as IL-6 and TNF-α in ALI. Our studies have found that inflammasome end product IL-1β have pathological function by altering epithelial barrier dysfunction. However, ablation of inflammasome blocks the IL-1β mediated alveolar permeability. These results suggest that under oxidative stress conditions, ceramide is released which causes inflammasome activation, which may be an adaptive immune response. In the present study we provide an understanding for the role of inflammasome in hyperoxia induced lung injury with specific focus on ceramide and its detrimental effects. Furthermore, our study provides a detailed view about the adaptive immune system in macrophages and how they contribute to the alveolar damage in ALI. Our studies show that inflammasome activation in macrophages influence inflammatory cytokine secretion and alveolar permeability. In addition, although the inflammasome has been investigated extensively in cells of the innate immune system, until now, there are no studies on functional effects of inflammasome on other resident cells. Here in our study, we analyzed the functional effects of inflammasome activation on epithelium, which is one of the key effectors in lung injury.
Inflammation is a major contributor to the pathogenesis of various lung diseases including ALI (Matthay and Idell, 2010, Matthay and Zemans, 2011). Previous studies suggest that inhibition of the inflammasome complex suppresses the inflammatory response after thromboembolic stroke in mice, and neutralizing Abs against the inflammasome successfully inhibits inflammasome activation in multiple models of CNS injury (Abulafia, et al., 2009). In our study, we show that inhibition of inflammasome suppresses ceramide inflammatory response. Therefore, inhibition of the inflammasome in ALI is a novel therapeutic strategy to inhibit caspase-1 activation and thereby protecting lung tissue from ceramide mediated damage in acute lung injury. This approach would also be useful in preventing ALI, which is one of the most common reasons for admission to critical care units.
Since ceramide release is an early event in ALI, we believe that therapeutic approaches for ameliorating disease progression can be a useful strategy. We demonstrate that ceramide activates NALP-3 complex formation that leads to caspase-1 activation and release of IL-1β production at early stage (Fig.5). Once IL-1β is released by inflammasome activation it damages the epithelium and leads to TNFα and IL-6 production, this is consistent with the time-course effects observed after ceramide treatment (Fig.2). The cytokine burst leads to cytokine-dependent inflammation and apoptosis with severe inflammation and injury at late stages of ALI. The pattern of injury observed in the mouse model (Fig.1) using hyperoxic insult is reflective of the ceramide mediated injury demonstrated by using the co-culture model.
Fig 5. Proposed scheme for the role of inflammasome in ceramide-induced alveolar permeability and pulmonary edema.

In hyperoxia, ceramide accumulation causes inflammasome activation and IL-1β production in macrophages. IL-1β further causes epithelial damage and releases other cytokines such as IL-6 and TNF-α which causes epithelial permeability and pulmonary edema. Silencing of NALP-3 within the inflammasome caused blunting of the cytokine production which leads to suppression of permeability and pulmonary edema.
In summary, we hypothesize that in early stages of ALI, ceramide activates NALP-3 complex formation that leads to caspase-1 activation and release of IL-1β production. Once IL-1β is released by inflammasome activation, it damages the epithelium and leads to production of TNF-α and IL-6. The cytokine burst further leads to cytokine-dependent inflammation, apoptosis and injury at late stages of ALI. This can be particularly relevant in ALI, in which inflammatory processes increase from the early stages to the late stages of ALI ((Matthay and Idell, 2010, Matthay and Zemans, 2011)). The cytokine burst also causes epithelial permeability, epithelial barrier dysfunction, and cell death. From a clinical perspective, the inhibition of inflammasome by pharmacologic approaches that disrupt inflammasome complex formation can provide a strategy for the protection of lung tissue from cellular injury during acute or chronic illness associated with ALI syndromes.
Acknowledgements
This work was supported by National Institutes of Health Grant R01 HL105932 and American Heart Association National Scientist Development Grant 09SDG2260957 to NK. The College of Pharmacy Dean’s Research Fund was also provided to SMT.
Contract grant sponsor: NIH, AHA
Contract grant numbers: R01 HL105932 and 09SDG2260957
REFERENCES
- Matthay MA, Idell S. Update on acute lung injury and critical care medicine 2009. Am J Respir Crit Care Med. 2010;181:1027–1032. doi: 10.1164/rccm.201001-0074UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Ceramide lances the lungs. Nat Med. 2004;10:130–131. doi: 10.1038/nm0204-130. [DOI] [PubMed] [Google Scholar]
- Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schutze S, Gulbins E, Uhlig S. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. 2004;10:155–160. doi: 10.1038/nm977. [DOI] [PubMed] [Google Scholar]
- Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812. doi: 10.1161/CIRCRESAHA.107.161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolliputi N, Shaik RS, Waxman AB. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J Immunol. 2010;184:5819–5826. doi: 10.4049/jimmunol.0902766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. Molecular mechanisms of ceramide-mediated telomerase inhibition in the A549 human lung adenocarcinoma cell line. J Biol Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- Milhas D, Clarke CJ, Hannun YA. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett. 2010;584:1887–1894. doi: 10.1016/j.febslet.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldkorn T, Filosto S. Lung injury and cancer: Mechanistic insights into ceramide and EGFR signaling under cigarette smoke. Am J Respir Cell Mol Biol. 2010;43:259–268. doi: 10.1165/rcmb.2010-0220RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Zhang Y, Delikat S, Mathias S, Basu S, Kolesnick R. Phosphorylation of Raf by ceramide-activated protein kinase. Nature. 1995;378:307–310. doi: 10.1038/378307a0. [DOI] [PubMed] [Google Scholar]
- Mathias S, Pena LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335(Pt 3):465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bismarck P, Wistadt CF, Klemm K, Winoto-Morbach S, Uhlig U, Schutze S, Adam D, Lachmann B, Uhlig S, Krause MF. Improved pulmonary function by acid sphingomyelinase inhibition in a newborn piglet lavage model. Am J Respir Crit Care Med. 2008;177:1233–1241. doi: 10.1164/rccm.200705-752OC. [DOI] [PubMed] [Google Scholar]
- Carpinteiro A, Dumitru C, Schenck M, Gulbins E. Ceramide-induced cell death in malignant cells. Cancer Lett. 2008;264:1–10. doi: 10.1016/j.canlet.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Lavnikova N, Prokhorova S, Helyar L, Laskin DL. Isolation and partial characterization of subpopulations of alveolar macrophages, granulocytes, and highly enriched interstitial macrophages from rat lung. Am J Respir Cell Mol Biol. 1993;8:384–392. doi: 10.1165/ajrcmb/8.4.384. [DOI] [PubMed] [Google Scholar]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through NALP-3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–8. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- Barker GF, Manzo ND, Cotich KL, Shone RK, Waxman AB. DNA damage induced by hyperoxia: quantitation and correlation with lung injury. Am J Respir Cell Mol Biol. 2006;35:277–288. doi: 10.1165/rcmb.2005-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taga S, Carlier K, Mishal Z, Capoulade C, Mangeney M, Lecluse Y, Coulaud D, Tetaud C, Pritchard LL, Tursz T, Wiels J. Intracellular signaling events in CD77-mediated apoptosis of Burkitt’s lymphoma cells. Blood. 1997;90:2757–2767. [PubMed] [Google Scholar]
- Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, Zhang X, Matthay MA, Ware LB, Homer RJ, Lee PJ, Geick A, de Fougerolles AR, Elias JA. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med. 2006;12:1286–1293. doi: 10.1038/nm1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V, Choo-Wing R, Homer RJ, Elias JA. Increased hyperoxia-induced mortality and acute lung injury in IL-13 null mice. J Immunol. 2007;178:4993–5000. doi: 10.4049/jimmunol.178.8.4993. [DOI] [PubMed] [Google Scholar]
- Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, Waxman AB, Elias JA. IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest. 2000;106:783–791. doi: 10.1172/JCI9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q, Zhu Z, Zhong M, Nakayama K, Nakayama KI, Homer R, Elias JA. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115:1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lin L, Lee SJ, Mo L, Cao J, Ifedigbo E, Jin Y. Deletion of caveolin-1 protects hyperoxia-induced apoptosis via survivin-mediated pathways. Am J Physiol Lung Cell Mol Physiol. 2009;297:L945–53. doi: 10.1152/ajplung.00081.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairbaurl H, Mayer K, Kim KJ, Borok Z, Bartsch P, Crandall ED. Hypoxia decreases active Na transport across primary rat alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2002;282:L659–65. doi: 10.1152/ajplung.00355.2001. [DOI] [PubMed] [Google Scholar]
- Uhlig S, Goggel R, Engel S. Mechanisms of platelet-activating factor (PAF)-mediated responses in the lung. Pharmacol Rep. 2005;57(Suppl):206–221. [PubMed] [Google Scholar]
- Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]